
Synthesis and biological evaluation of santacruzamate A and analogs as potential anticancer agents†
Qi Liu‡
ab,
Wenhua Lu‡a,
Mingzhe Maa,
Jianwei Liaoa,
A. Ganesanc,
Yumin Hua,
Shijun Wen*ab and
Peng Huanga
aSun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, 651 Dongfeng East Road, Guangzhou 510060, China. E-mail: wenshj@sysucc.org.cn
bSchool of Pharmaceutical Sciences, Sun Yat-sen University, 132 Waihuan East Road, Guangzhou 510006, China
cSchool of Pharmacy, University of East Anglia, Norwich NR4 7TJ, UK
First published on 26th November 2014
Abstract
Santacruzamate A, a recently discovered natural product from a Panamanian marine cyanobacterium Symploca sp., features a similar structure to the clinically used histone deacetylase (HDAC) inhibitor vorinostat (SAHA). We have synthesized the natural product and a small set of analogues for SAR studies. To our surprise, the synthetic natural product santacruzamate A (1a) and the analogues did not show an obvious inhibition even at 2 μM in HDAC enzyme assays while the IC50 value was 0.12 nM in the original report. However, a novel compound, 5, containing a terminal thiourea motif was found to inhibit the growth of malignant cells at submicromolar concentrations. Moreover, 5 was not cytotoxic to normal human colonic epithelial cells CCD841, suggesting that its cytotoxicity was specific to cancer cells. Further investigation indicated that the compound induced apoptosis, affected cell cycle progression and increased ROS production. We believe its mechanism of action is unrelated to HDAC inhibition and the original activity reported for santacruzamate needs to be reevaluated.
Tumors, the result of abnormal cells with uncontrolled, rapid and pathological proliferation, cause one of the most formidable afflictions globally.1,2 Apart from the use of surgical treatment and irradiation, chemotherapy still remains the main therapeutic strategy to treat cancer.3,4 However, one of the major hurdles in cancer chemotherapy is attributed to the prevalence of drug and multidrug resistance and the need for selectivity against normal cells.5–7 Therefore, considerable efforts have been made on the design and discovery of new anticancer agents, focusing on the search of novel chemical entities for the successful treatment of cancer.8,9
Nature has been continuously providing humans with important leads and natural product medicines for the treatment of a wide spectrum of diseases.10–12 Natural products have been a particularly important source of anticancer chemotherapeutic medicines including taxanes, Vinca alkaloids and camptothecin that act upon the mitotic spindle.13 This trend is likely to continue as natural products are identified that modulate specific signaling pathways in cells. One example is the relatively new field of epigenetics relating to chromatin modelling via structural modifications of the DNA and histone proteins. A variety of natural products have already been reported that inhibit the enzymes involved in epigenetics.14 Among these, the histone deacetylase (HDAC) family of enzymes has received much attention. HDACs are the enzymes that hydrolyse acetyl-lysine amino acid residues in proteins back to lysine and play crucial roles in diverse cellular functions, while their overexpression or mutation is widely observed in cancer cells.15–17 A number of potent natural product HDAC inhibitors such as trichostatin A and apicidin are used as biological tools while the depsipeptide FK228 (romidepsin) has received FDA approval for the treatment of cutaneous T-cell lymphoma (CTCL).
Recently, Balunas and co-workers reported the isolation of santacruzamate A (1a) from a marine Panamanian cyanobacterium resembling the genus Symploca.18 Santacruzamate A shares some structural similarity with the synthetic HDAC inhibitor vorinostat (SAHA), the first clinically approved drug in this class (Fig. 1). It was reported that santacruzamate A specifically inhibited the isoform HDAC2 with an IC50 of 0.12 nM. This was a surprising result given the established SAR of HDAC inhibitors in which a zinc-binding group, such as the hydroxamic acid in vorinostat, is important for reversible binding to the enzyme active site.19 Instead, santacruzamate A contains a carbamate and amide-functional groups that have not been previously associated with potent HDAC inhibition. The natural product azumamide A, for example, contains a carboxamide zinc-binding group and is only micromolar in HDAC inhibition.20,21 Furthermore, the high selectivity of santacruzamate A for HDAC2 was intriguing as SAHA itself is a non-selective inhibitor active against both Class I and Class II HDAC isoforms. With this background, it was of interest to synthesize santacruzamate A as well as a series of analogues to investigate the structure–activity relationships (SAR) of this new lead or the development of anticancer agents. This has led to the identification of a potent cytotoxic compound 5 that we nevertheless believe acts by a HDAC-independent mechanism of action.
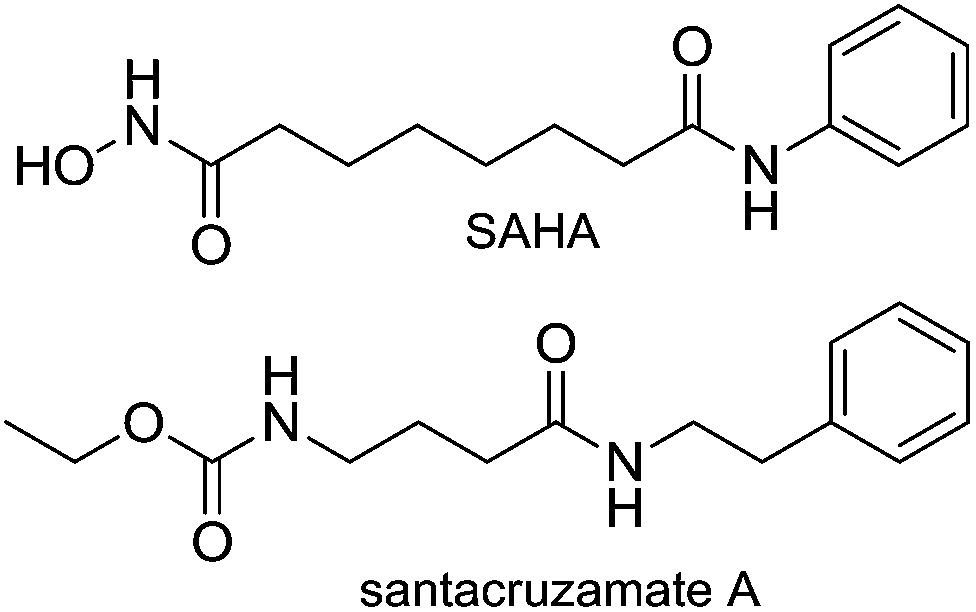 | ||
| Fig. 1 The structures of santacruzamate A and SAHA. | ||
To investigate the importance of the linker in santacruzamate A between the carbamate and amide structural features, our SAR strategy was to move around the position of the amide and also to replace the terminal ethoxycarbonyl group with other bioisosteric functional groups. Thus, a series of compounds were designed and prepared alongside with the natural product santacruzamate A itself (1a) (Fig. 2).
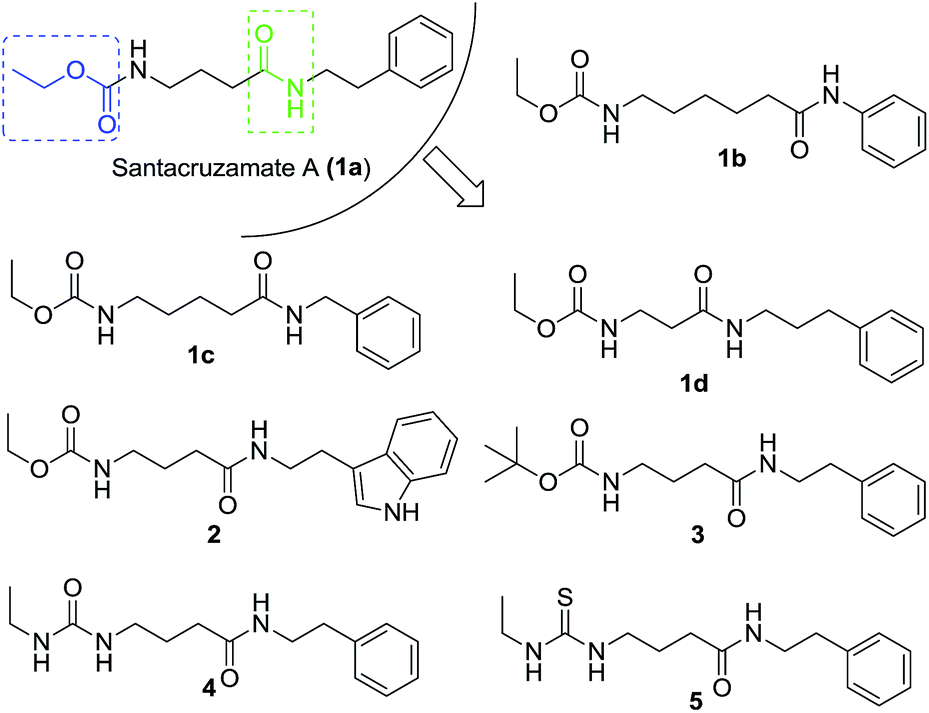 | ||
| Fig. 2 The strategy to design analogues of santacruzamate A. | ||
The synthetic route to obtain these compounds is shown in Scheme 1. Conventional acylation of the terminal amino groups of commercial available amino acids 6 with ethyl chloroformate gave compounds 7a–d, while protection of 6c with di(tert-butyl) carbonate (Boc2O) afforded 8. Amidation of the carboxylic acid groups of 7a–d and 8 with amines or aniline afforded the desired target products 1a–d, 2 and 3. After removal of the Boc group from 3 under acidic conditions, the obtained free amine 9 underwent subsequent treatment with either isocyanatoethane or ethyl isothiocyanate to give two additional analogues 4 and 5.
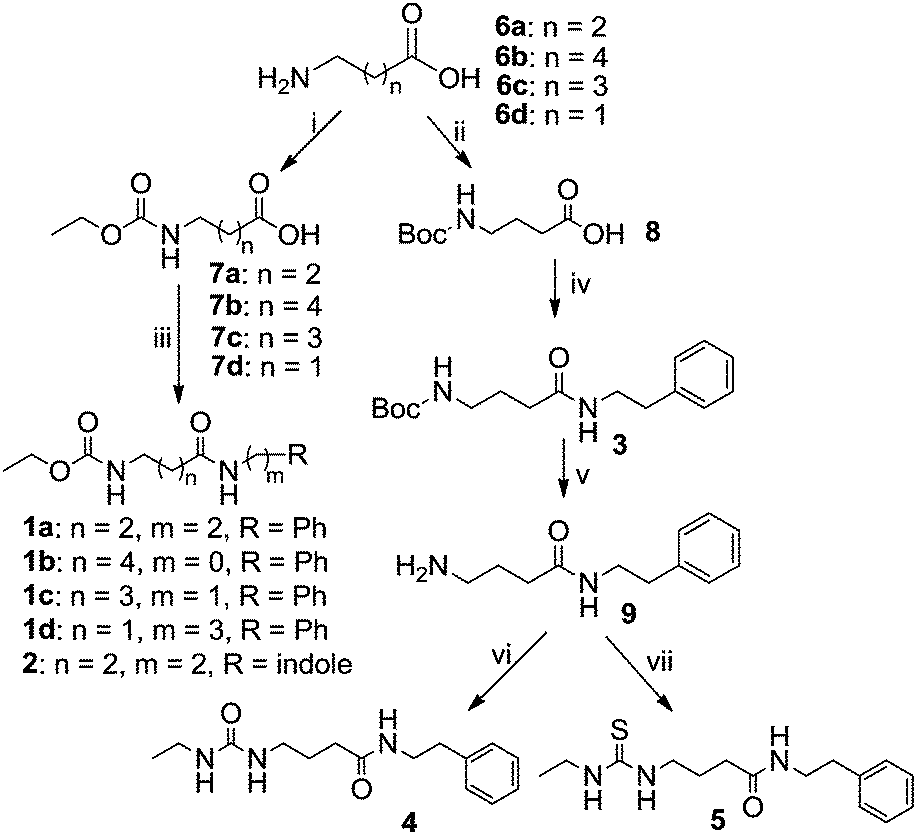 | ||
| Scheme 1 Synthesis of santacruzamate A (1a) and the analogues. Reagents and conditions: (i) ethyl chloroformate, K2CO3, THF/H2O, 0 °C to rt., (ii) Boc2O, NaOH, THF/H2O, 0 °C to rt., (iii) amine, Et3N, EDCI, cat. DMAP, CH2Cl2, 0 °C to rt., (iv) phenethylamine, Et3N, EDCI, cat. DMAP, CH2Cl2, 0 °C to rt., (v) TFA, CH2Cl2, rt., (vi) isocyanatoethane, THF, rt., (vii) isothiocyanate, THF, rt. Note: EDCI, 1-ethyl-3-(3-dimethyllaminopropyl)carbodiimide hydrochloride; DMAP, 4-dimethylaminopyridine. | ||
After the synthesis of the designed compounds 1a–d, and 3–5, they were subsequently screened for their cellular biological activity. To our surprise, synthetic santacruzamate A, i.e. 1a did not show cytotoxicity against human colon cancer cells HCT-116 even at 100 μM while most of the other synthetic analogues were inactive (Table 1). However, compound 5 inhibited the growth of HCT-116 cells and human myeloblastic leukemia cells ML-1 with IC50 values of 6.0 and 9.4 μM. To our delight, 5 was not cytotoxic to normal cells (CCD841) even at 100 μM. Indeed, it is high desirable to obtain a compound with a high selectivity to kill malignant cancer cells because most of clinical anticancer drug also kill normal cells during therapeutical treatment. For example, SAHA was very cytotoxic to CCD841 cells at 20 μM although with its lower IC50 value to cancer cells.
| Compound | IC50a (μM) ± SEM | ||
|---|---|---|---|
| HCT-116 | ML-1 | CCD841 | |
| a IC50 is the drug concentration effective in inhibiting 50% of the cell growth measured by the MTS assay. NT, not tested. | |||
| 1a | >100 | >100 | NT |
| 1b | 90.8 ± 6.9 | >100 | NT |
| 1c | >100 | >100 | NT |
| 1d | 94.3 ± 13.8 | >100 | NT |
| 2 | >100 | >100 | NT |
| 3 | 86.0 ± 9.0 | >100 | NT |
| 4 | >100 | 79.5 ± 3.8 | NT |
| 5 | 6.0 ± 1.2 | 9.4 ± 3.8 | >100 |
| SAHA | 1.4 ± 0.0 | 2.9 ± 1.1 | 20.8 ± 0.17 |
Next, we performed mechanistic enzyme assays against both total HDACs isolated from cell lysates and the individual recombinant isoform HDAC2, using SAHA as a positive control. When SAHA showed the expected activity with IC50 79.7 nM against HDAC2 (Fig. S1†), none of our synthesized compounds showed significant HDAC2 inhibition at the concentration of 2 μM while the IC50 value of santacruzamate A was reported to be 0.12 nM.18 The assay was repeated three times on different dates, and the results were consistent. We have also double checked the spectrum of 1H NMR of our synthetic santacruzamate A (1a), and verified that our synthetic sample matches the reported data for the natural product (Fig. 3). To exclude a possibility that 1a might be decomposed under the enzymatic assay conditions, a solution of 1a in DMSO was diluted in the buffer employed for the enzymatic assay and incubated for several hours. The sample was finally taken for mass spectroscopy detection, and the clean major peak (m/z 301.0) corresponded to 1a, indicating that 1a tolerated the enzymatic assay conditions (Fig. S2†). These solid results lead us to believe that the original report need to be further reexamined.
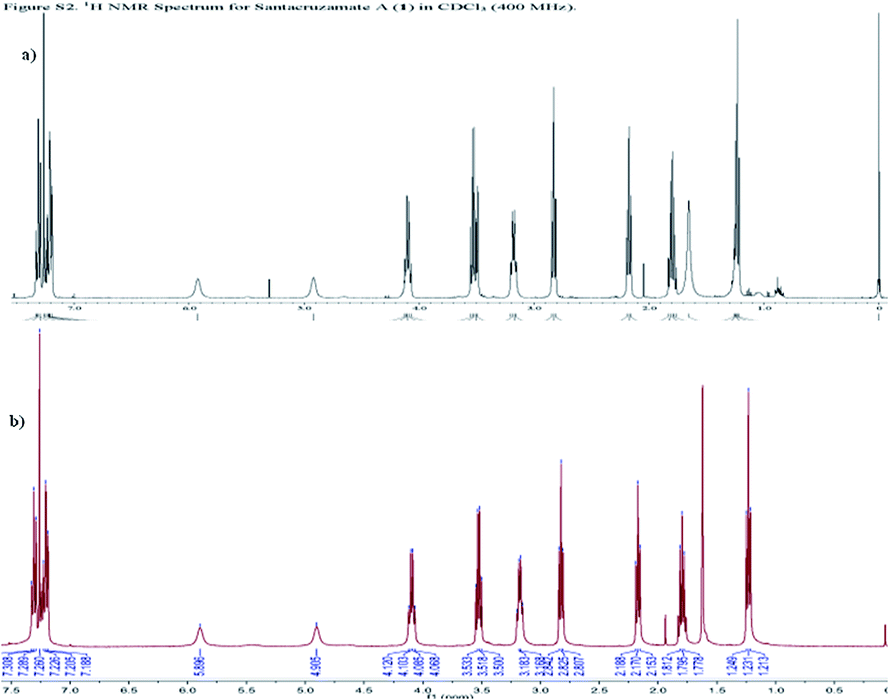 | ||
| Fig. 3 The 1HNMR spectra of (a) the reported santacruzamate A and (b) our synthetic 1a. | ||
While the mechanism of action of our compound 5 does not involve HDAC inhibition, further studies were carried out to profile its biological activity. At a concentration of 5 μM, 5 was able to effectively suppress colony formation of HCT-116 cells in a concentration dependent manner (Fig. 4) and to induce cell cycle arrest at the G2/M phase of HCT-116 cells but not ML-1 cells (Fig. 5).
 | ||
| Fig. 4 Inhibition of colony formation of HCT-116 cells by 5. | ||
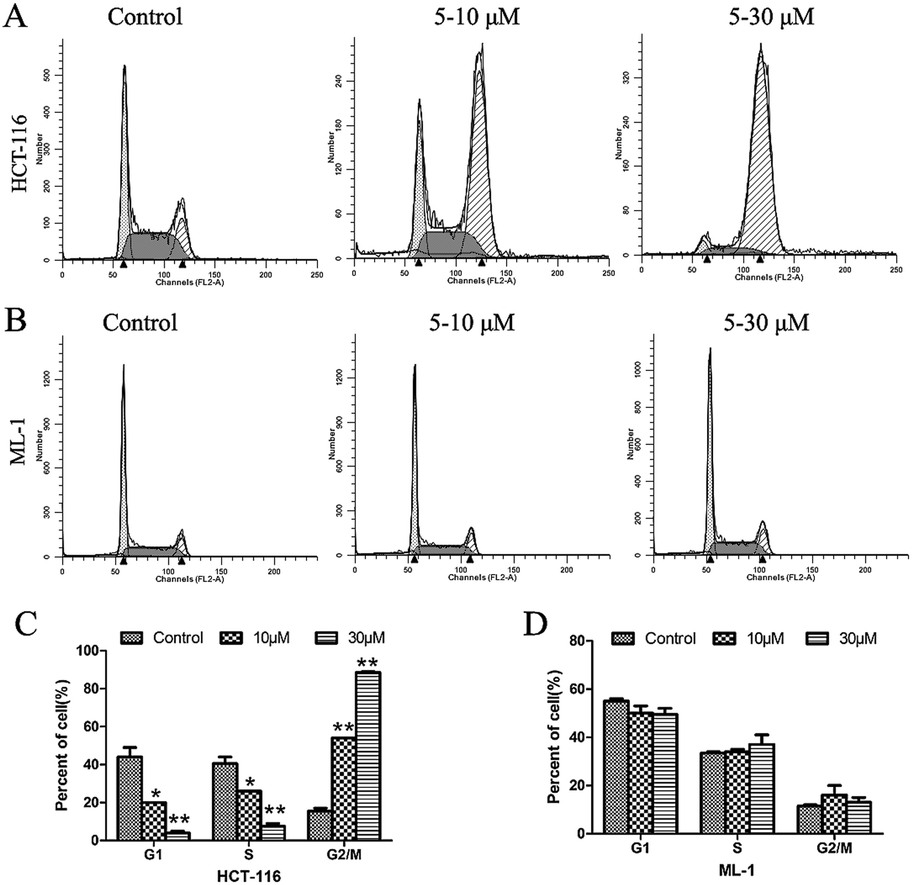 | ||
| Fig. 5 The effect of 5 on cell cycle progression of (A) HCT-116 cells and (B) ML-1 cells; (C) and (D) the normalization of the effects above. | ||
We then further explored whether this inhibition of cell growth and cell cycle arrest by 5 was attributed to the induction of apoptosis. Annexin V/PI double-staining assay was used to study whether 5 could directly induce apoptotic cell death in HCT-116 and ML-1 cells (Fig. 6). The results indicated that 5 significantly increased the percentage of apoptotic cells (Annexin-V-positive) in a dose-dependent manner. No obvious change was observed in necrotic cells (only PI stained) as compared to control at 48 h (data not shown).
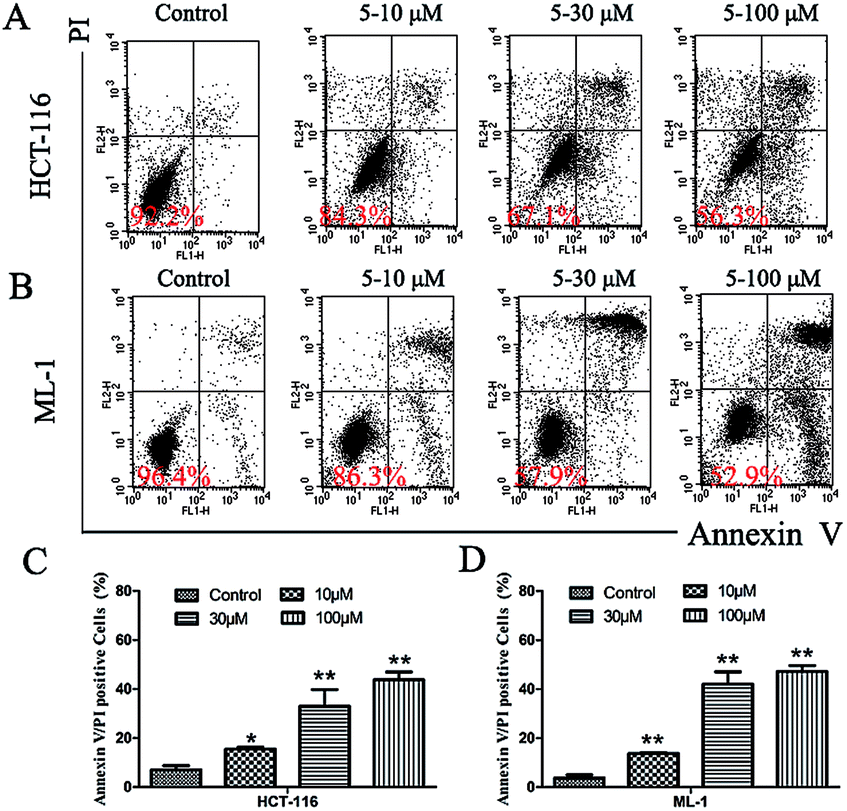 | ||
| Fig. 6 Appotosis of (A) HCT-116 and (B) ML-1 cells induced by 5; (C) and (D) the normalization of the induced appoptosis above. | ||
It has been widely recognized that increased endogenous reactive oxygen species (ROS) generation can selectively eliminate cancer cells, mainly by raising oxidative stress over the threshold of toxicity to abnormal cancer cells.22 Recently, Schreiber and co-workers have discovered that a natural product piperlongumine selectively killed cancer cells by targeting the stress response to ROS.23 Thus, it was of our interest to examine ROS level in HCT-116 and ML-1 cells treated with 5 (Fig. S3†). The data indicated that a treatment with 5 at 10 μM induced a significant increase in ROS levels both in HCT-116 and ML-1 cells (P < 0.05). Our results implied that selective killing of cancer cells but not normal cells by 5 might result from the ROS generation. Further study to investigate its exact mechanism is under way.
Conclusions
In summary, a novel series of compounds were designed and synthesized based on the natural product santacruzamate A. The SAR study demonstrated that most of these analogues as well as synthetic santacruzamate A showed weak cytotoxicity against the two tested cancer cell lines, HCT-116 and ML-1. It is noteworthy that synthetic santacruzamate A did not inhibit either total HDACs or HDAC2 in enzyme assays. While this is in stark contrast to the original publication, it is consistent with the known SAR of HDAC inhibitors and it is likely that the earlier report was in error.18 However, one analogue, 5 was found to exhibit anti-proliferative activity against HCT-116 (IC50 = 6.0 μM) and ML-1 (IC50 = 9.4 μM) cell lines. In addition, 5 did not cause damage to normal human colorectal cells, suggesting that 5 selectively killed the abnormal cancer cells. It is phenomenal that such a simple compound with a terminal thiourea has gained submicromolar anticancer activity with low toxicity to normal cells although the thiourea motif are reported in some biologically active compounds.24 Further studies showed that 5 inhibited colony formation of HCT-116, induced apoptosis of both cancer cells HCT-116 and ML-1, and arrested cell cycle of HCT-116 at G2/M phase. Finally, ROS generation was observed in both cancer cell lines HCT-116 and ML-1, implying that this might be the reason why 5 selectively eliminated cancer cells. Further study to investigate its exact mechanism of action is underway. Due to its simple structure and selective killing of cancer cells, 5 might provide a useful scaffold for anticancer drug development.Acknowledgements
The work was supported by Doctoral Program of Higher Education of China (grant 20110171120098), National Basic Research Program of China (973 Program grant 2012CB967004), and Guangdong Provincial International Project of Science and Technology (2013B051000034).Notes and references
- O. O. Fadeyi, S. T. Adamson, E. L. Myles and C. O. Okoro, Bioorg. Med. Chem. Lett., 2008, 18, 4172–4176 CrossRef CAS PubMed.
- S. A. F. Rostom, Bioorg. Med. Chem., 2006, 14, 6475–6485 CrossRef CAS PubMed.
- W. Liu, J. Zhou, T. Zhang, H. Zhu, H. Qian, H. Zhang, W. Huang and R. Gust, Bioorg. Med. Chem. Lett., 2012, 22, 2701–2704 CrossRef CAS PubMed.
- K. Juvale, J. Gallus and M. Wiese, Bioorg. Med. Chem., 2013, 21, 7858–7873 CrossRef CAS PubMed.
- C. P. Reyes, F. Muñoz-Martínez, I. R. Torrecillas, C. R. Mendoza, F. Gamarro, I. L. Bazzocchi, M. J. Núñez, L. Pardo, S. Castanys, M. Campillo and I. A. Jiménez, J. Med. Chem., 2007, 50, 4808–4817 CrossRef CAS PubMed.
- H. Y. Hung, E. Ohkoshi, M. Goto, K. F. Bastow, K. Nakagawa-Goto and K. H. Lee, J. Med. Chem., 2012, 55, 5413–5424 CrossRef CAS PubMed.
- X. Tang, X. Gu, Z. Ren, Y. Ma, Y. Lai, H. Peng, S. Peng and Y. Zhang, Bioorg. Med. Chem. Lett., 2012, 22, 2675–2680 CrossRef CAS PubMed.
- M. Nagaraju, E. Gnana Deepthi, C. Ashwini, M. V. P. S. Vishnuvardhan, V. Lakshma Nayak, R. Chandra, S. Ramakrishna and B. B. Gawali, Bioorg. Med. Chem. Lett., 2012, 22, 4314–4317 CrossRef CAS PubMed.
- D. Roell, T. W. Rösler, S. Degen, R. Matusch and A. Baniahmad, Chem. Biol. Drug Des., 2011, 77, 450–459 CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed.
- G. M. Cragg, P. G. Grothaus and D. J. Newman, J. Nat. Prod., 2014, 77, 703–723 CrossRef CAS PubMed.
- A. Ganesan, Curr. Opin. Chem. Biol., 2008, 12, 306–317 CrossRef CAS PubMed.
- A. Ganesan, The Impact of Natural Products Upon Cancer Chemotherapy, in Natural Products and Cancer Drug Discovery, ed. F. E. Koehn, Springer, Heidelberg, 2012, pp. 3–15 Search PubMed.
- F. L. Cherblanc, R. W. M. Davidson, P. D. Fruscia, N. Srimongkolpithak and M. J. Fuchter, Nat. Prod. Rep., 2013, 30, 605–624 RSC.
- M. Haberland, R. L. Montgomery and E. N. Olson, Nat. Rev. Genet., 2009, 10, 32–42 CrossRef CAS PubMed.
- P. Zhu, E. Martin, J. Mengwasser, P. Schlag, K. P. Janssen and M. Göttlicher, Cancer Cell, 2004, 5, 455–463 CrossRef CAS.
- A. Vaquero, R. Sternglanz and D. Reinberg, Oncogene, 2007, 26, 5505–5520 CrossRef CAS PubMed.
- C. M. Pavlik, C. Y. B. Wong, S. Ononye, D. D. Lopez, N. Engene, K. L. McPhail, W. H. Gerwick and M. J. Balunas, J. Nat. Prod., 2013, 76, 2026–2033 CrossRef CAS PubMed.
- M. Paris, M. Porcelloni, M. Binaschi and D. Fattori, J. Med. Chem., 2008, 51, 1505 CrossRef CAS PubMed.
- Y. Nakao, S. Yoshida, S. Matsunaga, N. Shindoh, Y. Terada, K. Nagai, J. K. Yamashita, A. Ganesan, R. W. M. van Soest and N. Fusetani, Angew. Chem., Int. Ed., 2006, 45, 7553–7557 CrossRef CAS PubMed.
- S. Wen, K. L. Carey, Y. Nakao, N. Fusetani, G. Packham and A. Ganesan, Org. Lett., 2007, 9, 1105–1108 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- L. Raj, T. Ide, A. U. Gurkar, M. Foley, M. Schenone, X. Li, N. J. Tolliday, T. R. Golub, S. A. Carr, A. F. Shamji, A. M. Stern, A. Mandinova, S. L. Schreiber and S. W. Lee, Nature, 2011, 475, 231–234 CrossRef CAS PubMed.
- (a) A. Solinas, H. Faure, H. Roudaut, E. Traiffort, A. Schoenfelder, A. Mann, F. Manetti, M. Taddei and M. Ruat, J. Med. Chem., 2012, 55, 1559–1571 CrossRef CAS PubMed; (b) A. Mishra and S. Batra, Curr. Top. Med. Chem., 2013, 13, 2011–2025 CrossRef CAS; (c) H. Nishiyama, M. Ono, T. Sugimoto, T. Sasai, N. Asakawa, S. Ueno, Y. Tominaga, T. Yaegashi, M. Nagaoka, T. Matsuzaki, N. Kogure, M. Kitajima and H. Takayama, Med. Chem. Commun., 2014, 5, 452–458 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of the compounds. See DOI: 10.1039/c4ra13889a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |