
1-Methyl-3-(propyl-3-sulfonic acid)imidazolium triflate supported on magnetic nanoparticles: an efficient and reusable catalyst for synthesis of mono- and bis-isobenzofuran-1(3H)-ones under solvent-free conditions†
Forouz Rastegari,
Iraj Mohammadpoor-Baltork*,
Ahmad R. Khosropour*,
Shahram Tangestaninejad,
Valiollah Mirkhani and
Majid Moghadam
Department of Chemistry, Catalysis Division, University of Isfahan, Isfahan 81746-73441, Iran. E-mail: imbaltork@sci.ui.ac.ir; arkhosropour@sci.ui.ac.ir; Fax: +98-031-36689732; Tel: +98-031-3793270
First published on 19th January 2015
Abstract
1-Methyl-3-(propyl-3-sulfonic acid)imidazolium triflate supported on magnetic nanoparticles ([HSO3PMIM]OTf–SiO2@MNPs) was prepared by immobilization of [HSO3PMIM]OTf onto the surface of silica-coated Fe3O4 nanoparticles and characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM), vibrating sample magnetometry (VSM), and FT-IR techniques. Efficient synthesis of mono- and bis-isobenzofuran-1(3H)-ones was performed in the presence of this catalyst under thermal conditions and MW irradiation. The catalyst could be easily separated by an external magnet and reused six times under thermal conditions and MW irradiation without significant loss of its activity.
Introduction
During recent years, ionic liquids (ILs) as environmentally-friendly reaction media or catalysts, have received increasing interest due to their distinctive physicochemical properties such as high thermal stability, negligible vapor pressure, non-flammability, and the ability to dissolve many organic and inorganic materials.1–3 In spite of extensive applications in organic reactions, they suffer from disadvantages such as difficult product separation and catalyst recovery and the use of large amounts of ILs. Such disadvantages limit their usefulness in large scale operation and lead to serious economical and environmental problems. However, these problems can be solved by immobilization ILs on solid supports such as organic polymers, silica, ZrO2, TiO2 and Al2O3 and mesoporous MCM-41.4–10 These heterogeneous IL catalysts have many advantages over their unsupported counterpart, such as separation, reusability, and the ability to provide practical conveniences in a continuous system. In addition, magnetic nanoparticles (MNPs) which have found many applications in the fields of biotechnology and biomedicine11–14 have emerged as viable alternatives to the conventional heterogeneous supports.15–25 The magnetic separation technology offers many advantages over conventional filtration and other purification methods in which the catalysts can be simply and efficiently recovered from reaction media with the external magnetic field.23,24,26–31 This can be considered as a green technology that avoids the consequences brought about by filtration steps.Isobenzofuran-1(3H)-ones are an important class of compounds possess a wide range of biological properties such as anti-bacterial, anti-convulsant, anti-HIV, anti-asthmatic, anti-tumor, anti-platelet activities32–37 as well as anesthesia prolongation, and PGF2α inhibitory properties.38–41 Because of the applications of these compounds in medicine and in the synthesis of natural products,42,43 several acidic and basic catalysts such as trifluoroacetic acid (TFA),44 trifluoromethanesulfonic acid (HOTf),45 montmorillonite K-10,46 NaOH,47 KOH,48 KF–Al2O3,49 silica-supported preyssler nanoparticles (H14[NaP5W30O110]/nano-SiO2)50 and ZrOCl2·8H2O51 have been reported for their synthesis.
In continuation of our research on the use of efficient catalytic systems in the synthesis of fine chemicals,52–57 herein, we wish to report a convenient method for the synthesis of mono- and bis-isobenzofuran-1(3H)-ones in the presence of 1-methyl-3-(propyl-3-sulfonic acid)imidazolium triflate supported on magnetic nanoparticles ([HSO3PMIM]OTf–SiO2@MNPs) as a highly reusable catalyst under thermal conditions and MW irradiation (Scheme 1). To the best of our knowledge, this is the first report on the application of MNPs supported IL catalyst in the synthesis of these important heterocyclic compounds.
 | ||
| Scheme 1 Synthesis of isobenzofuran-1(3H)-ones catalyzed by [HSO3PMIM]OTf–SiO2@MNPs. | ||
Results and discussion
The acidic IL supported on magnetic nanoparticles ([HSO3PMIM]OTf–SiO2@MNPs) was prepared according to the procedure shown in Scheme 2. The ionic liquid [HSO3PMIM]OTf was prepared by quaternization of 1-methyl-3H-imidazole with 1,3-propanesultone, followed by treatment with HOTf. The magnetite nanoparticles (MNPs) was easily prepared via the co-precipitation method described by Massart58 and protected with a layer of silica to prevent aggregation.59 The coating process was performed by ultrasonic suspending of MNPs in ethanol and added tetraethoxysilane (TEOS) to form a silica shell under basic conditions through a sol–gel method. Ultimately, a mixture of MNPs and [HSO3PMIM]OTf in dichloromethane was dispersed by sonication for 5 h. | ||
| Scheme 2 Preparation of [HSO3PMIM]OTf–SiO2@MNPs. | ||
The prepared catalyst was characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM), vibrating sample magnetometery (VSM) and FT-IR techniques. The XRD patterns of Fe3O4 magnetic nanoparticles and silica-coated Fe3O4 magnetic nanoparticles are shown in Fig. 1. These patterns show the characteristic peaks and relative intensity which are well-matched with those of standard Fe3O4 nanoparticles (JCPDS file no. 19-0629). Moreover, the broad peak from 2θ = 20° to 30° is consistent with an amorphous silica phase in the shell of the silica-coated Fe3O4 nanoparticles (Fig. 1a).60,61 The TEM image of [HSO3PMIM]OTf–SiO2@MNPs showed dark nano-Fe3O4 cores surrounded by grey silica shells (Fig. 2a). The TEM image of the reused catalyst reveals its stability during the reaction (Fig. 2b). The histogram of size distribution shows that the average diameter of nanoparticles is about 8–12 nm (Fig. 2c).
 | ||
| Fig. 1 X-ray diffraction pattern of: (a) SiO2@MNPs and (b) standard magnetite pattern (JCPDS no. 19-0629). | ||
 | ||
| Fig. 2 TEM images of: (a) [HSO3PMIM]OTf–SiO2@MNPs; (b) recovered [HSO3PMIM]OTf–SiO2@MNPs and (c) particle size distribution results for [HSO3PMIM]OTf–SiO2@MNPs. | ||
The magnetic properties of MNPs, SiO2@MNPs and [HSO3PMIM]OTf–SiO2@MNPs were measured at room temperature from −10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to +10000 Oe. The magnetization curves for these samples are shown in Fig. 3. The saturation magnetization (Ms) values of samples are 70, 35 and 20 emu g−1, respectively. The decreasing of Ms values from MNPs to [HSO3PMIM]OTf–SiO2@MNPs could be attributed to the coating of iron oxide magnetic nanoparticles with silica and IL. Furthermore, the magnetization curves of the magnetic nanoparticles display no hysteresis before and after functionalization. These results clearly demonstrate their superparamagnetic characteristics,58 which further reinforce by easy separation of these magnetic nanoparticles with an external magnetic field.
000 to +10000 Oe. The magnetization curves for these samples are shown in Fig. 3. The saturation magnetization (Ms) values of samples are 70, 35 and 20 emu g−1, respectively. The decreasing of Ms values from MNPs to [HSO3PMIM]OTf–SiO2@MNPs could be attributed to the coating of iron oxide magnetic nanoparticles with silica and IL. Furthermore, the magnetization curves of the magnetic nanoparticles display no hysteresis before and after functionalization. These results clearly demonstrate their superparamagnetic characteristics,58 which further reinforce by easy separation of these magnetic nanoparticles with an external magnetic field.
 | ||
| Fig. 3 The VSM result for: (a) MNPs; (b) SiO2@MNPs and (c) [HSO3PMIM]OTf–SiO2@MNPs. | ||
Further characterization of the catalyst was performed by FT-IR spectroscopy. The FT-IR spectra of blank SiO2@MNPs, [HSO3PMIM]OTf and [HSO3PMIM]OTf–SiO2@MNPs are presented in Fig. 4. All these samples show broad bands at around 3424 and 1630 cm−1, that are assigned to the Si–OH group and adsorbed water, respectively, in which band at 1630 cm−1 is overlapped with band at 1655 cm−1 (Fig. 4b and c). The spectra of the SiO2@MNPs and [HSO3PMIM]OTf–SiO2@MNPs exhibit the characteristic bands at about 1099, 948 and 484 cm−1, which are attributed to the stretching vibrations of Si–O–Si and Fe–O, respectively (Fig. 4a and c). Furthermore, in the FT-IR spectra of [HSO3PMIM]OTf and [HSO3PMIM]OTf–SiO2@MNPs, the typical bands at around 1655 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1572 cm−1 (CC), 1428 cm−1 (C–H), 1179 and 1030 cm−1 (SO) were observed (Fig. 4b and c). In addition, the characteristic band at 1247 cm−1 is attributed to the CF3 group. These results confirm that IL has been successfully supported on the surface of SiO2@MNPs (Fig. 4c).
N), 1572 cm−1 (CC), 1428 cm−1 (C–H), 1179 and 1030 cm−1 (SO) were observed (Fig. 4b and c). In addition, the characteristic band at 1247 cm−1 is attributed to the CF3 group. These results confirm that IL has been successfully supported on the surface of SiO2@MNPs (Fig. 4c).
 | ||
| Fig. 4 FT-IR spectra of: (a) SiO2@MNPs; (b) [HSO3PMIM]OTf, (c) [HSO3PMIM]OTf–SiO2@MNPs and (d) the 6-times reused catalyst. | ||
Synthesis of isobenzofuran-1(3H)-ones catalyzed by [HSO3PMIM]OTf–SiO2@MNPs
The condensation of phthalaldehydic acid (1 mmol) with acetophenone (1 mmol) was chosen as a model reaction for the optimization of parameters such as the amount of catalyst, temperature, and MW power. The results are summarized in Table 1. Initially, the reaction was examined in the absence of the catalyst at 100 °C; no desired product was obtained under this conditions even after 2.5 h. Then, the reaction was performed in the presence of SiO2@MNPs and [HSO3PMIM]OTf and the corresponding product was obtained in 8% and 69% yields, respectively. In order to find the optimum amount of [HSO3PMIM]OTf–SiO2@MNPs catalyst, the reaction was performed with different amounts of the catalyst (15, 20, 25, and 30 mg) under solvent-free conditions at 100 °C. The best yield of the desired product was achieved with 25 mg of [HSO3PMIM]OTf–SiO2@MNPs. Using lower amount of catalyst resulted in lower yield, while higher amount did not affect the reaction time and yield. To evaluate the influence of temperature, the model reaction was performed in the range of 80–110 °C. It was found that 100 °C was the optimal temperature and the reaction was incomplete at lower temperature. To find the optimized MW power, the model reaction was carried out in the presence of 25 mg catalyst at 550, 600 and 650 W, and the desired product was obtained in 80%, 95% and 95% yields, respectively. Therefore, 600 W and 100 °C was selected as the optimum power and temperature.
|
|||||
|---|---|---|---|---|---|
| Entry | Method | T (°C) | Catalyst amount (mg) | Time | Yielda (%) |
| a Isolated yield.b Reaction was performed in the presence of SiO2@MNPs.c Reaction was performed in the presence of 0.01 mmol [HSO3PMIM]OTf. | |||||
| 1 | Thermal | 100 | 0 | 2.5 h | 0 |
| 2b | Thermal | 100 | 25 | 2.5 h | 8 |
| 3c | Thermal | 100 | — | 2.5 h | 69 |
| 4 | Thermal | 100 | 15 | 2.5 h | 65 |
| 5 | Thermal | 100 | 20 | 2.5 h | 82 |
| 6 | Thermal | 100 | 25 | 2.5 h | 95 |
| 7 | Thermal | 100 | 30 | 2.5 h | 95 |
| 8 | Thermal | 80 | 25 | 2.5 h | 72 |
| 9 | Thermal | 90 | 25 | 2.5 h | 83 |
| 10 | Thermal | 110 | 25 | 2.5 h | 95 |
| 11 | MW (550 W) | 100 | 25 | 10 min | 80 |
| 12 | MW (600 W) | 100 | 25 | 10 min | 95 |
| 13 | MW (650 W) | 100 | 25 | 10 min | 95 |

The scope of this protocol was then extended by using a variety of acetophenones to synthesize isobenzofuran-1(3H)-ones. The results are summarized in Table 2. Unsubstituted acetophenone and acetophenones with electron-withdrawing or electron-donating substituents reacted very well with phthalaldehydic acid in the presence of [HSO3PMIM]OTf–SiO2@MNPs catalyst under thermal conditions, affording good to excellent yields of the corresponding isobenzofuran-1(3H)-one derivatives (Table 2, entries 1–12). Acid-sensitive ketones such as 2-acetylthiophene and 4-acetylpyridine were also reacted efficiently to give the desired products in high yields (Table 2, entries 13 and 14.). In order to investigate the effect of MW, the reaction of acetophenones with phthalaldehydic acid was performed under the optimized microwave irradiation. Under these conditions, the desired isobenzofuran-1(3H)-ones were obtained in 65–98% yields in very short reaction times (4–12 min) (Table 2). These results clearly demonstrated that the yields were slightly improved, but the reaction times were significantly reduced under microwave irradiation.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ketone | Product | Thermal | MW | ||
| Time (h) | Yieldb (%) | Time (min) | Yieldb (%) | |||
| a Reaction conditions: phthalaldehydic acid (1 mmol), acetophenone (1 mmol), catalyst (25 mg), thermal (100 °C) or MW (600 W, 100 °C).b Isolated yield. | ||||||
| 1 |  |
 |
2.5 | 95 | 10 | 95 |
| 2 |  |
 |
3 | 92 | 12 | 95 |
| 3 |  |
 |
1.5 | 60 | 8 | 65 |
| 4 |  |
 |
1.45 | 87 | 4 | 91 |
| 5 |  |
 |
3 | 85 | 12 | 90 |
| 6 |  |
 |
1.5 | 85 | 8 | 97 |
| 7 |  |
 |
1 | 90 | 8 | 98 |
| 8 |  |
 |
1.75 | 95 | 8 | 95 |
| 9 |  |
 |
2 | 92 | 7 | 93 |
| 10 |  |
 |
2 | 92 | 8 | 96 |
| 11 |  |
 |
2.5 | 65 | 12 | 75 |
| 12 |  |
 |
0.75 | 90 | 7 | 93 |
| 13 |  |
 |
0.75 | 90 | 7 | 93 |
| 14 |  |
 |
3 | 88 | 10 | 92 |

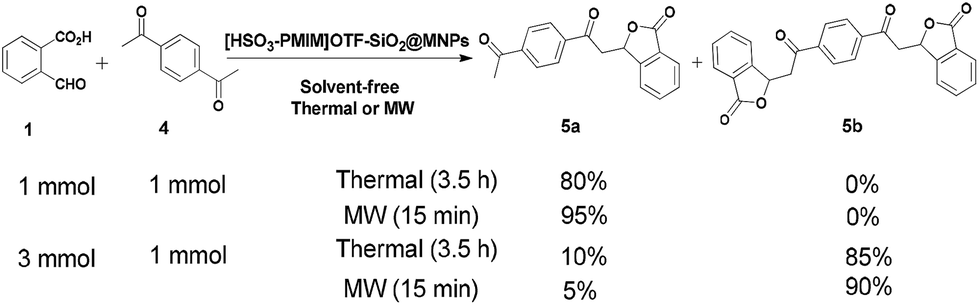
Another noteworthy advantage of this catalytic system lies in the selective reaction of 1,4-diacetylbenzene with phthalaldehydic acid. Using a 1:1 molar ratio of 1,4-diacetylbenzene to phthalaldehydic acid, only one acetyl group reacted selectively and the corresponding mono-isobenzofuran-1(3H)-one 5a was obtained in 80% and 95% yields under thermal conditions and MW irradiation, respectively. Whereas, with a 1:3 molar ratio of 1,4-diacetylbenzene to phthalaldehydic acid, both acetyl groups reacted and the corresponding bis-isobenzofuran-1(3H)-one 5b was obtained in 85% and 90% yields, under the above mentioned conditions (Scheme 3). To the best of our knowledge, this is the first report on the selective synthesis of mono- and bis-isobenzofuran-1(3H)-one, which shows the efficiency and applicability of this catalytic system.
 | ||
| Scheme 3 Selective synthesis of mono- and bis-isobenzofuran-1(3H)-ones catalyzed by [HSO3PMIM]OTf–SiO2@MNPs. | ||
A plausible mechanism for the formation of isobenzofuran-1(3H)-one is proposed in Scheme 4. Initially, [HSO3PMIM]OTf–SiO2@MNPs activates the formyl group of phthalaldehydic acid and also catalyzes the keto–enol tautomerization of acetophenone to give A and B, respectively. Nucleophilic attack of B to A furnishes the intermediate C. The intermediate C undergoes intramolecular nucleophilic cyclization in the presence of the catalyst giving the adduct D. Finally, dehydration of D in the presence of the catalyst produces the desired isobenzofuran-1(3H)-one 3 and releases the catalyst for the next run.
 | ||
| Scheme 4 Proposed mechanism for the synthesis of isobenzofuran-1(3H)-ones catalyzed by [HSO3PMIM]OTf-SiO2@MNP. | ||
Recyclability of the catalyst
Finally, we turned our attention to the possibility of recycling the catalyst, since the recovery and reuse of the catalyst are highly preferable for a green process. In this respect, the reusability of the catalyst was investigated in the reaction of phthalaldehydic acid with acetophenone under thermal conditions and MW irradiation. At the end of the reaction, the mixture was cooled to room temperature and chloroform (15 mL) was added. The catalyst was easily separated by a permanent magnet, dried and reused for subsequent reactions. The experimental results suggest that there is no appreciable loss in catalytic activity even after sixth recycle (Fig. 5). The comparison of FT-IR spectra of fresh and reused catalyst suggests the stability of the present catalyst during the reaction (Fig. 4d).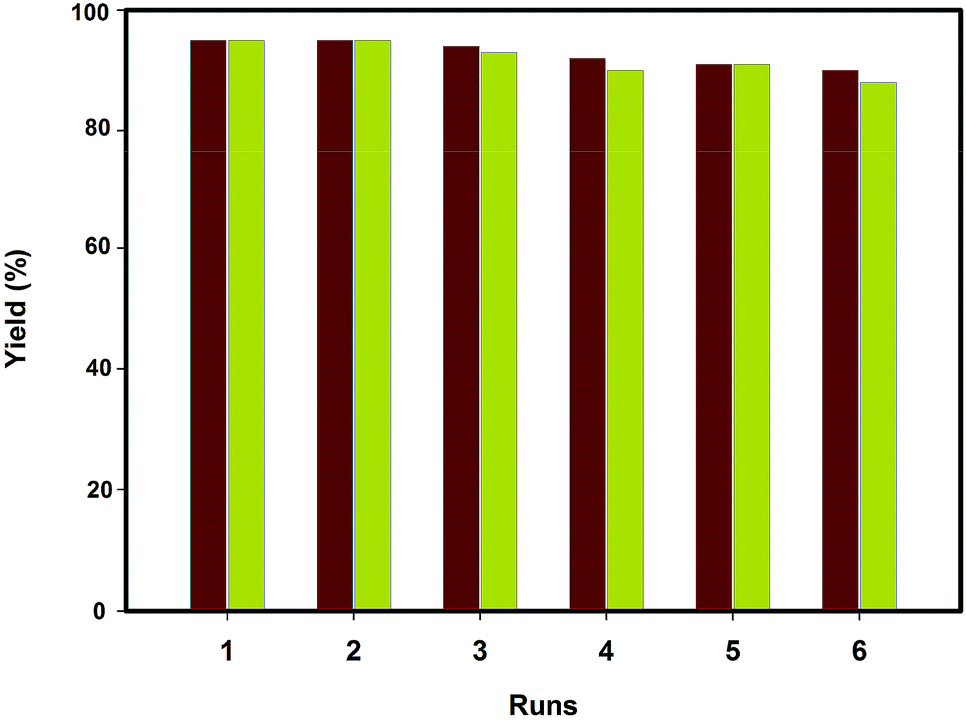 | ||
| Fig. 5 Recycling experiment of [HSO3PMIM]OTf–SiO2@MNPs under thermal conditions (red) and MW irradiation (green). | ||
Conclusions
In conclusion, we have developed a convenient, efficient, and environmentally benign procedure for the preparation of isobenzofuran-1(3H)-one derivatives in the presence of [HSO3PMIM]OTf–SiO2@MNPs as a highly recyclable catalyst under solvent-free thermal conditions and MW irradiation. Easy work-up, high yields, short reaction times, high atom-economy, environmental acceptability, and use of easy recoverable and reusable catalyst are the significant advantages of the present protocol in the synthesis of these important compounds.Experimental
General information
The chemicals used in this work were purchased from Fluka and Merck chemical companies. Melting points were determined with a Stuart Scientific SMP2 apparatus. FT-IR spectra were recorded on a Nicolet-Impact 400D spectrophotometer. 1H NMR (400 and 500 MHz) and 13C NMR (100 and 125 MHz) spectra were recorded on a Bruker Avance spectrometer using CDCl3 and DMSO-d6 as solvent. X-Ray diffraction (XRD) images were obtained from a Bruker XRD D8 Advance instrument with Co Kα radiation at 40 kV. The transmission electron microscopy (TEM) was carried out on a Philips CM10 transmission electron microscope operating at 200 kV. The magnetic measurements were performed with a vibrating sample magnetometer (VSM) at Meghnatis Daghigh Kavir Co. The microwave system used in these experiments includes the following items: Micro-SYNTH labstation, equipped with a glass door, a dual magnetron system with pyramid shaped diffuser, 1000 W delivered power, exhaust system, magnetic stirrer, ‘quality pressure’ sensor for flammable organic solvents, and a ATCFO fiber optic system for automatic temperature control.Synthesis of 1-methyl-3-(propyl-3-sulfonic acid)imidazolium triflate supported on magnetic nanoparticles ([HSO3PMIM]OTf–SiO2@MNPs)
General procedure for the synthesis of isobenzofuran-1(3H)-ones under thermal conditions and MW irradiation
A mixture of phthaladehydic acid (1 mmol), acetophenone (1 mmol) and [HSO3PMIM]OTf–SiO2@MNPs (25.0 mg) was stirred at 100 °C or subjected to MW irradiation (600 W, 100 °C) for the appropriate time according to Table 2. The progress of the reaction was monitored by TLC (eluent: petroleum ether–ethyl acetate, 5:2). After completion of the reaction, the mixture was cooled to room temperature, chloroform (15 mL) was added and the catalyst was easily separated by a permanent magnet. The filtrate was evaporated and the crude product was purified by chromatography on silica gel (eluent: petroleum ether–ethyl acetate, 5:2) or by recrystallization from acetone and ethanol (1:2) to afford the pure product.
Acknowledgements
The authors are grateful to the Research Council of the University of Isfahan for financial support of this work.Notes and references
- S. Chowdhury, R. S. Mohan and J. L. Scott, Tetrahedron, 2007, 63, 2363–2389 CrossRef CAS PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- W. Miao and T. H. Chan, Acc. Chem. Res., 2006, 39, 897–908 CrossRef CAS PubMed.
- H. Li, P. S. Bhadury, B. Song and S. Yang, RSC Adv., 2012, 2, 12525–12551 RSC.
- C. DeCastro, E. Sauvage, M. Valkenberg and W. Hölderich, J. Catal., 2000, 196, 86–94 CrossRef CAS.
- B. Gadenne, P. Hesemann and J. J. Moreau, Chem. Commun., 2004, 1768–1769 RSC.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217–3274 CrossRef CAS PubMed.
- K. B. Sidhpuria, A. L. Daniel-da-Silva, T. Trindade and J. A. Coutinho, Green Chem., 2011, 13, 340–349 RSC.
- Y. Yang, C. Deng and Y. Yuan, J. Catal., 2005, 232, 108–116 CrossRef CAS PubMed.
- Y. Yang, H. Lin, C. Deng, J. She and Y. Yuan, Chem. Lett., 2005, 34, 220–221 CrossRef CAS.
- L. Babes, B. t. Denizot, G. Tanguy, J. J. Le Jeune and P. Jallet, J. Colloid Interface Sci., 1999, 212, 474–482 CrossRef CAS PubMed.
- S. Gomez-Lopera, R. Plaza and A. Delgado, J. Colloid Interface Sci., 2001, 240, 40–47 CrossRef CAS PubMed.
- A. Jordan, R. Scholz, P. Wust, H. Fähling and R. Felix, J. Magn. Magn. Mater., 1999, 201, 413–419 CrossRef CAS.
- H. P. Khng, D. Cunliffe, S. Davies, N. A. Turner and E. N. Vulfson, Biotechnol. Bioeng., 1998, 60, 419–424 CrossRef CAS.
- R. Abu-Reziq, H. Alper, D. Wang and M. L. Post, J. Am. Chem. Soc., 2006, 128, 5279–5282 CrossRef CAS PubMed.
- A. Hu, G. T. Yee and W. Lin, J. Am. Chem. Soc., 2005, 127, 12486–12487 CrossRef CAS PubMed.
- D. Lee, J. Lee, H. Lee, S. Jin, T. Hyeon and B. M. Kim, Adv. Synth. Catal., 2006, 348, 41–46 CrossRef CAS.
- N. T. Phan and C. W. Jones, J. Mol. Catal. A: Chem., 2006, 253, 123–131 CrossRef CAS PubMed.
- P. D. Stevens, J. Fan, H. M. Gardimalla, M. Yen and Y. Gao, Org. Lett., 2005, 7, 2085–2088 CrossRef CAS PubMed.
- T.-J. Yoon, W. Lee, Y.-S. Oh and J.-K. Lee, New J. Chem., 2003, 27, 227–229 RSC.
- Y. Zheng, P. D. Stevens and Y. Gao, J. Org. Chem., 2006, 71, 537–542 CrossRef CAS PubMed.
- R. Abu-Reziq, D. Wang, M. Post and H. Alper, Chem. Mater., 2008, 20, 2544–2550 CrossRef CAS.
- L. M. Rossi, L. L. Vono, F. P. Silva, P. K. Kiyohara, E. L. Duarte and J. R. Matos, Appl. Catal., A, 2007, 330, 139–144 CrossRef CAS PubMed.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- S. Shylesh, J. Schweizer, S. Demeshko, V. Schünemann, S. Ernst and W. R. Thiel, Adv. Synth. Catal., 2009, 351, 1789–1795 CrossRef CAS.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- T. Hara, T. Kaneta, K. Mori, T. Mitsudome, T. Mizugaki, K. Ebitani and K. Kaneda, Green Chem., 2007, 9, 1246–1251 RSC.
- D. m. Lai, L. Deng, J. Li, B. Liao, Q. x. Guo and Y. Fu, ChemSusChem, 2011, 4, 55–58 CrossRef CAS PubMed.
- E. Rafiee and S. Eavani, Green Chem., 2011, 13, 2116–2122 RSC.
- H. Yang, Y. Wang, Y. Qin, Y. Chong, Q. Yang, G. Li, L. Zhang and W. Li, Green Chem., 2011, 13, 1352–1361 RSC.
- X. Zheng, S. Luo, L. Zhang and J.-P. Cheng, Green Chem., 2009, 11, 455–458 RSC.
- A. Arnone, G. Assante, G. Nasini, S. Strada and A. Vercesi, J. Nat. Prod., 2002, 65, 48–50 CrossRef CAS PubMed.
- P. Kulanthaivel, Y. F. Hallock, C. Boros, S. M. Hamilton, W. P. Janzen, L. M. Ballas, C. R. Loomis, J. B. Jiang and B. Katz, J. Am. Chem. Soc., 1993, 115, 6452–6453 CrossRef CAS.
- G. Lin, S. S.-K. Chan, H.-S. Chung and S.-L. Li, Stud. Nat. Prod. Chem., 2005, 32, 611–669 CAS.
- J. E. Baldwin and K. W. Bair, Tetrahedron Lett., 1978, 19, 2559–2562 CrossRef.
- K. Kim, M. Spatz and F. Johnson, Tetrahedron Lett., 1979, 20, 331–334 CrossRef.
- N. Narasimhan and R. Mali, Synthesis, 1975, 12, 796–797 CrossRef.
- S. F. Brady, M. M. Wagenaar, M. P. Singh, J. E. Janso and J. Clardy, Org. Lett., 2000, 2, 4043–4046 CrossRef CAS PubMed.
- K. Yoganathan, C. Rossant, S. Ng, Y. Huang, M. S. Butler and A. D. Buss, J. Nat. Prod., 2003, 66, 1116–1117 CrossRef CAS PubMed.
- Y. Ogawa, K. Hosaka, K. Kubota and M. Chin, Chem. Abstr., 1992, 117, 69721 Search PubMed.
- H. Sato, H. Yorozu and S. Yamaoka, Biomed. Res., 1993, 14, 385–390 CAS.
- X. Yang, T. Rotter, C. Piazza and P. Knochel, Org. Lett., 2003, 5, 1229–1231 CrossRef CAS PubMed.
- R. Karmakar, P. Pahari and D. Mal, Chem. Rev., 2014, 114, 6213–6284 CrossRef CAS PubMed.
- M. V. Paradkar, S. Y. Gadre, T. A. Pujari, P. P. Khandekar and V. B. Kumbhar, Synth. Commun., 2005, 35, 471–474 CrossRef CAS PubMed.
- R. Rendy, Y. Zhang, A. McElrea, A. Gomez and D. A. Klumpp, J. Org. Chem., 2004, 69, 2340–2347 CrossRef CAS PubMed.
- S. M. Landge, M. Berryman and B. Toeroek, Tetrahedron Lett., 2008, 49, 4505–4508 CrossRef CAS PubMed.
- D. C. Pinto, A. Silva, J. A. Cavaleiro and J. Elguero, Eur. J. Org. Chem., 2003, 747–755 CrossRef CAS.
- E. W. Bousquet, M. D. Moran, J. Harmon, A. L. Johnson and J. C. Summers, J. Org. Chem., 1975, 40, 2208–2211 CrossRef CAS.
- D. Villemin, N. Cheikh, B. Mostefa-Kara, N. Bar, N. Choukchou-Braham and M. A. Didi, Tetrahedron Lett., 2006, 47, 5519–5521 CrossRef CAS PubMed.
- M. M. Heravi, V. Rasmi, F. F. Bamoharram, S. Sadjadi, L. Fotouhi, S. Sadjadi and M. Bakavoli, Synth. Commun., 2009, 39, 4109–4116 CrossRef CAS.
- J. N. Sangshetti, S. A. M. Ansari and D. B. Shinde, Chin. Chem. Lett., 2011, 22, 163–166 CrossRef CAS PubMed.
- S. Anvar, I. Mohammadpoor-Baltork, S. Tangestaninejad, M. Moghadam, V. Mirkhani, A. R. Khosropour, A. Landarani Isfahani and R. Kia, ACS Comb. Sci., 2014, 16, 93–100 CrossRef CAS PubMed.
- M. Ghotbinejad, A. R. Khosropour, I. Mohammadpoor-Baltork, M. Moghadam, S. Tangestaninejad and V. Mirkhani, J. Mol. Catal. A: Chem., 2014, 385, 78–84 CrossRef CAS PubMed.
- A. Landarani Isfahani, I. Mohammadpoor-Baltork, V. Mirkhani, A. R. Khosropour, M. Moghadam, S. Tangestaninejad and R. Kia, Adv. Synth. Catal., 2013, 355, 957–972 CrossRef.
- I. Mohammadpoor-Baltork, A. R. Khosropour, M. Moghadam, S. Tangestaninejad, V. Mirkhani, M. Soltani and A. Mirjafari, J. Heterocycl. Chem., 2011, 48, 1419–1427 CrossRef CAS.
- M. Nasr-Esfahani, I. Mohammadpoor-Baltork, A. R. Khosropour, M. Moghadam, V. Mirkhani, S. Tangestaninejad and H. Amiri Rudbari, J. Org. Chem., 2014, 79, 1437–1443 CrossRef CAS PubMed.
- S. Safaei, I. Mohammadpoor-Baltork, A. R. Khosropour, M. Moghadam, S. Tangestaninejad, V. Mirkhani and R. Kia, RSC Adv., 2012, 2, 5610–5616 RSC.
- A. Bee, R. Massart and S. Neveu, J. Magn. Magn. Mater., 1995, 149, 6–9 CrossRef CAS.
- Y. P. M. Shokouhimehr, J. Kim, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 7039–7043 CrossRef PubMed.
- J. Lee, Y. Lee, J. K. Youn, H. B. Na, T. Yu, H. Kim, S. M. Lee, Y. M. Koo, J. H. Kwak and H. G. Park, Small, 2008, 4, 143–152 CrossRef CAS PubMed.
- X. Wang, P. Dou, P. Zhao, C. Zhao, Y. Ding and P. Xu, ChemSusChem, 2009, 2, 947–950 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14112a |
| This journal is © The Royal Society of Chemistry 2015 |