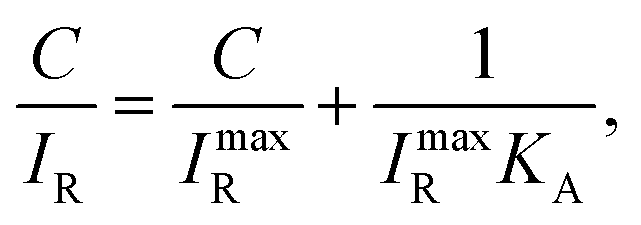DOI:
10.1039/C5RA00040H
(Paper)
RSC Adv., 2015,
5, 34720-34729
Label-free C-reactive protein SERS detection with silver nanoparticle aggregates†
Received
2nd January 2015
, Accepted 9th April 2015
First published on 9th April 2015
Abstract
In this work, we report a qualitative approach for detecting the adsorption of C-reactive protein on phosphocholine-terminated self-assembled monolayers without the use of any labels. An amplified plasmon of concentration-induced silver nanoparticle aggregates located ∼4.0 nm away from the C-reactive protein via the phosphocholine-terminated self-assembled monolayer linker is considered to be the source of the robust electromagnetic enhancement. The high level (109 to 1010 M−1) of apparent binding constant (KA) of C-reactive protein suggests that the immobilized surface was well-oriented without extreme random stacking. A Raman sensitivity toward the C-reactive protein around 2800–3000 cm−1 was noted, which gradually increased upon the addition of successive layers up to approximately 6–7 layers of phosphocholine-coated silver nanoparticle aggregates, with minimum detection amounts of ∼0.01 ng mL−1 in buffer and ∼0.1 ng mL−1 in 1% serum. A cross-reactivity test confirmed the excellent selectivity and specificity of the measured signals. A computational study based on the finite-difference-time-domain method successfully demonstrated the enhanced (∼1.1 × 106) electromagnetic field of the 2-D silver nanoparticle aggregates as compared with that of isolated particles, and was congruent with the analytical enhancement factor (1.7 × 105).
Introduction
Raman spectroscopy is a non-destructive and label-free modality for analytical science due to its ability to detect vibrational, rotational, and other low-frequency modes in a given system. However, it has limited applications owing to the low scattering cross-section.1 Several technical approaches have been developed to overcome this limitation, of which surface-enhanced Raman scattering (SERS) is a popular approach.2 Briefly, the SERS effect is obtained by positioning target molecules (or a single molecule) in the vicinity of a localized free electron-rich metallic surface (i.e., Au, Ag, and Cu) to compensate for the low Raman signal due to the effect of the electromagnetic field. SERS is often utilized in the design of nanoscale metallic structures for maximized field enhancements that are often highly dependent on the size, shape, and geometry of the nanomaterials. Notably, SERS was demonstrated with the use of honeycomb-shaped metallic island arrays by Hulteen and van Duyne in 1995,3 where periodic islands exhibited uniformly distributed Raman scatterings by the arbitrarily positioned illumination of excitation photons. Additionally, Nie and Emory reported single molecular Raman scattering events of rhodamine dye in mixtures with silver nanoparticles in atmosphere.4 Other studies subsequently disclosed that the gaps between metallic structures, so-called hotspots, are important in creating highly localized plasmons to boost the intended Raman scattering cross-sections. Therefore, subsequent endeavors to obtain stable Raman-enhancing substrates/chips have focused on improving the “quality” of hotspots.5
Clustered nanostructures are one of the most robust sources for stable and strong collective electromagnetic enhancements.6 The aggregation of nanostructures can occur randomly (e.g., theoretical affine fractals7) or by design (e.g., peptide-tethered branched nanoparticle clusters8). In regard to randomly generated structures, researchers are highly interested in maximizing localized surface plasmons within the focal volume of the Raman excitation laser beam, which leads to the fabrication of fractal nanoclusters,9 where excited localized dipoles assist in the indiscriminate field enhancement of Raman, Rayleigh, and even nonlinear optical scattering. However, low control over the number of hotspots and limited signal reproducibility associated with the randomly formed fractals hinder the potential for practical applications. Thus, engineered SERS substrates (as described in regard to nanosphere photolithography) have extensively evolved towards the design of metallic islands10 or nanoparticle arrays.11 However, the high price and time consumption inherent to engineered SERS substrates has led researchers to exploit coffee ring-like structures, which are formed at the edge of dried nanoparticle-analyte drops, as cheap and easily prepared SERS substrates.12
SERS has also been applied to the detection of biomolecules such as DNA oligomers,13 bacteria,14 and biofilms,15 all of which are feasible by the use of properly chosen optical dyes. As such, aggregates of noble nanometallic colloids are typically utilized to create hotspots of SERS signals, in some cases without the use of any Raman-specific labels. SERS research elucidated the vibrational fingerprints of analytes in contact with silver colloids, suggesting the morphology of the contacts between the molecules and metallic nanoparticles.16 However, direct quantitative monitoring of ligand–protein interactions using the vibrational modes of target molecules via SERS has been scarcely reported.17,18
C-reactive protein (CRP) is a ubiquitous biomarker found in the blood stream, and its levels are sensitive to diet, smoking, and mental condition. Less than 1 μg mL−1 is detected in healthy human blood, but increases 100-fold during infection and inflammation.19 Clinically, CRP is correlated with physiological mechanisms such as acute inflammation, diabetes, and cardiovascular diseases.20 CRP is a protein complex (∼125 kDa) composed of five identical subunits (∼25 kDa each) in a cyclic pentagon-shaped (i.e., pentraxin family) structure held together by non-covalent interactions.21 CRP binds to phosphocholine (PC) expressed on the surface of dying cells to accelerate necrosis, rendering PC one of the most eligible moieties for the detection of CRP.22 The detection of CRP has been established based on enzyme-linked immunosorbent assay (ELISA),23 surface plasmon resonance (SPR),24 and magnetic nanoparticles.25 Recently, SERS was used to detect CRP by the generation of colored labels through an enzymatic reaction on CRP antibodies, as in ELISA (detection limit: ∼5 ng mL−1) with a detection level of ∼0.3 ng mL−1.26 The direct detection of CRP in blood plasma was also demonstrated using near-IR Raman spectroscopy assembled with a post data processing algorithm, with a detection limit of ∼30 μg mL−1.27 However, the detailed procedure of a label-generating method required the use of gold nanoparticles and was not clearly discriminated from the complex surface chemistry of ELISA; the detectability of the near-IR Raman spectroscopy method was poor.
We have developed a label-free SERS experiment to monitor CRP-specific ligand–protein interactions. A reflection-free SERS chip using concentration-induced Ag nanoparticle aggregates (AgNAs) allowed for the qualitative detection of the PC–CRP interaction at the C–H stretching vibrational mode (2800–3000 cm−1); the resolvability of this mode was superior to that in the fingerprint (1000–1600 cm−1) and low frequency (∼900 cm−1) regions. The apparent binding constants calculated from the SERS measurements (109 to 1010 M−1) were comparable to those obtained via fluorescence assays and SPR measurements, and indicated a sensitivity of ∼0.01 ng mL−1 (100 fM). Moreover, this method can be applied to the detection of CRP in serum with a signal to noise level of 20. SERS depended on the thickness of the AgNAs, and finite-difference-time-domain (FDTD) calculations revealed that the collective enhancement factor from AgNA reached 5–6 orders of magnitude in well-aligned 2-D array structures.
Experiments
Fabrication of a capillary effect-driven PC-functionalized AgNA SERS chip
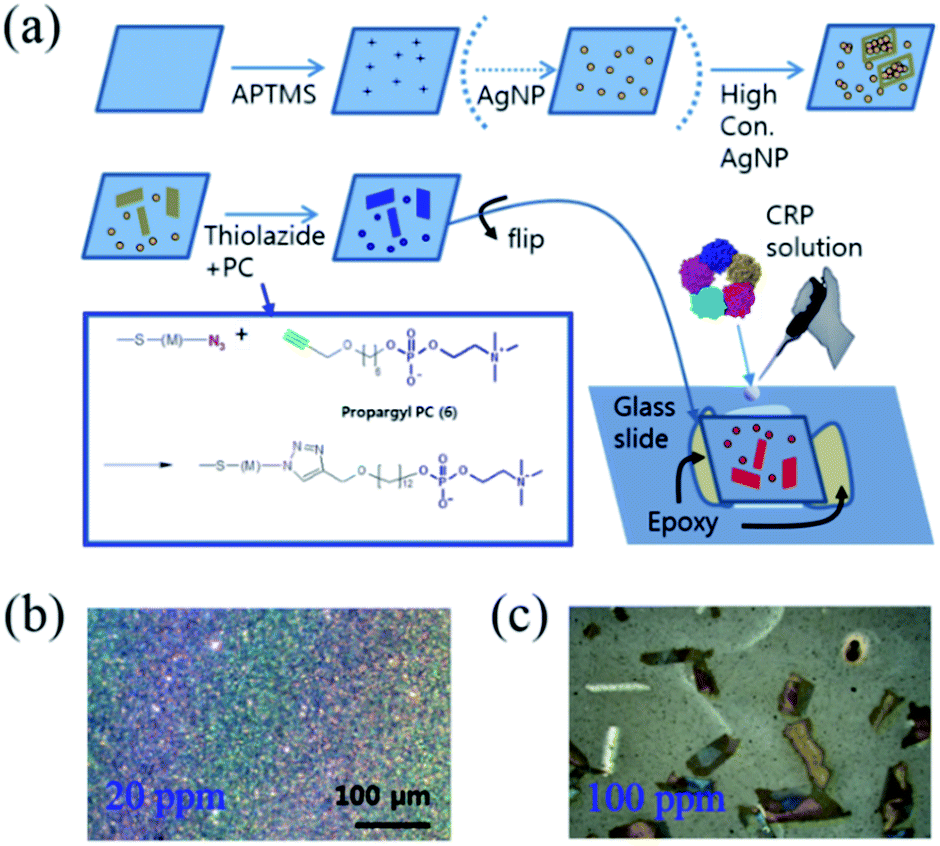
For SERS-based protein detection, microfluidics have been frequently adopted to control the reaction dynamics between the ligands and target proteins coupled with immunoassays, where one of the sandwiched antibodies holding the target molecule also contains a catalytic molecule to produce Raman labels for signal detection. In this case, SERS hotspots are created by bifunctional contacts with gold nanoparticles,28 or gold nanoarrays in the microfluidic device.29 It is unclear whether the aforementioned methods are more focused on the fluency of sample preparation or the efficient maximization of the SERS enhancement factor; the combination of both properties has not been reported in any SERS-related methods reported to date. Fig. 1(a) shows the stepwise functionalization of PC on the concentration-induced AgNAs and the creation of a capillary gap allowing for the controlled release of the protein-containing solution. The synthetic details regarding the chemical moieties are given in the ESI.† Briefly, commercially available thin cover slips (22 mm × 22 mm) were extensively cleaned with ethanol and water and dried prior to use. Then, the glass substrate was immersed in a piranha solution (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (v/v) concentrated sulfuric acid:30% hydrogen peroxide) for 30 min in order to hydroxylate the glass surface. The substrate was rinsed with water and coated with organosilane by immersing the glass substrate in a 2% 3-aminopropyltrimethoxysilane (APTMS) toluene solution overnight. Colloidal silver nanoparticles (730807, Sigma-Aldrich, Co., Ltd.) were centrifuged at 13000g and re-suspended in water (one tenth of the initial volume of the silver nanoparticle (AgNP) colloid solution). In this way, we achieved 200 ppm of concentration from the stock solution (0.02 mg mL−1, 20 ppm). The APTMS-modified glass substrate was immersed in variously concentrated AgNP solutions (10 ppm–100 ppm) for 24 h at room temperature. Note that the morphological variations of the coated AgNPs were determined by the concentration of the applied colloidal AgNP solution from the well-distributed monolayer films (Fig. 1(b)) to the scattered AgNAs (Fig. 1(c)), where aggregates were formed at concentrations higher than 50–60 ppm. To immobilize PC on the AgNP surface, 2-(2-(2-azidoethoxy)ethoxy)ethyl 11-thioundecanoate (azido-thiol) and 6-propargylhexyl-phosphorylcholine (propargyl-PC) were synthesized (Fig. S1–S3†) and conjugated using click chemistry. To prepare the PC-immobilized AgNAs, the glass substrate coated with concentration-induced Ag cluster was immersed in 2 mM ethanolic solution (2 mL) of azido-thiol for 12 h at room temperature. The substrate was thoroughly rinsed with ethanol several times and then dried in air. To modify the surface of AgNPs with PC, the click chemistry, 1,3-dipolar cycloaddition reaction, was used.30 It was performed by dropping 10 μL Tris buffer solution (0.1 M Tris, pH 8.0) containing 0.5 mM copper sulfate(II) pentahydrate, 1 mM sodium ascorbate, and 1 mM propargyl-PC on the glass substrate coated with the azido-thiol and incubated for 16 h at 4 °C. The molar ratio of copper sulfate:sodium ascorbate:propargyl-PC was 0.5:1:1. To prepare control surface without PC, propargyl-alcohol, instead of propargyl-PC, was introduced to the glass substrate as the same method described above. Finally the substrates were thoroughly washed with deionized water. As the last step, the PC-immobilized AgNAs-containing glass coverslips were flipped and adhered to the glass slide using epoxy bonds to create a small gap (thickness: <1 mm) between the coverslips and glass slides to permit the capillary immersion of the protein-containing solution. Note that the removal of the reacting solution was achieved by blowing the smeared solution with an N2 gas gun. The concentration-dependent formation of AgNAs are featured in the Fig. S4.† The collection of Raman spectra from the CRP-containing solutions of various concentrations was facilitated without the use of complicated microfluidic microelectromechanical systems (MEMS).
:3 (v/v) concentrated sulfuric acid:30% hydrogen peroxide) for 30 min in order to hydroxylate the glass surface. The substrate was rinsed with water and coated with organosilane by immersing the glass substrate in a 2% 3-aminopropyltrimethoxysilane (APTMS) toluene solution overnight. Colloidal silver nanoparticles (730807, Sigma-Aldrich, Co., Ltd.) were centrifuged at 13000g and re-suspended in water (one tenth of the initial volume of the silver nanoparticle (AgNP) colloid solution). In this way, we achieved 200 ppm of concentration from the stock solution (0.02 mg mL−1, 20 ppm). The APTMS-modified glass substrate was immersed in variously concentrated AgNP solutions (10 ppm–100 ppm) for 24 h at room temperature. Note that the morphological variations of the coated AgNPs were determined by the concentration of the applied colloidal AgNP solution from the well-distributed monolayer films (Fig. 1(b)) to the scattered AgNAs (Fig. 1(c)), where aggregates were formed at concentrations higher than 50–60 ppm. To immobilize PC on the AgNP surface, 2-(2-(2-azidoethoxy)ethoxy)ethyl 11-thioundecanoate (azido-thiol) and 6-propargylhexyl-phosphorylcholine (propargyl-PC) were synthesized (Fig. S1–S3†) and conjugated using click chemistry. To prepare the PC-immobilized AgNAs, the glass substrate coated with concentration-induced Ag cluster was immersed in 2 mM ethanolic solution (2 mL) of azido-thiol for 12 h at room temperature. The substrate was thoroughly rinsed with ethanol several times and then dried in air. To modify the surface of AgNPs with PC, the click chemistry, 1,3-dipolar cycloaddition reaction, was used.30 It was performed by dropping 10 μL Tris buffer solution (0.1 M Tris, pH 8.0) containing 0.5 mM copper sulfate(II) pentahydrate, 1 mM sodium ascorbate, and 1 mM propargyl-PC on the glass substrate coated with the azido-thiol and incubated for 16 h at 4 °C. The molar ratio of copper sulfate:sodium ascorbate:propargyl-PC was 0.5:1:1. To prepare control surface without PC, propargyl-alcohol, instead of propargyl-PC, was introduced to the glass substrate as the same method described above. Finally the substrates were thoroughly washed with deionized water. As the last step, the PC-immobilized AgNAs-containing glass coverslips were flipped and adhered to the glass slide using epoxy bonds to create a small gap (thickness: <1 mm) between the coverslips and glass slides to permit the capillary immersion of the protein-containing solution. Note that the removal of the reacting solution was achieved by blowing the smeared solution with an N2 gas gun. The concentration-dependent formation of AgNAs are featured in the Fig. S4.† The collection of Raman spectra from the CRP-containing solutions of various concentrations was facilitated without the use of complicated microfluidic microelectromechanical systems (MEMS).
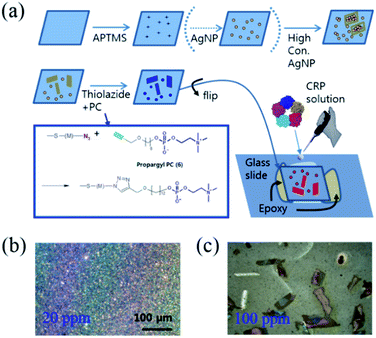 |
| | Fig. 1 (a) Diagram of the synthesis of the SERS substrate using 3-aminopropyltrimethoxysilane (APTMS), silver nanoparticles (AgNPs), phosphorylcholine (PC), and C-reactive protein (CRP). A capillary gap was created to control the flow-in/out of CRP solutions. Phosphocholine (PC) was linked to silver nanoparticle aggregates (AgNAs) using click chemistry. Optical images of silanized glass surfaces coated by a colloidal AgNP solution of (b) 20 ppm and (c) 100 ppm, respectively. The formed AgNAs are seen as ∼100 μm scale colored plates. | |
Results and discussion
SERS detection of C–H stretching modes of CRP
The high-frequency Raman range (2600–3100 cm−1), which corresponds to the vibrational overtones of the fingerprint region (1000–1700 cm−1),31 has recently garnered attention since the measured Raman signals are stronger and more spectrally resolvable than the fingerprint regions.32 For instance, the D′ band (∼2650 cm−1) in isolated graphene flakes was found to be intrinsically strong and highly sensitive to the number of fundamental layers.33 The C–H stretching of benzene rings (∼3050 cm−1) is a good Raman indicator to chemically identify polymers. Additionally, linear symmetric/asymmetric C–H stretching modes (2800–3000 cm−1) have been exploited in coherent Raman scattering (CRS) microscopy due to the signal sensitivity in lipids and proteins.34 Notably, SERS in the high-frequency Raman range was also employed to analyze the adsorption of small biomolecules such as amino acids and DNA bases on colloidal AgNPs35 and to obtain pictures of cell vibrations.36
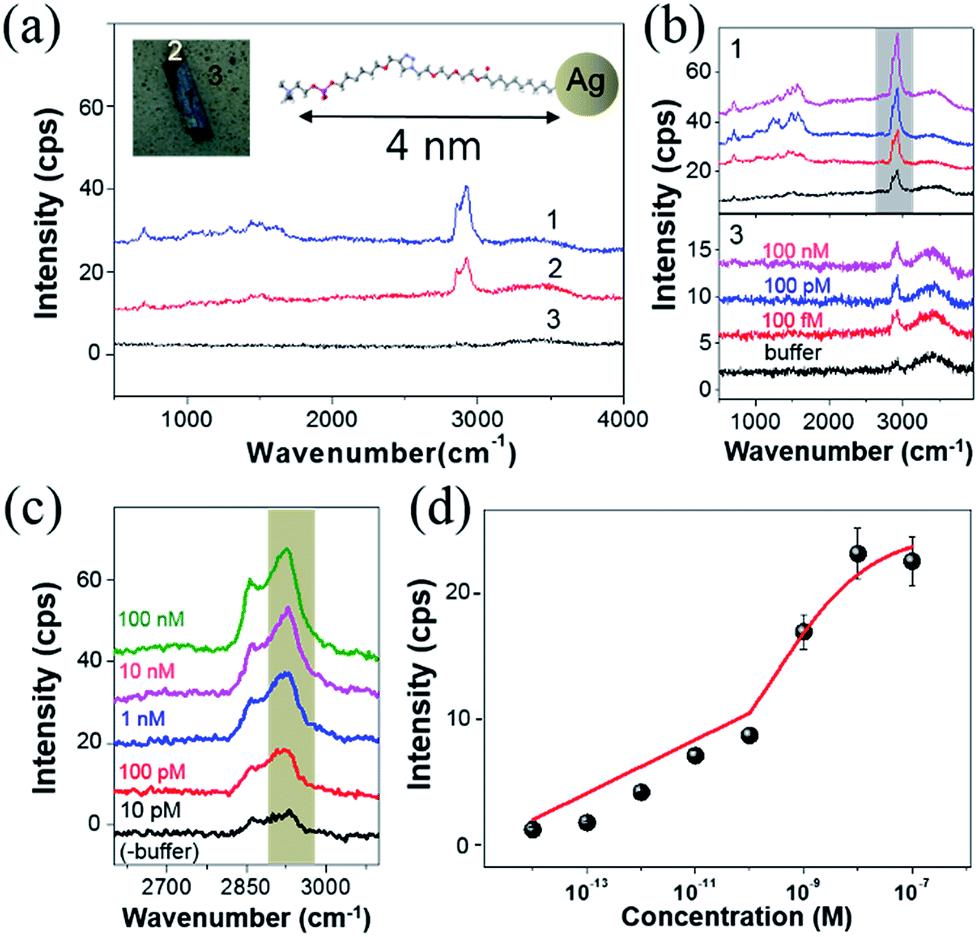
Fig. 2(a) shows the SERS spectra from three different regions around one of the AgNAs indicated with numbers (1–3). Note that all regions were functionalized with PC, as indicated in the figure inset, where differences originate from the density of the AgNPs (see also Fig. S5 and S6†). The overall Raman intensity was the highest for 3, and was the lowest and almost unrecognizable for 1. Fig. 2(b) shows the Raman intensity variation when the same substrate was treated with CRP solutions of various concentrations from Tris buffer (0.1 M) solution (control) to 100 nM. The concentration of Ca2+, which is known to be essential in PC–CRP conjugation, did not critically affect the reaction if it was in the test solution (as confirmed by SPR in Fig. S7†) and was fixed at 5 mM in all experiments. In spectra set 3 in Fig. 2(b), no discernible change was observed, while there was an overall increase in the Raman intensity when the concentration of the CRP solution was increased (spectra set 1). For a clearer understanding of the spectra, the C–H stretching region was zoomed in according to the CRP concentration, and each spectrum was subtracted from that of the buffer solution. The results for the C–H stretching region (grey shadow) are presented in Fig. 2(c). In this work, no clear evidence of the concentration dependence of the Raman signal increase in the fingerprint region (1000 cm−1 to 1700 cm−1) could be obtained. The collected signals fluctuated even with fixed concentrations according to the position-to-position variation of collected data, which was attributed to the photocarbonization effect frequently noted in SERS experiments.37,38 It has been suggested that the design of new AgNA hybrid structures with 2-D hexagonal atomic layered materials, such as graphene or MoS2, could drastically promote charge transfer between analytes and AgNAs and may allow for the measurement of the fingerprint region via chemical enhancement effects.39 In contrast, the Raman spectra of the C–H stretching region had a high S/N ratio (>10) and demonstrated a distinct change with increased concentrations of CRP. A drastic increase in the asymmetric C–H vibrational mode (∼2930 cm−1) was observed with increased CRP concentrations. Notably, this signal variation is known to be closely related to the amount of protein present.40 The Fig. 2(d) reveals that the concentration dependence of the Raman peak intensity followed a Langmuir isotherm-type trend with an apparent binding constant (KA) of 5.27 × 10−9 M−1 and high level of statistical tolerance (R2 = 0.97) (see also Fig. S6†). Overall, even if the performance of the PC–AgNA system as a SERS detector at the C–S stretching mode was a bit disappointing owing to the low signal increase upon the addition of CRP, a significant Raman signal enhancement was observed in the C–H stretching mode (2800–3000 cm−1) with a solid 100 fM (∼0.01 ng mL−1) detection level.
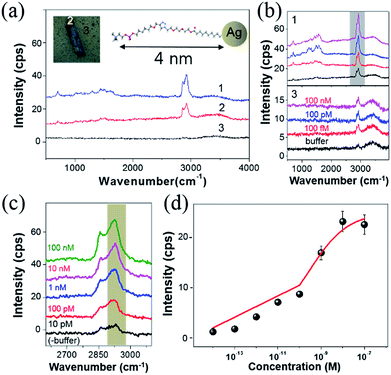 |
| | Fig. 2 (a) Raman spectra of PC-functionalized AgNAs for three different vibrational modes. Inset shows the functionalized PC on top of AgNAs in a dimensionally exaggerated manner. (b) Raman spectra taken from 1 and 3 after injecting CRP solutions of varying concentrations (from buffer to 100 nM). (c) Raman intensity increase in the C–H vibrational modes. (d) CRP-concentration dependent plot of the Raman signals for highlighted maximum peaks in (c). | |
Evaluation of the specificity of CRP–PC conjugation in SERS
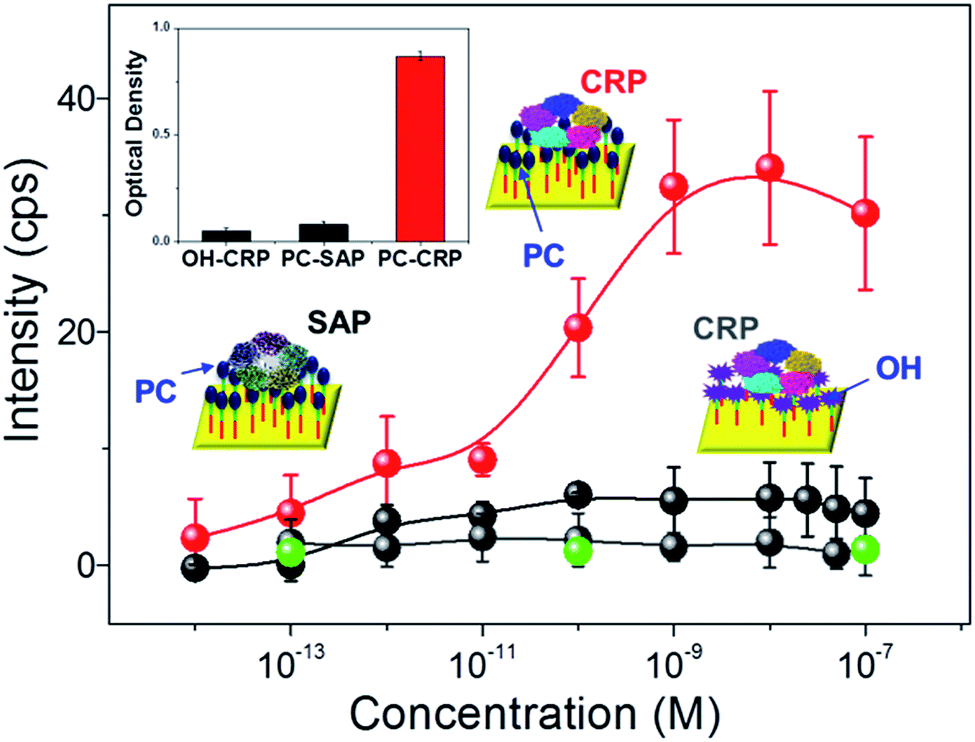
Reducing the non-specific protein binding to target ligand-functionalized surfaces is essential in producing selective biomolecule sensors and has been achieved using surface property-altering chemicals such as bovine serum albumin (BSA),41 and diverse SAMs.42 Notably, SAMs are often utilized due to their facile fabrication when combined with other moiety-specific complex surface modification processes, leading to the creation of hydrophilicity or hindering the formation of electrostatic forces that prevent the unwanted adsorption of non-targeted proteins. The tailored non-specific adsorption of CRP in a rationally designed SAM bridge allows for the creation of molecularly imprinted polymer (MIP) arrays on gold surfaces, which possess binding affinities 2–3 orders of magnitude higher than that of SPR and ELISA.43 Here, the specificity and selectivity of the reaction between the SAM–ligand treated surface and proteins in solution were examined through Raman spectroscopy. Fig. 3 summarizes the changes in Raman peak intensity at arbitrary PC/OH functionalized AgNAs (200 nm thick), where the Raman signals were well-resolved as a function of the reacting protein (CRP, serum amyloid P component (SAP), and phosphatidyl choline transfer protein (PCTP) concentration. SAP is another protein from the pentraxin family that consists of five subunits (25 kDa) and shows a ∼51% resemblance to CRP in regard to the protein sequence and selective reactivity with proline terminating groups, but not with PCs; thus, it is frequently selected for cross-reactivity tests of CRP.44 PCTP is not homologous to CRP in protein sequence, but it binds to PC head group of phosphatidyl choline.45 As the inset cartoon illustrates, the ligand bridges are composed of hydrophobic C–H chains connected to AgNAs and hydrophilic PC/OHs, where CRP and water can approach and the specificity of CRP binding can be unambiguously tested. First, a concentration-dependent plot of the attachment of CRP to the PC-functionalized surface was obtained at 2930 cm−1 in the Raman spectra; SAP, PCTP, and the OH-functionalized surface were used as controls for cross-comparison of the CRP link to PC. An increase in attachment was observed for CRP concentrations of 10−12 to 10−9 M, while that of SAP was not increased in the corresponding concentration regime, demonstrating that the PC–AgNA system had ultrahigh selective sensitivity. In addition, no significant binding of PCTP was observed on the PC-functionalized substrate, which indicated that the Raman spectroscopic analysis with the prepared substrates was highly specific to CRP detection. Furthermore, the attachment of CRP to the substrates was attributed to the PC functionalization on the surface, because CRP binding was strictly limited to OH surface compared to that of PC surface; the intensity of CRP to PC surface was increased by more than 1 order of magnitude as judged from the difference in the net Raman spectroscopy. Importantly, the results could be reproduced after the adsorbed CRPs were removed with ethylenediaminetetraacetic acid (EDTA) from the PC-functionalized surface (Fig. S8†). Optical absorption assays of the attachment of CRP to PC were also carried out to determine a qualitative reference for the Raman measurements. The inset in Fig. 3 shows the results of the immunoabsorbant assays (at 450 nm) on the SAM control (OH) and PC-functionalized chips, which were subsequently submerged in CRP and SAP solutions. The attachments of CRP were the most prominent for the PC-functionalized surfaces treated by CRP-only solutions, confirming that interaction between CRP and PC does occur.
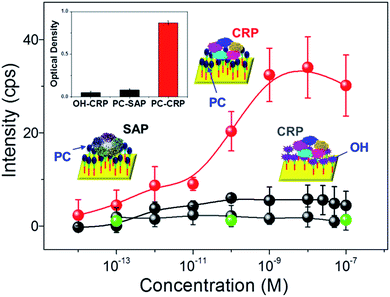 |
| | Fig. 3 Cross-reactivity of the functionalized AgNA substrate. Variations in the Raman intensity at 2930 cm−1 are shown in red (CRP) and black (SAP) with increasing concentrations of the reacting solutions applied to the PC-functionalized substrates, while that of the CRP solution interacting with the OH-functionalized substrates is shown in dark grey. Green dots correspond to the Raman intensity driven by the reaction of PCTP with PC-functionalized substrates. Inset shows the optical density measured at 450 nm in the immunocolorimetric assay (ICA). | |
SERS of CRP–PC–AgNA systems in serum
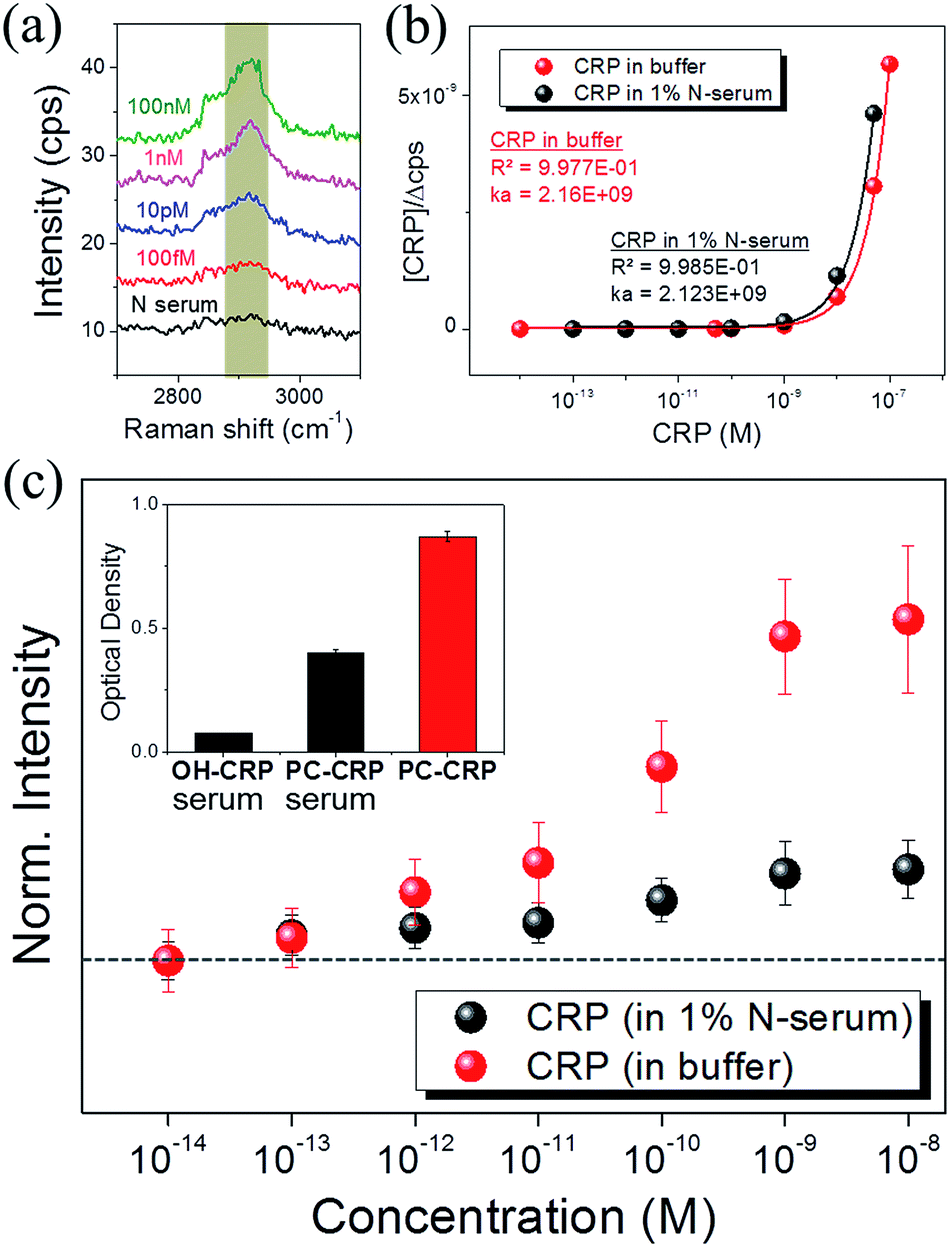
SERS of PC-functionalized AgNAs was also evaluated with 1% serum-containing CRP solutions. It is always challenging to optically monitor analytes in serum due to the presence of numerous biological light scatterers and/or absorbers that can interfere with the signal from target molecules; thus, most biosensors are reported to exhibit less molecular sensitivity in serum as compared with that in buffer systems. Fig. 4(a) shows the Raman spectra of the C–H stretching mode for various concentrations of CRP dissolved in 1% serum solution. The S/N ratio of the spectra was approximately 5, which was less than that of the spectra presented in previous figures; however, the intensity of the C–H mode (2930 cm−1) increased as the concentration of CRP in serum increased. Fig. 4(b) shows the calibration curve used to calculate the binding constant (KA) of CRP to PC in buffer and serum.
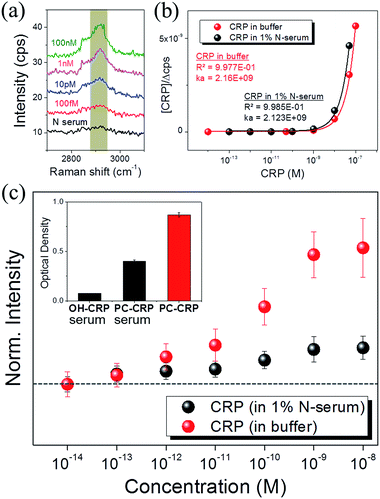 |
| | Fig. 4 (a) Variation in peak intensities (C–H stretching at ∼2930 cm−1) according to the CRP concentration dissolved in 1% N-serum measured by Raman spectroscopy. (b) Calibration curve calculated by the Langmuir isotherm to determine the binding constant of CRP in pure solution and 1% N-serum. (c) Normalized Raman peak increase versus CRP concentration in the pure solution and 1% N-serum. Inset shows CRP binding capacity in 1% N-serum determined by ICA analysis. | |
The Langmuir adsorption model allows for the calculation of KA through the following equation:46
| |
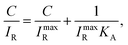 | (1) |
where
C is the concentration of CRP,
IR and
ImaxR are the Raman intensities when the concentration of CRP is specified and extrapolated to infinity, respectively, and
KA is the fitted binding constant. The
KAs were 2.16 × 10
9 and 2.12 × 10
9 M
−1 (
R2 > 0.99) for CRP to PC on AgNAs in buffer and serum, respectively, both of which were comparable to those obtained in previous reports,
47 indicating that the immobilized surface was well-oriented without extreme random stacking.
48 Notably, there was no significant difference in the
KA values obtained in buffer or serum. To qualitatively examine the performance of the SERS detection in buffer and serum, we compared the CRP concentration dependence of the CRP–PC reactivity (
Fig. 4(c)). The plot was normalized to the lowest concentration (10 fM) of added CRP; all data sets collected from each concentration were averaged. The log scale plot on the
x-axis nicely resolves the gradual increase in the Raman peak intensity even around 10–100 pM (12.5 ng mL
−1) where the surface coverage is drastically saturated for both cases, albeit the degree of signal enhancement was different by 3–4 times. Based on the measured and calculated information, the CRP sensitivity of our system reached ∼0.01 ng mL
−1 (100 fM) in buffer and ∼0.1 ng mL
−1 in 1% serum, where the signal was discernible with
S/
N ratio greater than 3 at the target concentration. The inset bar-diagram summarizes the immunoabsorbant assays of CRP reactivity with respect to the PC/OH-functionalized surface in serum as compared with the reactivity of CRP with PC in buffer. This data confirms that the CRP reactivity was moiety selective in serum, but was inferior to that in buffer by approximately 2 times.
AgNA thickness dependence of CRP–PC–AgNA in SERS
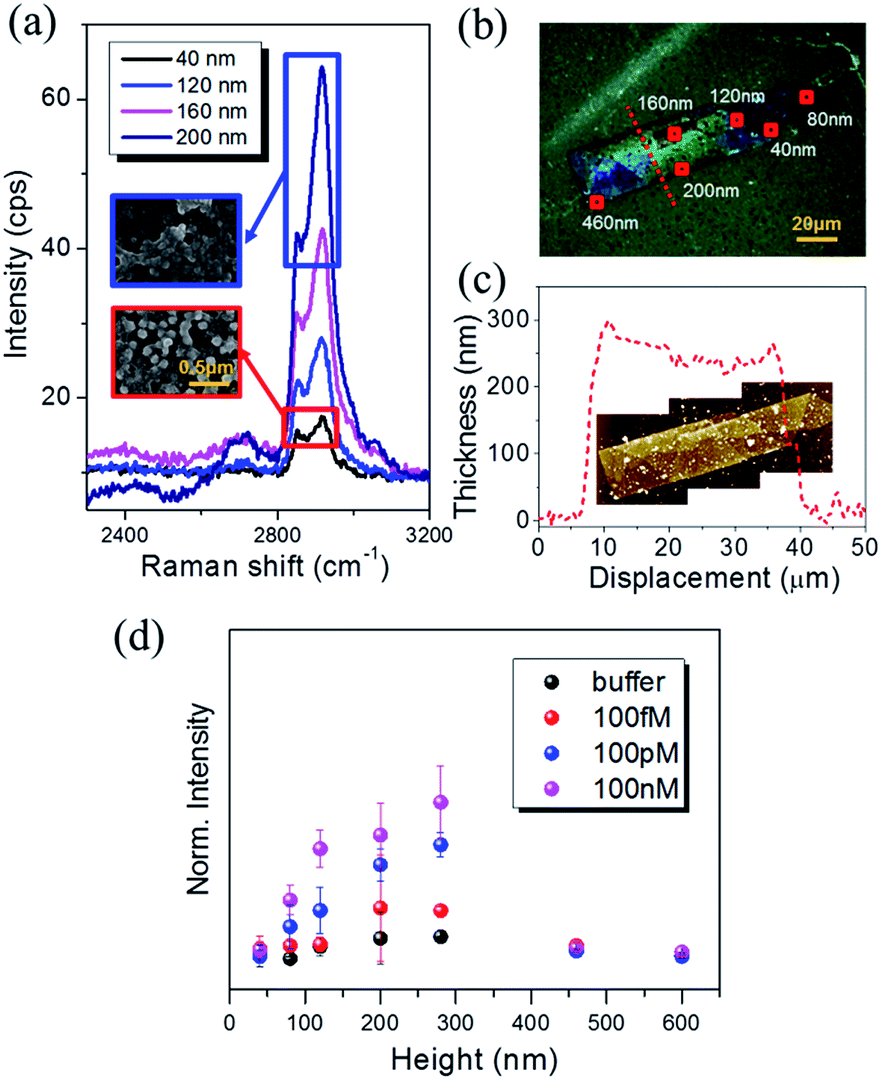
Fig. 5(a) summarizes the Raman spectra of CRP–PC–AgNA systems with varying thicknesses and clearly demonstrates that the peak intensity of 2930 cm−1 gradually increases as the thickness of AgNA increases up to 200 nm. The inset SEM images show the morphology of the vacuum-dried CRP–PC–AgNA surface after the CRP coating was formed over the AgNAs at two different thicknesses. The SEM reveals that CRP was densely adsorbed to the AgNAs (5 layers; 200 nm) as a thick biological film, whereas attachments were quite scarce; even individual nanoparticles were microscopically resolvable in the single-layered AgNA (40 nm) region. Abundant asymmetric C–H stretching modes (2930 cm−1) were observed. Moreover, the asymmetric mode drastically increased with an increase in AgNA thickness compared with the symmetric C–H stretching mode (∼2850 cm−1). The thickness of each position was assigned by correlating the optical microscopy image with the AFM image, as exemplified in Fig. 5(b) and the inset of (c). Fig. 5(c) shows the line profile for the dashed line in (b). Through a careful AFM study, some regions over 300 nm thick were identified, but were very rare except in some wall-like edges of the AgNAs. Fig. 5(d) summarizes the thickness dependence of the SERS signal at 2930 cm−1 for CRP aqueous solutions with concentrations ranging from 100 fM to 100 nM. Generally, the SERS signals increased with increasing AgNA thicknesses and gradually plateaued around 280 nm (7 layers). Notably, the SERS intensity in buffer showed a similar tendency, presumably induced by the SERS from the ligand group (SAM + PC) as shown in Fig. 2(a). Because the Raman signals from CRP adsorbed to one PC-treated AgNA system are proportional to the number of adsorbed molecules in the nanoparticle system, the peak intensities of the measured Raman spectra were saturated as the concentration of the added solution increased, following a Langmuir-isotherm-like surface coverage. Notably, the SERS is proportional to the number of hotspots, assuming that the correction factors can be ignored for each hotspot with reference to the variation in geometric factors and Raman cross-sections. The Raman signal increase with increased thickness may result from the CRP molecules near hotspots participating in Raman scattering. For thicknesses between 300–600 nm, a decrease in the transmittance of AgNAs must be adopted to assess the tailoring of SERS signals. Importantly, this concept has also been used to explain the decrease in the linear/nonlinear optical response above a critical thickness in planar structures such as multi-layered graphene49 or non-plasmonic TiO2 3-D frameworks.50
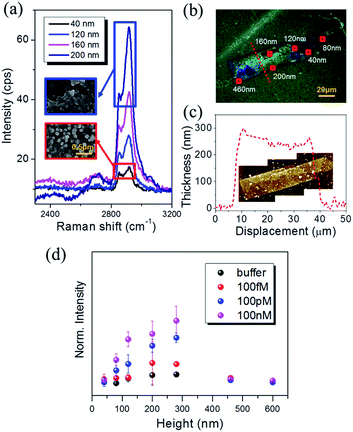 |
| | Fig. 5 (a) Raman spectra plotted versus the AgNA thickness with the addition of 100 pM CRP. Inset SEM images show the morphological difference of CRP coatings adsorbed on monolayer (red squared) and multiple-stack (blue squared) AgNA, respectively. The scale bar corresponds to 100 nm. (b) Optical microscopy image and (c) AFM line profile along the red dotted line in (b). Inset AFM image was obtained for the identical imaging area in (b). See also Fig. S9† for AFM measurement. (d) Variation in the normalized Raman peak intensity with increasing AgNA thickness. | |
Theoretical approach to AEF of SERS in the CRP–PC–AgNA system
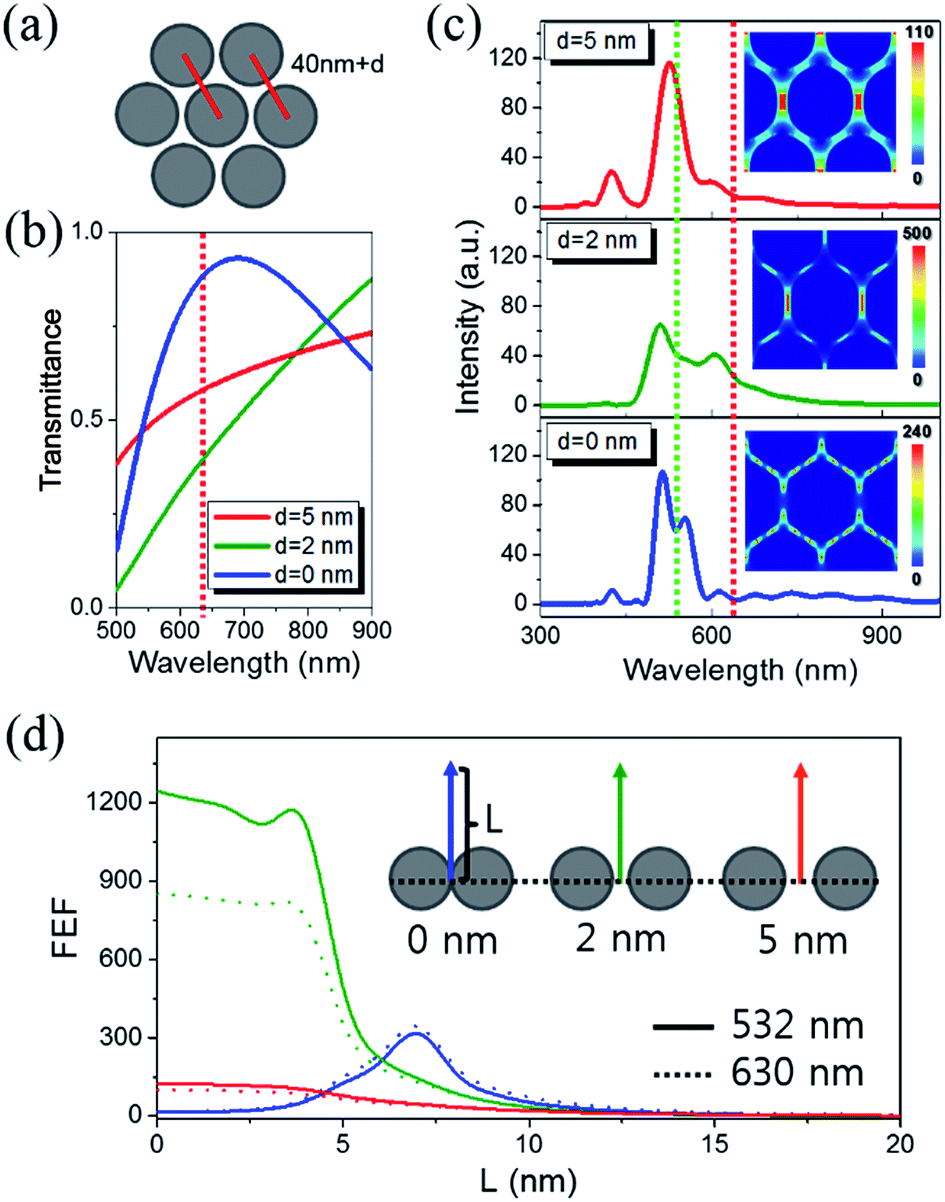
The analytical enhancement factor (AEF), defined as the ratio of the differential cross-sections of molecules in adsorbed and reference conditions,51 becomes the Raman intensity ratio of the molecules participating in the generation of Raman signals under each condition at a fixed collection time, similar signal collecting configurations, and known surface adsorption/coverage. To obtain the reference data, we acquired the Raman spectrum of 13 μM CRP using a 50 times higher laser power, resulting in a ∼5 cps signal after background subtraction. Using a molecular weight of 125 kDa and a 109 cm−2 surface coverage of CRP,52 the AEF value for saturated 100 nM detection (∼60 cps) on 200 nm-thick AgNA was ∼1.7 × 105. On the other hand, the structural SERS enhancement factor (gSERS) can be independently calculated from the contribution of the electromagnetic field enhancement for a certain nanostructure:53| | |
gSERS ∝ |FEF(λE)|2|FEF(λRS)|2.
| (2) |
where FEF(λE) and FEF(λRS) are the field enhancement factors created at the wavelength of the excitation beam and Raman scattered beam, respectively. The concept that the nonlinear optical quantity in a given metallic geometry is proportional to the multiplication of the square of the local field enhancement of the involved wavelengths of photons is also applied in second harmonic generation (SHG),54 two photon fluorescence (TPF),55 and four-wave mixing (FWM).56 If the given metallic geometry is confined to the tightly localized vicinity of hotspots, the gSERS of the region of interest can be approximated by multiplication of two FEF(λ)s, which can be calculated using FDTD simulations. Thus, we examined the difference in field enhancements generated from hotspots with differing configurations of nanoparticles, namely 2 or 4 particles, and 2-D arrays with variations in the interparticle distance, d. Fig. S10(a)† summarizes the calculated frequency-dependent electric field intensity and shows the resonance wavelengths of different array geometries. Since the reference fields for the three arranged states were different, the data was normalized after taking their ratio to correlate the spectral dependence. Accordingly, the surface plasmon resonance of the 2-D array was so widely distributed that it could be overlaid with the excitation wavelength (λL = 532 nm) and scattered wavelength of the C–H stretching mode at 2930 cm−1 (λRS = 630 nm). Fig. 6(b) shows the transmittance for 2-D arrayed AgNAs as a function of d; the geometry is described in Fig. 6(a). The transmittance calculated for interparticle distances of 0, 2, and 5 nm at 630 nm (wavelength of C–H stretching) was ∼0.85, ∼0.35, and ∼0.6, respectively. Thus, the light transmittance of AgNAs with more than 10 layers would be practically zero regardless of d, which is in close agreement to the tailored nature of the thickness dependence above 300 nm shown in Fig. 5(c). Fig. 6(c) illustrates the frequency-domain field intensity spectra for 2-D array structures when d is 0, 2, and 5 nm, showing that the 2 nm periodic 2-D arrays resonates the most at 532 and 630 nm and maximally enhances the electromagnetic field as shown in the inset 2-D contour diagrams. Note that the calculated values are presented as the intensity, i.e., the square of the field. Thus, the electromagnetic field enhancement factor in a defined gap geometry can be achieved if we take |FEF(λ532)|2 and |FEF(λ630)|2 as 14 and 16 for d = 0 nm, 1310 and 867 for d = 2 nm, and 290 and 193 for d = 5 nm; the gSERS is then ∼2.1 × 102 (∼2.1 × 104 at 7 nm), ∼1.1 × 106, and ∼5.6 × 104, respectively. For more detailed discussion, we also calculated FEF as a function of the distance from the center of the inter-particle gap (L). As in Fig. 6(d), we can clearly observe that the level of FEF is retained until L is increased to 4 nm for each inter-particle spacing condition and even increased more by one order of magnitude (at ∼7 nm) when the particles are contacted. See also Fig. S10(b)† to have some hints of the actual size comparison between CRP's and adjacent AgNP. To theoretically emulate the experimental results regarding the AgNA thickness dependence, evolution of the electromagnetic field around the hotspots in the 3-D AgNP arrayed frameworks should be achieved. However, due to a technical limit in our program, we could not obtain the gSERS of 3-D arrayed structures in this work. Regarding this issue, Lee and Irudayaraj claimed that metallic nanoparticle clusters formed by DNA linkers can enhance the field by a factor of ∼107, which is ∼10 times higher than that of 2-D arrays.57 The use of this 10-fold enhancement is especially relevant when forming 3-D network structures to explain the thickness dependence of the signal enhancement, after adjusting the signal for the disturbance induced by the augmented surface plasmon modes (Fig. 6(c)) upon the increase in the thickness, and also explains the peak intensity plateau for thicknesses greater than 300 nm. Under the assumption that this 10-fold enhancement according to the dimensional change can be applied to our system, the gSERS of 3-D arrayed structures in this work will be ∼2.1 × 104 (∼2.1 × 106 at 7 nm), ∼1.1 × 108, and ∼5.6 × 106, respectively. These values might reasonably explain the AEF value (1.7 × 105) quite well.
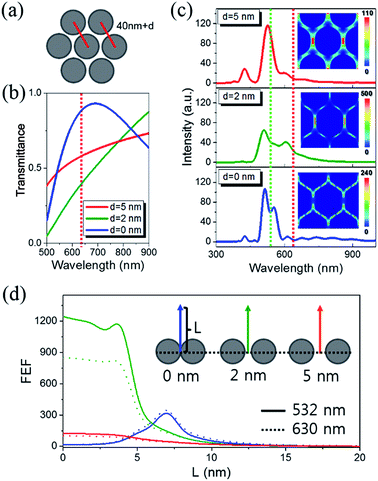 |
| | Fig. 6 (a) A simple representation of a hypothetical single-layered AgNA with an inter-particle distance of d. (b) Optical transmittance and (c) frequency-domain electric field intensity for 2-D arrays. Insets in (c) show the field enhancements calculated for the hotspots when the excitation beam (horizontal polarization) was 532 nm. The green dotted line marks 532 nm, and the red dotted line marks 630 nm. (d) Calculated field enhancement factors (FEF) as a function of the distance from the center of inter-particle gap for several AgNP array conditions. | |
Conclusions
In summary, we carried out the label-free SERS detection of CRP in a more qualitative fashion without the necessity of multi-step, antibody-architect chemistry. The SERS chip was fabricated using 3-D AgNAs formed by depositing a highly concentrated AgNP solution (40 nm in size) on a silanized coverslip, followed by PC functionalization to selectively capture CRP and consequently increase the SERS efficiency. The chip contained a capillary-gap where the injection of the reacting solution could be easily controlled. An inherently short distance (<4.0 nm) between the surface of the AgNAs and CRP is responsible for the high sensitivity shown in this work, induced by the large localized plasmon resonance, which could further shorten due to the protein weight and movement. Notably, a small length difference between the radius of the AgNP (∼20 nm) and size of CRP (∼12 nm in diameter) allows a higher chance of CRP attachment to the AgNP interparticle gap, subsequently promoting possible maximized field enhancements. The sensitivity of the detection was maximized at the asymmetric C–H stretching mode at 2930 cm−1. The cross-reactivity test using SAP- and OH-functionalized AgNAs revealed that CRP to PC binding is highly selective. Given the small dynamic range (from ∼1 fM to ∼1 nM) inherent to surface coverage-dependent biosensors, the detectability of CRP was 100 fM (0.01 ng mL−1) in buffer and 1 pM in serum when 200–300 nm thick AgNAs (6–7 layers) were utilized. A 1.7 × 105 field enhancement was achieved from 40 nm thick AgNA structures using AEF calculations, which were closely reproduced by FDTD simulations (104 to 106) for various 2-D AgNA models. Furthermore, the thickness dependence was ascribed to the ensemble field enhancement (∼10 times) accompanied by the dimensional increase after being adjusted with the abated signal collection efficiency induced by the enhanced light absorption with increased thicknesses.
Methods
All chemicals used for synthesis were purchased from Sigma-Aldrich or Fluka. Phosphate buffered saline (PBS, HyClone®) was obtained from Thermo Scientific Co., Ltd. (Rochester, USA). Human plasma CRP, monoclonal mouse anti-human CRP (anti-CRP), bovine serum albumin (BSA), and phosphocholine chloride calcium salt tetrahydrate were obtained from Sigma-Aldrich Co., Ltd. (St. Louis, USA). Polyclonal goat anti-human CRP-horseradish peroxidase conjugate (anti-CRP HRP) and tetramethylbenzidine (TMB) were obtained from Koma Biotech Inc. (Seoul, Korea). Human CRP-negative serum samples and the components of the CRP latex test set were purchased from Cenogenics Corp. (Morganville, USA). All other chemicals were of analytical grade and used as received. Details of the chemical structures and physical properties of chemicals are presented in the ESI.† 1H NMR and 13C NMR spectra were obtained in CDCl3 using a Bruker NMR spectrometer (AVANCE 400). All Raman experiments were carried out using a Nicolet spectrometer (Almeca XR). FT-IR spectra were measured on a ThermoNicolet IR spectrometer (IR380). Electrospray ionization mass spectrometry was performed on a Waters Micromass ZQ (MM1) mass spectrometer. Immunoabsorbance was read with a Thermo automatic ELISA reader (Multiskan EX). Computational simulations of CRP–PC binding were carried out using Scigress Explorer (v7.7.0, Fujitsu). Scanning electron microscopy (SEM) combined with electron dispersive spectroscopy (EDS) was performed using a Hitachi SU-70 with an acceleration voltage of 15 kV. Atomic force microscopy (AFM) was carried out in non-contact mode to examine the thickness of AgNAs on a high-dimension analytical AFM (Park systems, NX-10). All theoretical simulations based on the FDTD method for electromagnetic field evolution around the 2-D AgNA systems were performed using a commercial package (Lumerical Solutions, Vancouver).
Acknowledgements
H. K, E. K and S. W. J acknowledge the provision of the basic research programs (15-NB-04, 15-NB-01, 13-NB-01)) and start-up fund (H. K.) for new researchers through the Daegu Gyeongbuk Institute of Science and Technology (DGIST). C. H. C acknowledges the Basic Science Research Program (NRF-2013R1A1A1009552) and the DGIST R&D Program (15-BD-0401) funded by the Ministry of Science, ICT, and Future Planning of Korea.
Notes and references
- D. A. Gardiner and P. R. Graves, Practical Raman Spectroscopy, Springer-Verlag, New York, 1988 Search PubMed.
- A. J. Otto, Surface-Enhanced Raman Scattering of Adsorbates, J. Raman Spectrosc., 1991, 22, 743–752 CrossRef CAS.
- J. C. Hulteen and R. P. Van Duyne, Nanosphere Lithography: A Materials General Fabrication Process for Periodic Particle Array Surfaces, J. Vac. Sci. Technol., A, 1995, 13, 1553–1558 Search PubMed.
- S. Nie and S. R. Emory, Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- C. Won Joon, K. Youngsuk and K. Jin Kon, Ultrahigh-Density Array of Silver Nanoclusters for SERS Substrate with High Sensitivity and Excellent Reproducibility, ACS Nano, 2012, 6, 249–255 CrossRef PubMed.
- E. S. Shibu, K. Kimura and T. Pradeep, Gold Nanoparticle Superlattices: Novel Surface Enhanced Raman Scattering Active Substrates, Chem. Mater., 2009, 21, 3773–3781 CrossRef CAS.
- J. A. Sánchez-Gil and J. V. García-Ramos, Local and Average Electromagnetic Enhancement in Surface-Enhanced Raman Scattering from Self-Affine Fractal Metal Substrates with Nanoscale Irregularities, Chem. Phys. Lett., 2003, 367, 361–366 CrossRef.
- V. Voliani, F. Ricci, S. Luin and F. Beltram, Peptidic coating for gold nanospheres multifunctionalizable with photostable and photolabile moieties, J. Mater. Chem., 2012, 22, 14487–14493 RSC.
- V. M. Shalaev, R. Botet, D. P. Tsai, J. Kovacs and M. Moskovits, Fractals: Localization of Dipole Excitations and Giant Optical Polarizabilities, Phys. A, 1994, 207, 197–207 CrossRef.
- C. Y. Chen and E. Burstein, Giant Raman Scattering by Molecules at Metal-Island Films, Phys. Rev. Lett., 1980, 45, 1287–1291 CrossRef CAS.
- E. M. Hicks, S. Zou, G. C. Schatz, K. G. Spears and R. P. Van Duyne, Controlling Plasmon Line Shapes through Diffractive Coupling in Linear Arrays of Cylindrical Nanoparticles Fabricated by Electron Beam Lithography, Nano Lett., 2005, 5, 1065–1070 CrossRef CAS PubMed.
- W. Wang, Y. Yin, Z. Tana and J. Liu, Coffee-Ring Effect-Based Simultaneous SERS Substrate Fabrication and Analyte Enrichment for Trace Analysis, Nanoscale, 2014, 6, 9588–9593 RSC.
- D.-K. Lim, K.-S. Jeon, J.-H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J.-M. Nam, Highly Uniform and Reproducible Surface-Enhanced Raman Scattering from DNA-Tailorable Nanoparticles with 1-nm Interior Gap, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS PubMed.
- W. R. Premasiri, D. T. Moir, M. S. Klempner, N. Krieger, G. Jones II and L. D. Ziegler, Characterization of the Surface Enhanced Raman Scattering (SERS) of Bacteria, J. Phys. Chem. B, 2005, 109, 312–320 CrossRef CAS PubMed.
- Y. Chao and T. Zhang, Surface-Enhanced Raman Scattering (SERS) Revealing Chemical Variation during Biofilm Formation: from Initial Attachment to Mature Biofilm, Anal. Bioanal. Chem., 2012, 404, 1465–1475 CrossRef CAS PubMed.
- X. S. Shen, G. Z. Wang, X. Hong and W. Zhu, Nanospheres of silver nanoparticles: agglomeration, surface morphology control and application as SERS substrates, Phys. Chem. Chem. Phys., 2009, 11, 7450–7454 RSC.
- X. X. Han, G. G. Huang, B. Zhao and Y. Ozaki, Label-Free Highly Sensitive Detection of Proteins in Aqueous Solutions Using Surface-Enhanced Raman Scattering, Anal. Chem., 2009, 81, 3329–3333 CrossRef CAS PubMed.
- M. Kahraman, I. Sur and M. Çulha, Label-Free Detection of Proteins from Self-Assembled Protein-Silver Nanoparticle Structures using Surface-Enhanced Raman Scattering, Anal. Chem., 2010, 82, 7596–7602 CrossRef CAS PubMed.
- C. Gabay and I. Kushner, Acute-Phase Proteins and Other Systemic Responses to Inflammation, N. Engl. J. Med., 1999, 340, 448–454 CrossRef CAS PubMed.
- E. T. H. Yeh and J. T. Willerson, Coming of Age of C-Reactive Protein Using Inflammation Markers in Cardiology, Circulation, 2003, 107, 370–371 CrossRef PubMed.
- M. B. Pepys and G. M. Hirschfield, C-Reactive Protein: A Critical Update, J. Clin. Invest., 2003, 111, 1805–1812 CrossRef CAS PubMed.
- D. Thompson, M. B. Pepys and S. P. Wood, The Physiological Structure of Human C-Reactive Protein and Its Complex with Phosphocholine, Structure, 1999, 7, 169–177 CrossRef CAS PubMed.
- R. Dominici, P. Luraschi and C. Franzini, Measurement of C-Reactive Protein: Two High Sensitivity Methods Compared, J. Clin. Lab. Anal., 2004, 18, 280–284 CrossRef CAS PubMed.
- W. P. Hu, H. Y. Hsu, A. Chiou, K. Y. Tseng, H. Y. Lin, G. L. Chang and S. J. Chen, Immunodetection of Pentamer and Modified C-Reactive Protein Using Surface Plasmon Resonance Biosensing, Biosens. Bioelectron., 2006, 21, 1631–1637 CrossRef CAS PubMed.
- M. H. F. Meyer, M. Hartmann, H.-J. Krause, G. Blankenstein, B. Mueller-Chorus, J. Oster, P. Miethe and M. Keusgen, CRP Determination Based on A Novel Magnetic Biosensor, Biosens. Bioelectron., 2007, 22, 973–979 CrossRef CAS PubMed.
- F. M. Campbell, A. Ingram, P. Monaghan, J. Cooper, N. Sattar, P. D. Eckersall and D. Graham, SERRS Immunoassay for Quantitative Human CRP Analysis, Analyst, 2008, 113, 1355–1357 RSC.
- M. S. Bergholt and S. Hassing, Quantification of C-Reactive Protein in Human Blood Plasma Using Near-Infrared Raman Spectroscopy, Analyst, 2009, 134, 2123–2127 RSC.
- V. Voliani, S. Luin, F. Ricci and F. Beltram, Single-step bifunctional coating for selectively conjugable nanoparticles, Nanoscale, 2010, 2, 2783–2789 RSC.
- M. Lee, K. Lee, K. H. Kim, K. W. Oh and J. Choo, SERS-Based Immunoassay Using A Gold Array-Embedded Gradient Microfluidic Chip, Lab Chip, 2012, 12, 3720–3727 RSC.
- S. Qiu, S. Gao, Q. Liu, Z. Lin, B. Qiu and G. Chen, Biosens. Bioelectron., 2011, 26, 4326–4330 CrossRef CAS PubMed.
- N. Uzunbajakava, A. Lenferink, Y. Kraan, E. Volokhina, G. Vrensen, J. Greve and C. Otto, Nonresonant Confocal Raman Imaging of DNA and Protein Distribution in Apoptotic Cells, Biophys. J., 2003, 84, 3968–3981 CrossRef CAS PubMed.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic Press, San Diego, 1990 Search PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Raman Spectrum of Graphene and Graphene Layers, Phys. Rev. Lett., 2006, 97, 187401–187404 CrossRef CAS PubMed.
- D. Fu, F.-K. Lu, X. Zhang, C. Freudiger, R. Pernik Douglas, G. Holtom and X. S. Xie, Quantitative Chemical Imaging with Multiplex Stimulated Raman Scattering Microscopy, J. Am. Chem. Soc., 2012, 134, 3623–3626 CrossRef CAS PubMed.
- J. S. Suh and M. Moskovits, Surface-Enhanced Raman Spectroscopy of Amino Acids and Nucleotide Bases Adsorbed on Silver, J. Am. Chem. Soc., 1986, 108, 4711–4718 CrossRef CAS.
- A. F. Palonpon, J. Ando, H. Yamakoshi, K. Dodo, M. Sodeoka, S. Kawata and K. Fujita, Raman and SERS Microscopy for Molecular Imaging of Live Cells, Nat. Protoc., 2013, 8, 677–692 CrossRef CAS PubMed.
- R. P. Cooney, M. R. Mahoney and M. W. Howard, Intense Raman Spectra of Surface Carbon and Hydrocarbons on Silver Electrodes, Chem. Phys. Lett., 1980, 76, 448–452 CrossRef CAS.
- W. Xu, X. Ling, J. Xiao, M. S. Dresselhaus, J. Kong, H. Xu, Z. Liu and J. Jin Zhang, Surface Enhanced Raman Spectroscopy on A Flat Graphene Surface, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9281–9286 CrossRef CAS PubMed.
- X. Ling, W. Fang, Y.-H. Lee, P. T. Araujo, X. Zhang, J. F. Rodriguez-Nieva, Y. Lin, J. Zhang, J. Kong and M. S. Dresselhaus, Raman Enhancement Effect on Two-Dimensional Layered Materials: Graphene, h-BN and MoS2, Nano Lett., 2014, 14, 3033–3040 CrossRef CAS PubMed.
- D. Fu, G. Holtom, C. Freudiger, X. Zhang and X. S. Xie, Hyperspectral Imaging with Stimulated Raman Scattering by Chirped Femtosecond Lasers, J. Phys. Chem. B, 2013, 117, 4634–4640 CrossRef CAS PubMed.
- S. H. Kim, Y. K. Shin, K. M. Lee, J. S. Lee, J. H. Yun and S. M. Lee, An Improved Protocol of Biotinylated Tyramine-based Immunohistochemistry Minimizing Nonspecific Background Staining, J. Histochem. Cytochem., 2003, 51, 129–132 CrossRef CAS PubMed.
- A. Jain, R. Liu, Y. K. Xiang and T. Ha, Single-Molecule Pull-Down for Studying Protein Interactions, Nat. Protoc., 2012, 7, 445–452 CrossRef CAS PubMed.
- E. Kim, H.-C. Kim, S. G. Lee, S. J. Lee, T.-J. Go, C. S. Baek and S. W. Jeong, C-Reactive Protein-Directed Immobilization of Phosphocholine Ligands on A Solid Surface, Chem. Commun., 2011, 47, 11900–11902 RSC.
- N. R. Ling, D. Elliott and J. Lowe, Modulation of the murine immune response to human IgG by complexing with monoclonal antibodies. I. Antibody Responses to Determinants on the Constant Region of Light Chains and Gamma Chains, Immunology, 1987, 62, 17–22 Search PubMed.
- S. L. Roderick, W. W. Chan, D. S. Agate, L. R. Olsen, M. W. Vetting, K. R. Rajashankar and D. E. Cohen, Structure of human phosphatidylcholine transfer protein in complex with its ligand, Nat. Struct. Mol. Biol., 2002, 9, 507–511 CAS.
- Y. Kwon, Z. Han, E. Karatan, M. Mrksich and B. K. Kay, Antibody Arrays Prepared by Cutinase-Mediated Immobilization on Self-Assembled Monolayers, Anal. Chem., 2004, 76, 5713–5720 CrossRef CAS PubMed.
- B. A. Rosenzweig, N. T. Ross, D. M. Tagore, J. Jayawickramarajah, I. Saraogi and A. D. Hamilton, Multivalent Protein Binding and Precipitation by Self-Assembling Molecules on a DNA Pentaplex Scaffold, J. Am. Chem. Soc., 2009, 131, 5020–5021 CrossRef CAS PubMed.
- N. Tajima, M. Takai and K. Ishihara, Significance of Antibody Orientation Unraveled: Well-Oriented Antibodies Recorded High Binding Affinity, Anal. Chem., 2011, 83, 1969–1976 CrossRef CAS PubMed.
- E. Hendry, P. J. Hale, J. Moger, A. K. Savchenko and S. A. Mikhailov, Coherent Nonlinear Optical Response of Graphene, Phys. Rev. Lett., 2010, 105, 097401–097404 CrossRef CAS PubMed.
- I. Alessandri, Enhancing Raman Scattering Without Plasmons: Unprecedented Sensitivity Achieved by TiO2 Shell-Based Resonators, J. Am. Chem. Soc., 2013, 135, 5541–5544 CrossRef CAS PubMed.
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, Surface Enhanced Raman Scattering Enhancement Factors: A Comprehensive Study, J. Phys. Chem. C, 2007, 111, 13794–13803 CAS.
- E. Kim, S. G. Lee, H. C. Kim, S. J. Lee, C. S. Baek and S. W. Jeong, Protein-Directed Immobilization of Phosphocholine Ligands on a Gold Surface for Multivalent C-Reactive Protein Binding, Curr. Top. Med. Chem., 2013, 13, 519–524 CrossRef CAS PubMed.
- Y. R. Shen, The principles of nonlinear optics, Wiley-interface, New York, 2002 Search PubMed.
- C. K. Chen, T. F. Heinz, D. Ricard and Y. R. Shen, Surface-Enhanced Second-Harmonic Generation and Raman Scattering, Phys. Rev. B: Condens. Matter Mater.
Phys., 1983, 27, 1965–1979 CrossRef CAS.
- H. Kim, C. Xiang, A. G. Güell, R. M. Penner and E. O. Potma, Tunable Two-Photon Excited Luminescence in Single Gold Nanowires Fabricated by Lithographically Patterned Nanowire Electrodeposition, J. Phys. Chem. C, 2008, 112, 12721–12727 CAS.
- S. Zou and G. C. Schatz, Silver Nanoparticle Array Structures that Produce Giant Enhancements in Electromagnetic Fields, Chem. Phys. Lett., 2005, 403, 62–67 CrossRef CAS.
- K. Lee and J. Joseph Irudayaraj, Periodic and Dynamic 3-D Gold Nanoparticle–DNA Network Structures for Surface-Enhanced Raman Spectroscopy-Based Quantification, J. Phys. Chem. C, 2009, 113, 5980–5983 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details concerning syntheses, Raman spectroscopy, FT-IR, SEM, AFM, molecular dynamics, and FDTD calculation results are enclosed. See DOI: 10.1039/c5ra00040h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.