
One-pot conversion of carbohydrates into gamma-valerolactone catalyzed by highly cross-linked ionic liquid polymer and Co/TiO2
Huacong Zhou,
Jinliang Song,
Xinchen Kang,
Jiayin Hu,
Yingying Yang,
Honglei Fan,
Qinglei Meng and
Buxing Han*
Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: Hanbx@iccas.ac.cn; Fax: +86-10-62562821; Tel: +86-10-62562821
First published on 15th January 2015
Abstract
The acid catalytic conversion of carbohydrates into levulinate esters followed by metal catalytic hydrogenation is an important approach to obtain gamma-valerolactone (GVL) from biomass. In this work, we prepared the highly cross-linked polymer of divinylbenzene with acid ionic liquid (PDVB-IL) and supported Co on TiO2 (Co/TiO2), which were characterized by Fourier transform infrared (FTIR) spectroscopy, scanning electron microscopy (SEM), transmission electron microscopy (TEM), and elemental analysis. It was demonstrated that the PDVB-IL polymer could efficiently catalyze the esterification reaction of furfuryl alcohol (FAL), 5-hydroxymethylfurfural (HMF), and fructose with ethanol to produce the intermediate ethyl levulinate (EL), and the EL in the reaction mixture was directly hydrogenated to GVL over the Co/TiO2 without requiring purification and with high yields.
Introduction
With increasing demand for energy and limited planetary reserves of traditional fossil resources, exploring sustainable carbon sources becomes a critical issue for human society.1,2 Biomass is a well-accepted green and sustainable carbon source and can overcome the above problem.3–5 Lignocellulose, consisting mainly of cellulose, hemicellulose, and lignin, is an abundant and renewable carbon source and can be biologically or chemically converted into various bulk chemicals, materials, and fuel additives.1,6–13 A useful chemical obtained from lignocellulose is gamma-valerolactone (GVL), which has wide applications including fuel additive, solvent, liquid fuel, and a precursor for the production of more valuable chemicals.1,14–19One of the approaches to obtain GVL from biomass is the C5 sugar route with hemicellulose-derived furfuryl alcohol (FAL) as the initial or intermediate molecule.1,20 Tandem acid catalytic alcoholysis of FAL and the subsequent metal catalytic hydrogenation reaction is a key process in this route.21–23 Although inorganic mineral acids have a high efficiency, their corrosion and recovery problems drive researchers to investigate more economically and environmentally feasible heterogeneous acid catalysts. Lange et al. have proven that strongly acidic resins and zeolites were effective catalysts for the alcoholysis of FAL into ethyl levulinate (EL).24 Zhang et al. developed a novel organic–inorganic hybrid solid acid catalyst for the conversion of FAL into alkyl levulinates.25 Comparatively, the one-pot conversion directly from FAL to GVL is more attractive because no separation of the intermediate alkyl levulinates was needed. Rode et al. conducted the first attempted direct conversion of FAL to GVL by the combination of acid ionic liquids (ILs) and different supported noble metal catalysts.20 The combination of acid resin Amberlyst-15 and RANEY® Ni was also reported to be efficient for the tandem conversion of FAL.18
Although much progress has been achieved as stated above, there are still many problems remaining to be solved for the direct conversion of FAL to GVL. For example, for the homogeneous catalysts, the most efficient catalyst reported up to date except for the mineral acids is sulfonic acid-functionalized ionic liquid. Its homogeneous property makes the recovery of the catalyst troublesome and requires consumption of large amounts of organic solvents, water, and energy.20 For the reported heterogeneous catalysts, the humin byproducts formed in the acid catalytic process could easily be adsorbed on the surface of the catalysts and deactivate them.24–26 The commonly used method to regenerate the catalysts is coke burn-off, but it is not suitable for temperature-sensitive materials such as resins.24 Most of the reported metal catalysts are metal-based, including Ru, Rh, Pd, Ag, and Ir, which are expensive with very limited reserve. The only reported non-noble metal was RANEY® Ni, but it required the purification of the intermediate EL.18 Obviously, more efforts are still desirable to improve the above aspects of catalysts for GVL production.
Considering the above problems, in this work, we designed a novel catalytic system consisting of the highly cross-linked polymer of divinylbenzene with acid ionic liquid (PDVB-IL) and supported Co on TiO2 (Co/TiO2) for the one-pot conversion of FAL into GVL (Scheme 1), which consisted of esterification of the carbohydrates with ethanol to EL and hydrogenation reaction of EL to GVL. It was demonstrated that this catalytic system could efficiently catalyze the conversion of FAL, HMF, and fructose into GVL without the purification of the EL intermediate. The ILs could be easily recycled due to their heterogeneous property. The PDVB-IL polymer poorly adsorbed humin, giving the catalyst better reusability than that of the commercial Amberlyst-15.
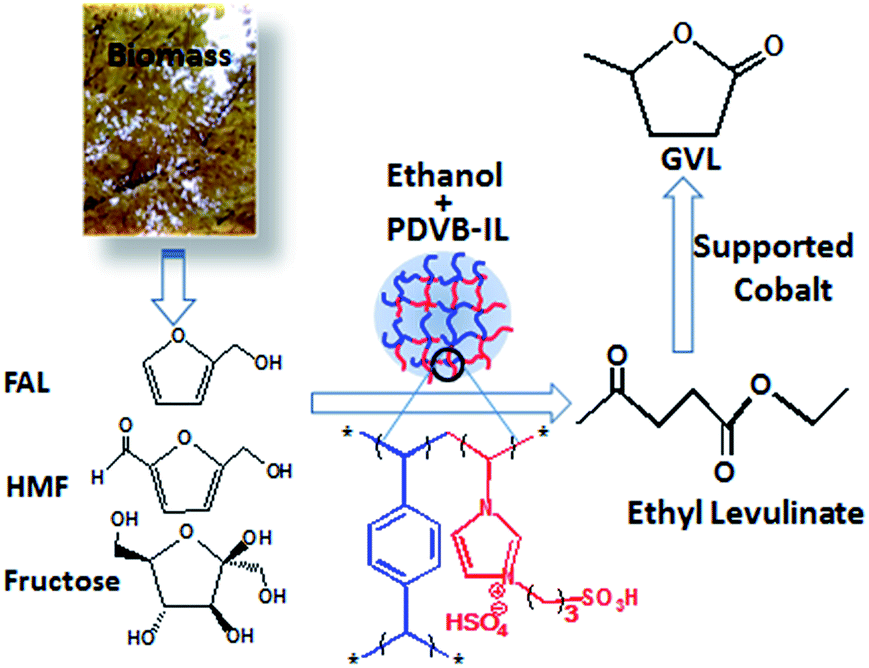 | ||
| Scheme 1 Schematic diagram of the tandem conversion of FAL, HMF, and fructose into GVL by the PDVB-IL polymer and supported cobalt catalytic system. | ||
Experimental
Materials
Furfuryl alcohol (98%), 1-vinylimidazole (99%), silicon(IV) oxide (amorphous fumed, S.A. 130–170 m2 g−1), Amberlyst-15 (H) ion exchange resin, molecular sieve (zeolite Y, hydrogen), and Celite (HyfloSupercel) were from Alfa Aesar. Divinylbenzene (DVB, m- and p-mixture, 55% in EVB + DEB, stabilized with TBC), benzoyl peroxide (BPO, 98%), 1,4-butane sultone (99%), ethyl levulinate (98%), gamma-valerolactone (98%), and dodecane (99%) were purchased from J & K Chemical. Titanium(IV) oxide Aeroxide® P25 was from Acros Organics. Cobalt nitrate hexahydrate, sodium borohydride, MgO, ZrO2, and other chemicals were A. R. grade and provided by Sinopharm Chemical Reagent Co. Ltd. H2 (99.99%) was provided by the Beijing Analytical Instrument Company.Preparation of IL precursor
Sulfonic acid-functionalized ionic liquid precursor was synthesized according to previously reported procedures with modification.20 In the experiment, 5.40 g (40 mmol) 1,4-butane sultone and 0.94 g (10 mmol) 1-vinylimidazole were added to a 25 mL one-necked flask equipped with a magnetic stirrer. After reflux for 10 h at 70 °C under N2 atmosphere with stirring, the reaction mixture was cooled, and the solid product was recovered by filtration. The product was washed with diethyl ether to completely remove the unreacted reactants and then dried overnight under vacuum. A white zwitterion product was obtained with a yield of 98%. A stoichiometric amount of sulfonic acid was subsequently added dropwise to the zwitterionic solution, and the mixture was stirred at 80 °C for 6 h. After removal of water via evaporation, the viscous IL was obtained and dried overnight under vacuum. The prepared IL was confirmed by 1H and 13C NMR. 1H NMR (400 MHz, D2O): δ 8.84 (s, 1H), 7.58 (s, 1H), 7.40 (s, 1H), 6.93 (dd, J = 15.6, 8.7 Hz, 1H), 5.59 (dd, J = 15.6, 2.7 Hz, 1H), 5.21 (dd, J = 8.7, 2.7 Hz, 1H), 4.08 (t, J = 7.1 Hz, 2H), 2.88–2.63 (m, 2H), 2.03–1.73 (m, 2H), 1.56 (dt, J = 18.3, 7.8 Hz, 2H). 13C NMR (100 MHz, D2O): δ 134.30, 128.13, 122.72, 119.49, 109.25, 49.95, 49.18, 27.83, 20.82.Preparation of the PDVB-IL polymer
The PDVB-IL polymer was prepared according to a previous method with modification.27 The synthesized IL was copolymerized with DVB to form highly cross-linked polymer. In a typical polymerization, 0.34 g prepared IL, 1.40 g DVB, and 40 mL ethanol or ethanol/chloroform mixtures with different ratios were added to a 100 mL three-necked round flask and heated to 70 °C. The flask was purged with nitrogen, and then 0.08 g benzoyl peroxide was added under stirring. The polymerization was allowed to proceed under N2 atmosphere for 24 h. After cooling, the product was separated by centrifugation, washed thoroughly with ethanol and water, and then dried under vacuum for use.Preparation of supported cobalt
The supported cobalt was prepared by the impregnation and NaBH4 reduction method.20 In a typical procedure, 0.05 g support was suspended in a solution containing a calculated amount of Co(NO3)2·6H2O under ultrasonification, and the mixture was stirred at room temperature for 2 h. Then 0.075 g NaBH4 dissolved in 10 mL water was added dropwise, and the mixture was stirred for half an hour. The supported cobalt was then separated via centrifugation, washed by ethanol, and kept in ethanol for use.Catalyst characterization
The PDVB-IL polymer was characterized by scanning electron microscopy (SEM, HITACHI S-4800), Fourier transform infrared (FTIR) spectroscopy (Bruker Tensor 27 spectrometer), and elemental analysis (EA, Flash EA 1112). The Co/TiO2 was characterized by transmission electron microscopy (TEM, JEOL JEM-1011).Reaction
For the conversion of FAL into EL, typically, 0.25 g (2.4 mmol) FAL, 0.18 g PDVB-IL polymer, and a certain quantity of ethanol were added to a 10 mL Teflon-lined stainless steel autoclave equipped with a magnetic stirrer. After using a vacuum pump to remove the air, the reactor was charged with 1 atm of nitrogen and then placed in a constant temperature oil bath at the desired temperature. After the reaction, the reactor was cooled in ice water, and the organic phase was extracted from the mixture with ethyl acetate. The sample was analyzed by gas chromatograph (GC, HP 4890) equipped with a flame ionization detector (FID), and dodecane was used as the internal standard. The products and reactants were identified using a GC-MS (SHIMADZU-QP2010) as well as by comparing the retention times to respective standards in GC traces. The results are expressed in mol% based on the total FAL loaded. In tandem reaction, the catalyst in each step was separated via centrifugation and washed once with ethanol for reuse. HMF and fructose were converted using the same procedures as those for FAL.The activity of the supported cobalt was evaluated via the hydrogenation of EL. In a typical experiment, 1 g EL and 0.05 g of the prepared catalyst were introduced into the reactor. The reaction was conducted under 130 °C and 3 MPa of hydrogen pressure. The product was analysed as described above.
Results and discussion
Preparation and characterization of the PDVB-IL polymer
The morphologies of the PDVB-IL polymer varied from dense microspheres to an irregular porous gel, depending strongly on the solvent system (Fig. 1). The average sizes of the samples prepared in mixed solvents with ethanol/chloroform ratios (v/v) of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 30:10, and 20:20 were 360 nm, 500 nm, and 2.8 μm, respectively (Fig. 1–c). The morphology change can be explained by the competition between the polymer chain growth rate and the phase separation rate.27 Ethanol is a much poorer solvent for the polymer than chloroform. Once the polymer was formed, the phase separation quickly occurred, and the polymer shrunk and grew into a dense microsphere in ethanol; however, this process was limited by the increasing chloroform content, leading to the formation of larger aggregated spheres or an irregular porous structure.
:0, 30:10, and 20:20 were 360 nm, 500 nm, and 2.8 μm, respectively (Fig. 1–c). The morphology change can be explained by the competition between the polymer chain growth rate and the phase separation rate.27 Ethanol is a much poorer solvent for the polymer than chloroform. Once the polymer was formed, the phase separation quickly occurred, and the polymer shrunk and grew into a dense microsphere in ethanol; however, this process was limited by the increasing chloroform content, leading to the formation of larger aggregated spheres or an irregular porous structure.
 | ||
| Fig. 1 SEM images and size distributions of the PDVB-IL polymer obtained in different solvents: (a) neat ethanol and ethanol/chloroform mixtures with volume (mL) ratios of (b) 30/10, (c) 20/20, and (d) 10/30. | ||
FTIR spectroscopy and elemental analysis were carried out to confirm the existence of IL segments in the polymer. As shown in Fig. 2, the peaks at 1654 cm−1, 1174 cm−1, and 1041 cm−1 attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations of imidazole rings, and asymmetrical and symmetrical stretching of SO3, respectively, appeared in the spectrum of the PDVB-IL polymer, proving the existence of the IL structure.27–29 Elemental analysis showed that the content of IL segments increased as the chloroform content increased (Fig. 3). This may be attributed to a higher chloroform concentration that facilitated the diffusion of IL in the polymer matrix and enhanced the probability of the copolymerization of IL and DVB.27 When the obtained polymers were used to convert FAL, the conversion of FAL decreased with the increase in IL content, and the EL yield reached the highest value at the IL content of approximately 3.1 wt% (Fig. 3). The decrease in FAL conversion may be attributed to a slower phase separation rate at a higher chloroform content, which resulted in the embedding of more IL segments into the matrix, leading to a decreased accessibility of the acid sites.27 Thus, the PDVB-IL polymer prepared in ethanol/chloroform with a ratio of 30:10 (v/v) was used to study the effects of various conditions on the reaction.
N stretching vibrations of imidazole rings, and asymmetrical and symmetrical stretching of SO3, respectively, appeared in the spectrum of the PDVB-IL polymer, proving the existence of the IL structure.27–29 Elemental analysis showed that the content of IL segments increased as the chloroform content increased (Fig. 3). This may be attributed to a higher chloroform concentration that facilitated the diffusion of IL in the polymer matrix and enhanced the probability of the copolymerization of IL and DVB.27 When the obtained polymers were used to convert FAL, the conversion of FAL decreased with the increase in IL content, and the EL yield reached the highest value at the IL content of approximately 3.1 wt% (Fig. 3). The decrease in FAL conversion may be attributed to a slower phase separation rate at a higher chloroform content, which resulted in the embedding of more IL segments into the matrix, leading to a decreased accessibility of the acid sites.27 Thus, the PDVB-IL polymer prepared in ethanol/chloroform with a ratio of 30:10 (v/v) was used to study the effects of various conditions on the reaction.
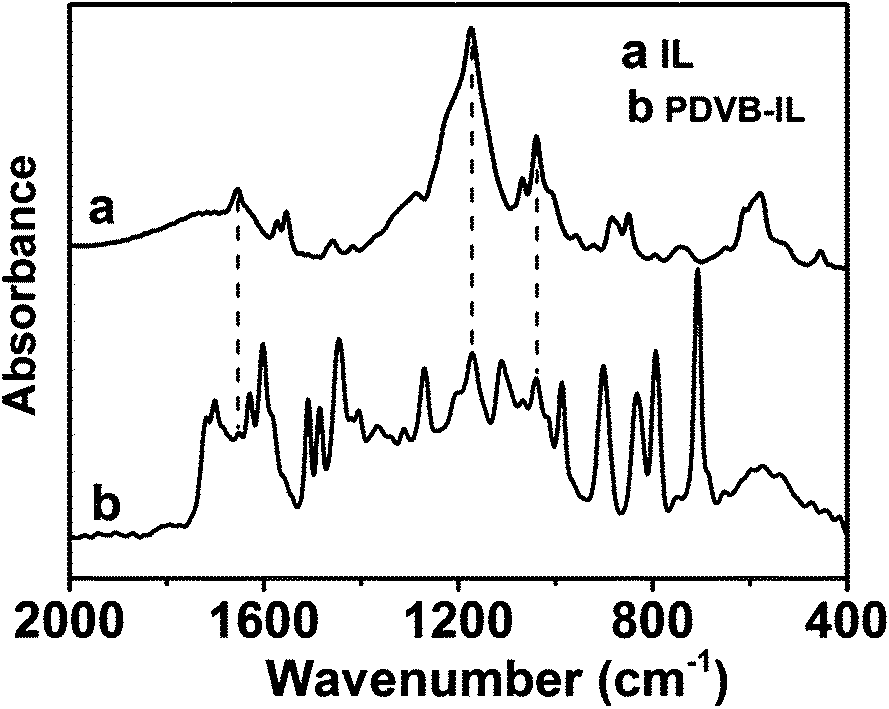 | ||
| Fig. 2 FTIR spectra of the prepared IL and PDVB-IL polymer obtained in ethanol/chloroform with a volume ratio of 30/10. | ||
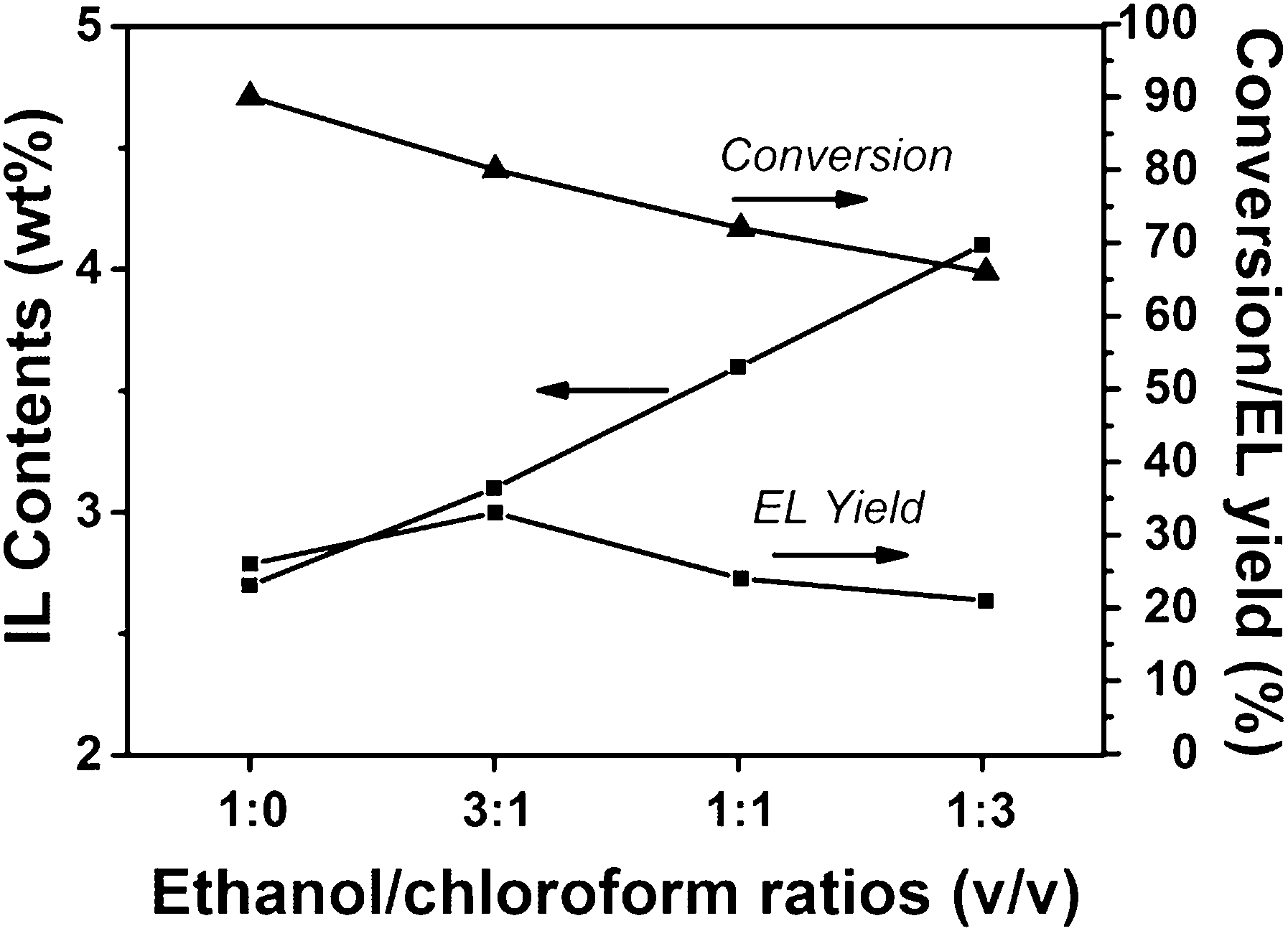 | ||
| Fig. 3 The IL content and activity of PDVB-IL polymers obtained under different ethanol/chloroform ratios. Reaction conditions: ethanol, 4 mL; FAL, 0.25 g; PDVB-IL polymer, 0.18 g; N2, 1 atm; 130 °C, 3 h. | ||
Conversion of FAL and optimization
The effects of the catalyst/substrate ratio, the amount of ethanol, the concentration of substrate, and temperature upon the reaction were studied, and the results are presented in Fig. 4. It can be seen from Fig. 4a that increasing the catalyst/substrate mole ratio from 0.3% to 1.3% significantly improved the EL yield from 14% to 38% within 12 h, while further increasing the catalyst/substrate ratio led to a slight decrease in product yield, resulting from the fast formation of humin byproducts. Increasing the volume of the solvent improved the EL yield, while increasing the concentration of FAL at a constant solvent volume resulted in a decrease of the product yield (Fig. 4b and c), which was consistent with a previous report describing the use of other catalysts.20 Temperature is also an important factor influencing the EL yield (Fig. 4d). A moderate EL yield of 33% was obtained even under 90 °C, and the yield increased up to 48% at the temperature of 130 °C. Further increasing the temperature to 150 °C resulted in an obvious drop in EL yield, which may be attributed to side reactions that were also accelerated under higher temperatures. Furthermore, in our experiments, we found that the only co-product was the intermediate methoxyfurfuryl alcohol formed by the reactant and ethanol, which was only detected under lower temperature or shorter reaction time, and no other side-products except humin were detected.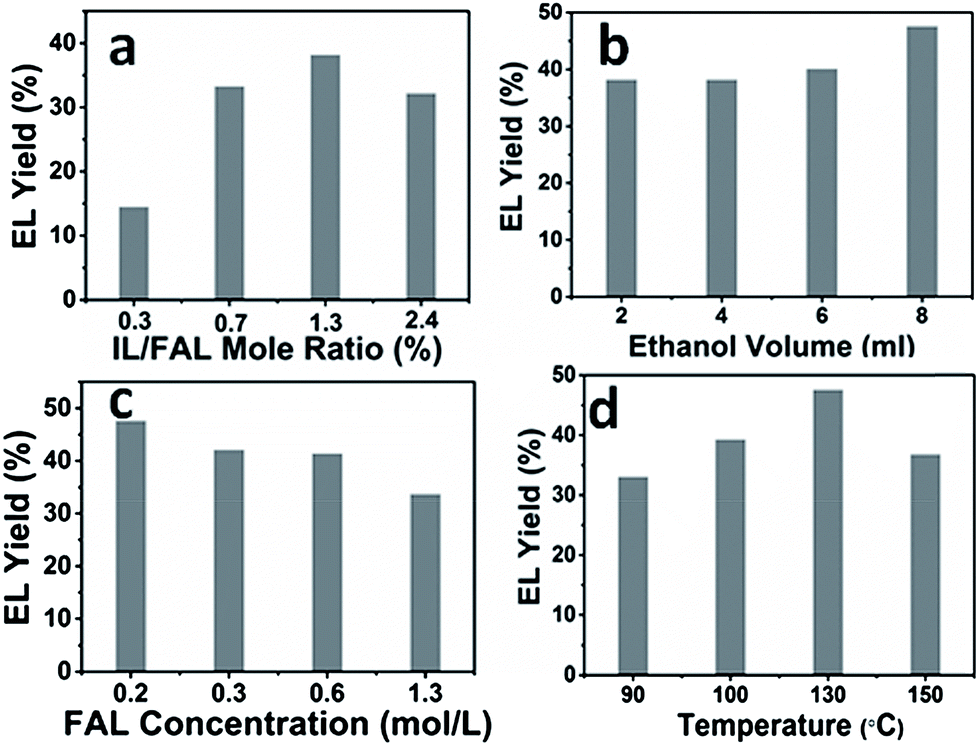 | ||
| Fig. 4 Effects of (a) the catalyst/substrate mole ratios, (b) the volume of ethanol, (c) the concentration of FAL, and (d) temperature on the EL yield. The catalyst/substrate mole ratios are defined as the mole ratios of IL/FAL. Typical reaction conditions were as follows unless otherwise stated in the above figures: ethanol, 8 mL; FAL, 0.12 g; PDVB-IL polymer, 0.18 g; N2, 1 atm; 130 °C, 12 h. | ||
The above results indicated that the formation of humin limited the further improvement of the EL yields. It has been reported that some organic solvents inhibited the formation of humin to some extent.26,30,31 Thus, several common solvents were used in an attempt to further improve the EL yields. The results are shown in Fig. 5. For all cases, FAL was completely converted but with different EL yields. Unlike the previously reported results,30,31 yields in acetonitrile and toluene were lower than that in pure ethanol, and the yield in THF was similar to that in pure ethanol. Interestingly, the yield of the product in DMSO reached 60% under the same conditions. During the experiments, we found that the intermediate ethoxy FAL was not totally converted into EL after reaction at 130 °C for 12 hours. Increasing the reaction time to 24 hours failed to fulfil the total conversion of the intermediate, while increasing the reaction temperature to 150 °C succeeded in total conversion of the intermediate within 12 hours. The EL yield was increased to 75%, which was higher than that obtained with common solid acids.24 As reported in the literature, DMSO stabilizes the furan structure in the reactants and suppresses the condensation of the reactants and formation of byproducts, thus improving the selectivity of EL.32,33
 | ||
| Fig. 5 Effects of solvents on the performance of the PDVB-IL polymer. EtOH, ethanol; Ace, acetonitrile; Tol, toluene; THF, tetrahydrofuran; DMSO, dimethyl sulfoxide. Reaction conditions: ethanol, 2 mL; solvent, 6 mL; FAL, 0.12 g; PDVB-IL polymer, 0.18 g; N2, 1 atm; 130 °C, 12 h. DMSO130 and DMSO150 denote that the reaction was conducted at 130 and 150 °C, respectively. | ||
In our experiments, it is interesting to find that the PDVB-IL polymer did not adsorb as much of the humin byproducts on its surface compared with the commercial acid resins, such as Amberlyst-15. We detected the dry mass change of Amberlyst-15 and the PDVB-IL polymer, and found that a mass increase of approximately 5% after two subsequent reactions occurred for Amberlyst-15, but there was no obvious mass change for the PDVB-IL polymer after reaction. The humin adsorption of the two catalysts could also be seen simply through the colour change after the reaction (Fig. 6a and b). The reusability comparison between our PDVB-IL polymer and Amberlyst-15 showed that the PDVB-IL polymer exhibited a better performance in both FAL conversion and EL yield than that of Amberlyst-15 within 5 cycles (Fig. 6c). This indicated that weak adsorption for humin is favourable to maintain the activity of the catalyst. We speculated that the strong adsorption of Amberlyst-15 for the humin could be related to its porous structures, which resulted in the accumulation of byproducts. It is worth noting that the PDVB-IL polymer also decreased in activity at a speed slower than that of Amberlyst-15. Experiments were conducted to detect the sulphur content in the reaction solutions with pure ethanol as the solvent and without DMSO. No obvious sulphur content was detected after the first catalysis reaction, while approximately 100 ppm of sulphur was detected after the fifth run. Thus, we speculated that the partial leaching of IL segments from the bulk polymer phase was the main reason for the activity decrease of the catalyst. We compared the FTIR spectra of the virgin and recycled PDVB-IL catalysts after they were used three times and no obvious differences were found, indicating that the main structures of PDVB-IL were well maintained after the reaction (Fig. 7).
 | ||
| Fig. 6 (a) Optical images of the Amberlyst-15 resin and (b) PDVB-IL polymer before and after reaction and (c) their reusability. The catalysts were washed once with ethanol before the photographs were taken. Reaction conditions: ethanol, 2 mL; DMSO, 6 mL; FAL, 0.12 g; PDVB-IL polymer, 0.18 g or Amberlyst-15, 0.2 g; N2, 1 atm; 130 °C; 12 h. The catalysts were washed once with ethanol before the next run. | ||
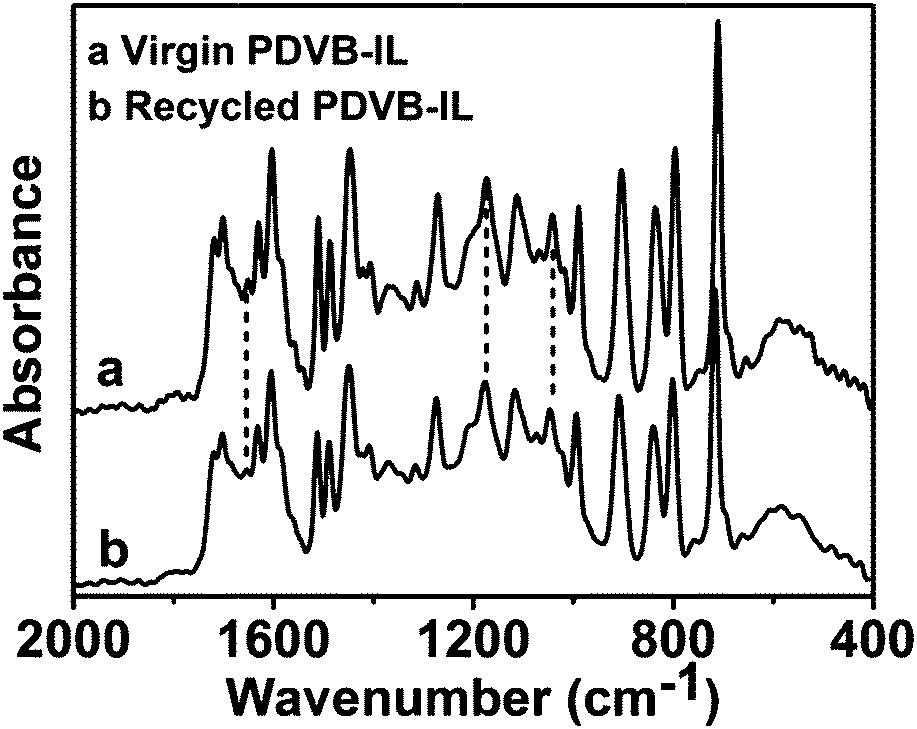 | ||
| Fig. 7 Comparison of FTIR spectra of the virgin and recycled PDVB-IL catalysts after being used three times. | ||
One-pot conversion of carbohydrates into GVL
After optimizing the reaction conditions of the PDVB-IL polymer for catalytic conversion of FAL into EL, we conducted a one-pot conversion of FAL into GVL. Here, we used the supported cobalt for the first time in the conversion of FAL into GVL. The impregnation and NaBH4 reduction method was used to load cobalt on various materials, and the hydrogenation of EL to GVL was used to evaluate the activity (Table 1). Among the materials investigated, TiO2 gave the highest GVL yield (entry 8). Increasing the loading of cobalt on TiO2 significantly increased the GVL yield (entries 8–10). TEM characterization of Co/TiO2 (20 wt% Co) showed that the Co particles with a diameter of 50–150 nm were uniformly dispersed in the TiO2 matrix without obvious aggregation (Fig. 8).| Entry | Supports | Conversion/% | Selectivity/% | Yield/% |
|---|---|---|---|---|
| a Typical reaction conditions: EL, 1.0 g; supported cobalt (5 wt%), 0.05 g; H2, 3 MPa; 130 °C; 3 h.b Cobalt loading: 10 wt%.c Cobalt loading: 20 wt%. | ||||
| 1 | Control | 8 | 84.3 | 6.8 |
| 2 | MgO | 16.8 | 94.1 | 15.8 |
| 3 | ZrO2 | 31 | 94.4 | 29.2 |
| 4 | Zeolite | 14.1 | 94.6 | 13.3 |
| 5 | SiO2 | 15.7 | 94.1 | 14.8 |
| 6 | Amberlyst-15 | 22.8 | 70.4 | 16 |
| 7 | Celite | 35.2 | 97.9 | 34.5 |
| 8 | TiO2 | 45.2 | 92.1 | 41.7 |
| 9 | TiO2b | 64.9 | 91.0 | 59.1 |
| 10 | TiO2c | 78.8 | 91.8 | 72.3 |
 | ||
| Fig. 8 TEM image of Co/TiO2 with a Co loading of 20 wt%. | ||
The tandem conversion of FAL into GVL is shown in the diagram in Table 2. It can be seen that the catalytic system is very efficient in the conversion of FAL, with an intermediate EL yield of 75% and a final GVL yield up to 69%, which was even higher than that of the combination of noble Ru/C and [BMIm-SH][HSO4].20 The activity was well maintained in the second run (entries 1–2). A decrease in the GVL yield was observed from the third recycle onward (entries 3–6). We detected the cobalt content in the reaction solutions. No obvious cobalt was found after the first two recycles, but a content of approximately 70 ppm was detected after the fifth run. Thus, the deactivation of the catalytic system may be attributed to the leaching of IL segments, as analyzed above, and subsequent partial cobalt dissolution. We expanded our catalytic system to the conversion of other carbohydrates, including HMF and fructose (entries 7 and 8), and GVL yields of 62% and 55% were obtained, respectively, which were comparable to the reported results using Amberlyst-15 and RANEY® Ni as the catalyst system.18
|
||||
|---|---|---|---|---|
| Entry | Substrate | Conversion/% | EL yield/% | GVL yield/% |
| a Reaction conditions: the first step, carbohydrate (FAL, HMF, or fructose), 1 mmol; PDVB-IL polymer, 0.18 g; ethanol, 2 mL, DMSO, 6 mL; N2, 1 atm; 150 °C; 12 hours. In the second step, Co/TiO2, 0.05 g; H2, 3 MPa; 130 °C; 6 hours. Yields were all given as the mole percent of the initiate substrates.b Entries 1–6 reusability of the catalytic systems for FAL: run times 1–6.c 24 hours for the hydrogenation reaction. | ||||
| 1b | FAL | >99 | 75 | 69 |
| 2b | FAL | >99 | 74 | 66 |
| 3b | FAL | 97 | 66 | 56 |
| 4b | FAL | 93 | 61 | 48 |
| 5b | FAL | 90 | 52 | 33 |
| 6b | FAL | 70 | 35 | 18 |
| 7 | HMF | >99 | 73 | 62c |
| 8 | Fructose | >99 | 65 | 55 |
Considering that the raw carbohydrate for the reaction may come from aqueous systems, we further investigated the effect of water on our catalytic system. The results showed that severe coking and no obvious products were detected in pure water due to the instability of FAL in water. Thus, we turned to a mixture of water and ethanol/DMSO and found that a water content of 10% (v/v) had no obvious influence on the high EL and GVL yields.
Conclusions
In this work, we have developed an efficient catalytic system consisting of the PDVB-IL polymer and Co/TiO2 for the one-pot conversion of FAL, HMF, and fructose into GVL via EL. Under optimized conditions, GVL yields can reach 69%, 62%, and 55% from FAL, HMF, and fructose, respectively. The PDVB-IL polymer is facile to recycle due to its heterogeneous property, and its lengthier reusability as compared to that of acid resins (Amberlyst-15) is due to its weak adsorption for humin. The obvious advantages of this catalytic system, including high efficiency, facile recovery, and use of low-cost and abundant cobalt as a metal catalyst, make it a promising candidate for biomass utilization.Acknowledgements
The authors thank the National Natural Science Foundation of China. This work was supported by the National Natural Science Foundation of China (21173234, 21133009, 21021003), and Chinese Academy of Sciences (KJCX2.YW.H30).Notes and references
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584 RSC.
- L. Hu, G. Zhao, W. W. Hao, X. Tang, Y. Sun, L. Lin and S. J. Liu, RSC Adv., 2012, 2, 11184 RSC.
- M. Y. He, Y. H. Sun and B. X. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed.
- M. S. Singhvi, S. Chaudhari and D. V. Gokhale, RSC Adv., 2014, 4, 8271 RSC.
- J. L. Song, H. L. Fan, J. Ma and B. X. Han, Green Chem., 2013, 15, 2619 RSC.
- B. B. Zhang, J. L. Song, G. Y. Yang and B. X. Han, Chem. Sci., 2014, 5, 4656 RSC.
- D. M. Alonso, J. M. R. Gallo, M. A. Mellmer, S. G. Wettstein and J. A. Dumesic, Catal. Sci. Technol., 2013, 3, 927 CAS.
- H. C. Zhou, J. L. Song, H. L. Fan, B. B. Zhang, Y. Y. Yang, J. Y. Hu, Q. G. Zhu and B. X. Han, Green Chem., 2014, 16, 3870 RSC.
- W. W. Zhu, H. M. Yang, J. Z. Chen, C. Chen, L. Guo, H. M. Gan, X. G. Zhao and Z. S. Hou, Green Chem., 2014, 16, 1534 RSC.
- H. B. Xie, H. W. Shen, Z. W. Gong, Q. Wang, Z. B. K. Zhao and F. W. Bai, Green Chem., 2012, 14, 1202 RSC.
- H. B. Xie, Z. B. K. Zhao and Q. Wang, ChemSusChem, 2012, 5, 901 CrossRef CAS PubMed.
- M. Dashtban, A. Gilbert and P. Fatehi, RSC Adv., 2014, 4, 2037 RSC.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657 CrossRef CAS PubMed.
- E. I. Gurbuz, J. M. Gallo, D. M. Alonso, S. G. Wettstein, W. Y. Lim and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 1270 CrossRef PubMed.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110 CrossRef CAS PubMed.
- X. L. Du, L. He, S. Zhao, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2011, 50, 7815 CrossRef CAS PubMed.
- Z. Yang, Y. B. Huang, Q. X. Guo and Y. Fu, Chem. Commun., 2013, 49, 5328 RSC.
- P. A. Son, S. Nishimura and K. Ebitani, RSC Adv., 2014, 4, 10525 RSC.
- A. M. Hengne, S. B. Kamble and C. V. Rode, Green Chem., 2013, 15, 2540 RSC.
- H. Gómez Bernal, L. Bernazzani and A. M. RaspolliGalletti, Green Chem., 2014, 16, 3734 RSC.
- R. F. Perez and M. A. Fraga, Green Chem., 2014, 16, 3942 RSC.
- T. Thananatthanachon and T. B. Rauchfuss, ChemSusChem, 2010, 3, 1139 CrossRef CAS PubMed.
- J. P. Lange, W. D. van de Graaf and R. J. Haan, ChemSusChem, 2009, 2, 437 CrossRef CAS PubMed.
- Z. Zhang, K. Dong and Z. K. Zhao, ChemSusChem, 2011, 4, 112 CrossRef CAS PubMed.
- F. Liu, M. Audemar, K. De Oliveira Vigier, J.-M. Clacens, F. De Campo and F. Jérôme, Green Chem., 2014, 16, 4110 RSC.
- B. J. Hu, T. B. Wu, K. L. Ding, X. S. Zhou, T. Jiang and B. X. Han, J. Phys. Chem. C, 2010, 114, 3396 CAS.
- Y. Hu, S. S. Tang, L. Jiang, B. Zou, J. Yang and H. Huang, Process Biochem., 2012, 47, 2291 CrossRef CAS PubMed.
- S. Ramesh and S.-C. Lu, J. Appl. Polym. Sci., 2012, 126, E484 CrossRef CAS.
- M. Chidambaram and A. T. Bell, Green Chem., 2010, 12, 1253 RSC.
- W. R. Yang and A. Sen, ChemSusChem, 2010, 3, 597 CrossRef CAS PubMed.
- E. Lam, E. Majid, A. C. W. Leung, J. H. Chong, K. A. Mahmoud and J. H. T. Luong, ChemSusChem, 2011, 4, 535 CrossRef CAS PubMed.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342 RSC.
| This journal is © The Royal Society of Chemistry 2015 |