
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrically conductive polymers and composites for biomedical applications
Gagan
Kaur
 *,
Raju
Adhikari
,
Peter
Cass
,
Mark
Bown
and
Pathiraja
Gunatillake
*
*,
Raju
Adhikari
,
Peter
Cass
,
Mark
Bown
and
Pathiraja
Gunatillake
*
CSIRO Manufacturing Flagship, Bayview Avenue, Clayton, VIC 3168, Australia. E-mail: gagan.kaur@csiro.au; thilak.gunatillake@csiro.au
First published on 16th April 2015
Abstract
Electrically conductive polymeric materials have recently attracted considerable interest from academic and industrial researchers to explore their potential in biomedical applications such as in biosensors, drug delivery systems, biomedical implants and tissue engineering. Conventional conductive homopolymers such as polypyrrole and PEDOT show promising conductivity for these applications, however their mechanical properties, biocompatibility and processability are often poor. This has led to more recent attention being directed towards conductive polymeric composites comprised of biostable/biocompatible polymers with dispersed conductive fillers such as graphene, carbon nanotubes and metallic nanoparticles. The major objective of this paper is to provide an up to date review of the recent investigations conducted in the development of conductive polymer composites focussing on the methods of their preparation, underlying concepts of their conductivity and the ways to tailor their properties. Furthermore, recent progress made in conventional conducting polymers and their composites/blends for biomedical applications is also discussed.
 Gagan Kaur | Gagan Kaur received her MSc Hons. (Chemistry) from Guru Nanak Dev University, India (1997) and her PhD (Polymer Chemistry) from Monash University, Australia (2013). Gagan is currently working as a Postdoctoral Fellow at Commonwealth Scientific and Industrial Research Organisation, Melbourne, Australia. Gagan's research interests lie in the synthesis and development of novel polymeric materials for biomedical, nanoscience and industrial applications. Her research interests also lie in the electroactive biomaterials, living radical polymerizations and drug delivery. |
 Raju Adhikari | Raju Adhikari completed his PhD in organic chemistry from University of Delhi. He did his post-doctoral studies at CSIRO Molecular Science in 1992 and later at Universitat of Hohenheim, Germany and rejoined CSIRO in 1995. He is one of the inventors of two platform technologies Elast-Eon™ and NovoSorb™ which are a family of biostable and biodegradable polyurethanes. Beside biomaterials, he has worked in OLED and Agriculture polymer areas and contributed significantly to technology and product development in both fields. He is currently working as a Principal Research Scientist and holds an Adjunct A/Professor position at RMIT University. |
 Peter Cass | Peter Cass received his PhD in polymer chemistry from Swinburne University, Australia in 2000. Peter did his Postdoctoral Fellowship at Melbourne University, Australia from 2001 to 2003. He is currently working as Research Scientist at Commonwealth Scientific and Industrial Research Organisation, Melbourne, Australia. His research interests lie in polymer chemistry, industrial polymers, adhesives, drug delivery and biomaterials. |
 Mark Bown | Mark Bown received the BSc degree in chemistry from Imperial College, London, U.K., and the PhD degree from the University of Leeds, U.K., respectively. He is currently the Team Leader with the Industrial Innovation Program, Commonwealth Scientific and Industrial Research Organisation, Melbourne, Australia, where he is responsible for organic light-emitting diode research. |
 Pathiraja Gunatillake | Pathiraja Gunatillake has a PhD in polymer chemistry from the City University of New York. He joined CSIRO in 1988 and has successfully made the transition from basic discovery research through the product development pipeline, and contributed to deliver products into the medical device market. AorTech and PolyNovo are two spin-off companies established from the technology developed under his leadership. He is a Fellow of the Biomaterials Science and Engineering (FBSE) and the American Institute of Medical and Biological Engineering (FAIMBE). He has received international recognition through technology innovations and literature contributions to the biomaterials filed and currently a Chief Research Scientist in CSIRO. |
1. Introduction
Since the discovery of intrinsically conducting polymers, researchers have explored their unusual electronic properties for a wide range of applications. Due to the presence of a conjugated π-electron backbone these polymers exhibit electronic properties such as low energy optical transmission, low ionization potential and high electron affinities. These unique properties make these materials suitable for applications as thin film transistors, organic light emitting diodes, sensors, supercapacitors, organic solar cells and electrochromic displays. Many research groups have extensively investigated conducting polymers for these applications and a number of excellent reviews are available.1–12More recently, conducting polymers and electroactive polymers have received the attention of researchers to explore their potential in biomedical applications. This new generation of “smart” biomaterials have been investigated for applications in biosensors; coatings on conventional electrodes used in neural sensing and stimulation; electrically induced drug release and delivery systems; modulators of activities of nerve, cardiac, skeletal muscle, and bone cells; and in emerging technologies such as tissue engineering.13 The most widely investigated conducting polymers for biomedical applications include polypyrrole, polyaniline, polythiophene and its derivatives such as poly(3,4-ethylenedioxythiophene).14–17Fig. 1 presents an overview of a broad conductivity range of conducting polymers and conductive polymeric composites.
 | ||
| Fig. 1 Conductivity range of conducting polymers and conductive polymeric composites. | ||
Most studies have focused on investigating the interaction of these polymers with biological tissues using in vitro assays and strategies to improve biocompatibility. Tailoring conducting polymers to have appropriate mechanical properties, electrical conductivity, processability as well as acceptable biocompatibility has been the major challenge in application of this class of polymers for clinically useful biomedical implants and devices. Development of composites of conducting polymers with conducting nanoparticles along with non-conducting polymers to improve mechanical performance and biocompatibility has been one of the recent approaches in attempting to overcome some of these limitations. The major focus of this paper is to provide a comprehensive review of the investigations conducted over the last decade in the development of conducting composites. A brief introduction to the chemistry and properties of the well known conducting polymers followed by review of the recent literature on conductive composites is provided.
2. Conducting polymers
The derivatives of polypyrrole with resistivities as low as 1 Ω cm were first reported in 1963 by Australian scientists Bolto and Weiss, and their coworkers.18 The discovery of polyacetylene and its high conductivity upon doping in by Shirakawa and coworkers in the 1970s further helped to advance the field of conductive polymers.19 Polypyrrole (PPy), polyaniline (PAni), polythiophene (PTh), and poly(3,4-ethylenedioxythiophene) (PEDOT) are most promising conducting polymers (CPs) for use in biomedical applications.4,14,15,20,21 CPs exhibit electrical and optical properties similar to those of metals and semiconductors, and offer advantages of conventional polymers such as ease of synthesis.14,15,22 As an electrode for stimulation and recording, conducting polymers are attractive due to the possibility of chemical surface modification with physiologically active species to enhance the biocompatibility and the functionality of the electrodes.23,24 This unique combination of properties makes CPs potential candidates for various biomedical applications such as biosensors, neural probes, neural prostheses, drug-delivery devices, tissue-engineering scaffolds, and bio-actuators.4,14,15,20,21,25–31The presence of conjugated double bonds (Fig. 2) along the backbone gives rise to the conductivity in CPs.22 The π electrons in the conjugated backbone are available to delocalize into a conduction band and in the idealized situation of a uniform chain, the resulting conduction band would give rise to metallic behaviour. However, such a system is unstable with respect to bond alternation, which causes the formation of an energy gap in the electronic spectrum.19 Dopant ions are introduced to the structure to overcome the energy gap and hence, to impart conductivity to these polymers. The dopant ions carry charge in the form of extra electrons to neutralise the unstable backbone of the polymer in its oxidised state by donating or accepting electrons.18,19 On application of a potential across the CP film, a charge is passed through the film as a result of a flux of ions either in or out of the film, dependent on dopant charge and mobility, causing a disruption to the polymer backbone.18,19 CPs can be doped with both p- and n-type dopants using a variety of molecules, such as small salt ions (Cl−, Br−, or NO3−), and larger dopants such as hyaluronic acid, peptides or polymers.14,20
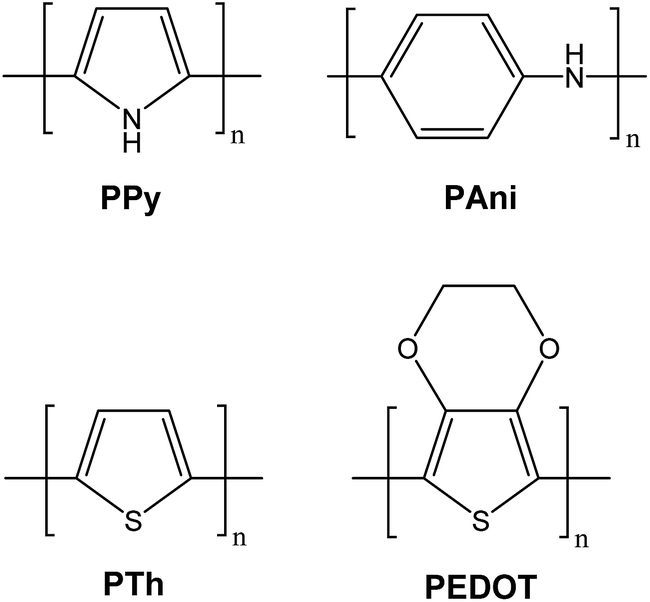 | ||
| Fig. 2 Structures of common CPs investigated for biomedical applications. | ||
CPs can be synthesized either chemically or electrochemically. Chemical methods of CP synthesis either use condensation polymerization or addition polymerization. While chemical synthesis provides many different possible routes to synthesize a variety of CPs and also permits the scale-up of these materials, electrochemical synthesis is relatively straightforward and hence, is most commonly used for making CPs.32,33 The advantages of electrochemical synthesis include ease of synthesis, simultaneous doping and entrapment of molecules during synthesis, however the films are difficult to remove from electrodes and post-synthesis covalent modification of CP is difficult.14 On the other hand, chemical synthesis offers more options to modify CP backbone covalently and makes the post-synthesis covalent modification possible, although this method is often more complicated.14 Another significant difference between electrochemical and chemical methods of CP synthesis is that electrochemical method can produce very thin CP films (of the order of 20 nm), whereas powders or very thick films are usually produced with chemical polymerization.14,17 Furthermore, electrochemical synthesis is limited to those systems in which the monomer can be oxidized upon application of potential to form reactive radical ion intermediates for polymerization.14 The common CPs (e.g. PPy, PTh, PAni, PEDOT) can be polymerized both chemically and electrochemically; however, several novel CPs with modified monomers can only be synthesised using chemical polymerization.14,15
Table 1 presents the properties and electrical conductivity of some conventional conducting polymers investigated for biomedical applications.
| Polymer | Conductivity (S cm−1) | Type of doping | Properties | Limitations | Ref. |
|---|---|---|---|---|---|
| Polypyrrole | 10–7.5 × 103 | p | High electrical conductivity, ease of preparation and ease of surface modification | Rigid, brittle and insoluble | 16, 34–36 |
| Polyaniline | 30–200 | n, p | Diverse structural forms, environmentally stable, low cost | Hard to process, non-biodegradable, limited solubility | 16, 35 and 36 |
| Polythiophene | 10–103 | P | High electrical conductivity, ease of preparation, good optical property | Hard to process | 16, 34–36 |
| Poly(3,4-ethylene dioxythiophene) | 0.4–400 | n, p | Transparent conductor, environmentally and electrochemically stable | Limited solubility | 37–39 |
2.1 Polypyrrole (PPy)
Doped PPy has been the most thoroughly investigated conductive polymer for biomedical applications because of its high electrical conductivity, ease of preparation, and ease of surface modification.17,20 PPy exhibits excellent environmental stability and has been shown to have the ability to support cell adhesion and growth of a variety of cell types.40–42 PPy has been explored for a number of biomedical applications including tissue engineering,17,40 biosensors,26 drug delivery,43 and bioactuators.16,31,40,41,44,45 PPy can easily be synthesised in large quantities at room temperature in a variety of common organic solvents and also in water.46–49 The conductivity of PPy films can be achieved up to ∼103 S cm−1 depending on the type and amount of dopant.16,34–36 Due to its molecular structure, a limitation of pure PPy is that once synthesised, it is hard to process it further as it is crystalline, mechanically rigid, brittle and insoluble, making unmodified PPy poorly suited to most biomedical applications such as tissue engineering.16,17Schmidt and coworkers reported the synthesis and physicochemical characterization of poly(1-(2-carboxyethyl)pyrrole) (PPyCOOH), a PPy derivative that contains a chemical group that can be easily modified with biological moieties at the N-position of the polymer backbone, enhancing the biomaterial–tissue interface and promoting desired tissue responses.50 Human umbilical vascular endothelial cells (HUVECs) cultured on PPyCOOH films surface-modified with the cell-adhesive Arg-Gly-Asp (RGD) motif demonstrated improved attachment and spreading (Fig. 3).
 | ||
Fig. 3 Typical fluorescent (top) and phase-contrast (bottom) images of the labelled PPyCOOH films (A) and powders (B), the control PPy films (C), cells (HUVECs) on the RGD-grafted PPyCOOH films (D), and on the control PPy films (E) cultured for 6 h at an initial density of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2. Reprinted with permission from ref. 50, copyright 2006, American Chemical Society. 000 cells per cm2. Reprinted with permission from ref. 50, copyright 2006, American Chemical Society. | ||
In a study by Richardson and co-workers, PPy coated electrodes were used for the delivery of charge and neurotrophins in order to reduce the degeneration of spiral ganglion neurons (SGNs) associated with cochlear implant use.43 The electrically conducting polypyrrole/para-toluene sulfonate containing neurotrophin-3 (PPy/pTS/NT3) was applied to cochlear implant electrodes. The in vivo studies on guinea pigs showed the use of the cochlear implant to deliver neurotrophic agents to SGNs in a safe and controlled manner over the short-term, in addition to electrical stimulation for enhanced preservation of SGNs after hearing loss.
There are a number of literature reports on the biocompatibility studies of PPy.44,51–54 A recent in vitro study has reported that PPy nanoparticles fabricated using oxidative polymerization route are cytotoxic at high concentrations.53 These nanoparticles negatively affected the cell viability/proliferation, and this effect was directly dependent on the nanoparticle concentration. But lower concentrations of PPy nanoparticles (<9.7 μg ml−1) were not found to affect cell viability/proliferation. The same group had also reported previously the results of an in vivo study showing that chemically synthesized PPy particles exhibited good biocompatibility in mice over a 6 weeks period of treatment with these particles.51 Furthermore, in a study by Martin et al., PPy and a synthetic peptide were co-deposited on an electrode surface by electrochemical polymerization.44 The stability of PPy/peptide coatings was tested using in vitro soaking experiments whilst their effect on the brain tissue response and neural recording was examined in vivo. For in vivo studies, the electrodes were implanted and evaluated for maximum of a 3 weeks period.44 The results showed that PPy/peptide coating promoted the neuron attachment and good recordings were obtained from the coated sites that had neurons attached. In another in vivo study,54 a PPy-silicone tube was synthesized electrochemically and was used to bridge across 10 mm sciatic nerve gap in rats. The regenerated tissues were observed by electrophysiological and histological techniques 24 weeks after the operation. PPy extraction solution showed no evidence of acute and subacute toxicity, pyretogen, hemolysis, allergen, and mutagenesis, but there was a mild inflammation observed.
In summary, despite PPy's attractive properties and reports, it is worth pointing out that the in vivo studies on PPy have been limited and mainly focused on short term toxicity evaluation only. Therefore, considering its drawbacks such as its poor solubility and rigidity, more in vivo studies are required to confirm the viability of PPy as a biomaterial.
2.2 Polyaniline (PAni)
PAni is the second most investigated conducting polymer with many advantages such as its diversity of structural forms, high environmental stability, low cost and the ability to electrically switch between its conductive and resistive states by doping/dedoping process.20,55–58 It exists in various forms based on its oxidation level i.e. the fully oxidized pernigraniline base, half-oxidized emeraldine base, and fully reduced leucoemeraldine base.16,59,60 PAni emeraldine form is the most stable and conductive.16,59 PAni is also difficult to process due to its poor solubility in most of the available solvents.58In a study by Humpolicek and coworkers,61 both the non-conducting PAni (emeraldine base) and its conducting form (PAni hydrochloride), were tested for their biocompatibility in terms of skin irritation, sensitization and cytotoxicity. The skin irritation and sensitization testing was done in vivo, while cytotoxicity testing was performed in vitro on human immortalized non-tumorigenic keratinocyte and human hepatocellular carcinoma cell lines. The results showed that both PAni hydrochloride and PAni base, have excellent biocompatibility properties in terms of dermal irritation and sensitization.61 However, both polymers showed considerable cytotoxicity, which was higher for PAni hydrochloride compared with PAni base. Furthermore, the polymer purification via reprotonation/deprotonation cycle resulted in significant reduction in cytotoxicity showing that the low molecular weight reaction residues or by-products, rather than PAni alone, are also likely to be responsible for observed cytotoxicity.
The main challenge for using PAni and its derivatives for biological applications arises from its poor cell compatibility, poor processibility, lack of flexibility, and non-biodegradability.56,62 Nevertheless, PAni has been investigated for use in biomedical applications such as biosensors, neural probes, controlled drug delivery, and tissue engineering applications with promising outcomes.59,61
2.3 Polythiophene (PTh) and derivatives
Polythiophenes have properties similar to, and in some cases superior to, those of PPy.63,64 Polythiophene and its derivatives have been explored for electroactive scaffolds for cell culture, biosensors, and neural probes.65–68 Poly(3,4-ethylenedioxythiophene) (PEDOT) is considered the most successful PTh derivative due to its higher electrical conductivity and chemical stability which allows its use in biomedicine and biotechnology.69,70 As compared to PPy and PAni, the investigations on PEDOT are relatively recent. The biocompatibility of PEDOT has been well established.71,72PEDOT can be synthesized in various forms such as nanofilms, nanorod arrays and nanofiber mats.73–75 Free-standing conductive ultra-thin nanofilms based on PEDOT and polystyrene sulfonic acid (PSS) were fabricated by Mattoli and coworkers using a process based on a modified supporting layer technique.73 The work demonstrated that the PEDOT:PSS nanofilms could be manipulated, folded and unfolded in water many times without suffering from cracks, disaggregation or from loss of conductive properties giving it potential applications in the field of sensing and actuation, as well as in the biomedical field, e.g. as smart substrates for cell culturing and stimulation.73 The same research group also fabricated a bending actuator by depositing a thin conductive polymer layer of PEDOT:PSS over the surface of a polysiloxane-based monodomain nematic liquid single crystal elastomer (LSCE) film.76 The mechanical properties of PEDOT:PSS, being better matched with LSCE than with metals or inorganic nanoparticles, allowed the development of an all-polymer reliable millimetre-scale actuating composite.76 Carmena and coworkers have explored the use of PEDOT (doped with PSS) coated microelectrodes for use as cortical neural prostheses.72In vivo chronic testing of microelectrode arrays implanted in rat cortex revealed that PEDOT coated Pt–Ir electrodes showed higher signal-to-noise recordings and superior charge injection compared to Pt–Ir electrodes.72 In a study by Feng et al., PEDOT nanofiber mats were fabricated by electrospinning combined with in situ interfacial polymerization using FeCl3 as an oxidant.75 The PEDOT nanofiber mats displayed good mechanical properties, flexibility, and achieved an electrical conductivity of 7.8 ± 0.4 S cm−1 and similar biocompatibility to tissue culture plates.75
Tarabella and coworkers employed organic electrochemical transistors (OECTs), based on the PEDOT:PSS, as sensors of liposome-based nanoparticles in electrolyte solutions to assess sensitivity and monitoring capabilities based on ion-to-electron amplified transduction.77 In an another study carried by Sui et al., PEDOT:PSS coatings incorporated with dopamine were fabricated on platinum electrodes and their electrochemical properties and dopamine delivery capacities were evaluated in vitro and in vivo.78 For in vivo studies, the PEDOT:PSS/dopamine coated electrodes were implanted into brain striatum area of rats. The results demonstrated that the PEDOT:PSS/dopamine coatings on platinum electrodes could reduce electrode impedances, increase charge storage capacities, and release significant levels of dopamine upon electrical stimulation of these electrodes. These results indicated a potential application of PEDOT:PSS/dopamine-coated implantable electrodes in the treatment of some diseases associated with dopamine deficits, such as Parkinson's disease.78
The use of biologically active dopants allows the CPs to have features of a multiple stimuli responsive material, and hence makes them more attractive as biomaterials for biomedical applications.42 In particular, electrical and biological cues are important factors to include in interfaces with neurons for applications such as nerve conduits and neural probes. The incorporation of nerve growth factor (NGF) as a co-dopant in the electrochemical deposition of conductive polymers, PPy and PEDOT, has been evaluated for its ability to draw forth specific biological interactions with neurons.42,79 These studies revealed that PC12 (rat pheochromacytoma) cells adhered to the NGF-modified substrate and extended neurites on both PPy and PEDOT, indicating that the NGF in the polymer film is biologically active. This approach can be used to fabricate materials capable of both biological as well as electrical stimulation for biomedical applications.79
2.4 Summary
In summary, CPs have several attractive properties that include, good stability, sufficiently high electrical conductivity, ability to entrap, and release biomolecules. Furthermore, they can be potentially modified for electrical, chemical, physical, and biocompatibility properties to better suit a specific application.14,15 However, their use in biomedical applications is limited because CPs are often brittle and difficult to handle and the use of larger dopants can further aggravate this effect.14,80 One way to overcome the shortcomings of a CP is to use it together with another polymer in the form of a blend or composite in order to combine the useful properties of both materials. The composites of CPs can provide the increased solubility and better mechanical properties necessary for various biomedical applications without significant compromise of their conductivity and other properties as discussed in later sections.3. Conductive polymer composites
3.1 Composites/blends based on conjugated conducting polymers
An effective way to improve mechanical properties of CPs is to create their composites or blends with other polymers that have better mechanical properties for the intended application. Conducting polymers like PPy and PAni have also been explored as conductive fillers, especially with natural polymers, in order to overcome the poor processability of these conducting polymers as well as to impart conductivity to otherwise insulating polymers.80–85 Doping with large molecules can also be used to prepare conducting polymer composites with improved mechanical properties. However, these processes unfortunately may cause interference with electron conjugation within the CP due to the presence of insulating molecules.14Ma and co-workers fabricated synthetic nerve conduits by dip coating from PPy/poly(D,L-lactic acid) (PDLLA) composite solution obtained as a result of emulsion polymerization of ppy in PDLLA solution. Aqueous FeCl3 solution was used to initiate the oxidative polymerization.92 PC12 cells were used to assess the in vitro cell compatibility, which exhibited more and longer neurites on composite than on PDLLA conduits after being stimulated with 100 mV for 2 h. The 5% PPy/PDLLA composite was also used to fabricate nerve conduits to bridge a 10 mm defect in the sciatic nerve of a rat. After 6 months, the rats with the PPy/PDLLA conduits showed functional recovery similar to that of the gold standard autologous nerve graft and significantly better than that of the PDLLA conduits. The authors suggested that such a conduit can potentially be used in nerve tissue regeneration eliminating the drawbacks associated with using an autologous graft, including limited donor source, donor site morbidity, multiple surgery sites, and possible size mismatch.
Ferraz et al. prepared composites of nanocellulose and PPy using FeCl3 as an oxidant and the effect of processing parameters such as rinsing and extraction, as well as aging, on electroactivity and cytotoxicity was examined.95 These studies showed that while the composites need to be thoroughly rinsed to remove impurities, reactants, and shorter oligomers to obtain a non-cytotoxic material, such processing has a negative effect on the electrochemical ion exchange capacity of the material. Aging of the PPy composites was also found to have a pronounced negative effect on the biocompatibility of the composite. In a recent study by Kobayashi and co-workers, conductive PPy-cellulose acetate films were prepared from cellulose acetate (CA) solution of pyrrole (Py) using wet cast methods.85 The PPy-CA composite films containing different concentrations of Py were prepared by casting a Py viscous solution of CA on glass plate and immersing it in FeCl3 aqueous solution. The resultant composite films showed maximum electrical conductivity of 3.6 × 101 S cm−1 with 4.7 wt% loading of PPy.85 In another study, composites of PPy and chitosan with radical scavenger activity were produced for antioxidant applications in food packaging and biomedical applications.84 The composites were synthesized by the chemical polymerization of pyrrole in chitosan solution using ammonium persulfate (APS) as the oxidant.84 In order to optimize the activity and stability of the composites, a range of ratios of APS to PPy in composite was investigated. The FTIR and UV-Vis measurements identified an attachment of PPy to chitosan in the chitosan–PPy composites, which were formed as membranes (coatings) with conductivity in the range of 10−7 to 10−3 S cm−1.
In an investigation by Kim et al., hybrid composites of PAni nanofibers and collagen with various ratios of well dispersed PAni nanofibers in a collagen matrix were fabricated.86 The PAni nanofiber-collagen composite film, doped in HCl solution, remained electronically conductive, although conductivity decreased significantly with decreasing amounts of PAni in the composite. The conductivity of a neat PAni sample was 3.0 S cm−1 and the sample with 7:1 ratio of PAni to collagen showed highest conductivity (0.27 S cm−1) among the composite films. The prepared composites showed a rather high percolation threshold value while samples with PAni content lower than 50% in a collagen matrix did not show any measurable conductivity. However, the PAni nanofiber–collagen composite film was found to be well suited for cell culture and was claimed as a potential candidate for use as scaffold material for biomedical applications.86 Wallace and coworkers used vacuum vapour phase polymerization to produce conductive PEDOT composites incorporated with triblock polymer poly(ethylene glycol-propylene glycol-ethylene glycol) (glycol) for implantable devices.87 Iron(III) tosylate was used as an oxidant in the polymerization. The PEDOT–glycol composites were found to have a maximum conductivity of 1486 S cm−1 being achieved at a glycol loading of 48 wt%. The results also indicated that cell attachment and proliferation depended on the individual cell lines used and that the impact of glycol within the PEDOT composite was negligible.87
Schmidt et al. synthesised conductive composites of PPy using biologically active polysaccharide hyaluronic acid (HA) as the dopant in order to create biomaterials for tissue engineering and wound-healing applications.80 These conductive, HA-containing PPy films retained HA on their surfaces for several days in vitro and promoted vascularisation in vivo and hence, were claimed as promising candidates for tissue engineering and wound-healing applications benefitting from both electrical stimulation and enhanced vascularisation. However, the PPy/HA films were more brittle, less conductive and exhibited a more nodular surface appearance when compared to PPy:PSS films. These differences were attributed to the diffusion limitations in the more viscous HA solution resulting in the inhomogeneous growth of the PPy/HA films. In a similar study,96 heparin (HE) was used as a dopant to simultaneously improve the electrical stability and cell adhesion to PPy, because HE is both a polyanion and an important glycosaminoglycan in cell membranes and extracellular matrix. PPy particles doped with HE were synthesized through emulsion polymerization using Fenton's reagent as an oxidant. Conductive biodegradable membranes of resistivity of 102 to 103 Ω sq−1 were prepared from PPy (5% wt) with various amounts of HE and 95 wt% poly(L,L-lactide) (PPy/PLLA). The results showed that HE was incorporated into the PPy particles as counter ions and was present on the particle surface. The conductive membranes containing HE-doped PPy particles recorded enhanced electrical stability, cell adhesion (human skin fibroblasts), and cell growth.
Combining the characteristics of a conducting polymer such as PPy with an elastomeric material, such as polyurethane (PU), may yield a composite with electrical activity and significantly improved biocompatibility and mechanical resilience. A series of electrically conducting PPy nanoparticle and PU composites with different ratios were prepared by Broda et al. via an in situ polymerization of Py using FeCl3 as an oxidant in a PU emulsion.81 The polymerization resulted in a composite with a principle base of PU interspersed with an electrically percolating network of PPy nanoparticles. As the mass ratio of PPy to PU increased so did electrical conductivity of the composites. In addition, as the mass ratio of PPy to PU increased, the stiffness of the composite increased while the maximum elongation decreased. The PPy–PU composites exhibited elastomeric properties as well as conductivity, and were shown to be cytocompatible with C2C12 myoblast cells. The composite with ratio of 1:5 of PPy:PU was found to have the highest conductivity (2.3 × 10−6 S cm−1) while the composite with ratio 1:100 was least conductive (1.0 × 10−10 S cm−1).81 Perez-Madrigal and co-workers prepared polythiophene derivative/thermoplastic PU nanomembranes for tissue engineering applications.90 The conductivity values determined for the nanomembranes ranged from 5.19 × 10−6 to 2.23 × 10−5 S cm−1.90 In another study, a polymer-based stretchable electrode fabricated from a blend of PEDOT:PSS and an aqueous polyurethane dispersion (PUD) was reported by Park and coworkers.93 The blend containing 73 wt% of non-conductive PUD exhibited an electrical conductivity of ∼120 S cm−1. Ionic liquid(IL) based IL/PU/PEDOT:PSS composites were fabricated by Okuzaki and co-workers by sandwiching the IL/PU gel between two conductive polymer films made of PEDOT:PSS as soft and flexible electrodes.91 The electrical conductivity was found to increase from 3.1 × 10−5 S cm−1 to 8.8 × 10−5 S cm−1 with increasing the IL content from 0 wt% to 40 wt%.
Degradable polymers exhibiting conductivity have also recently gained considerable attention.15 The electrically conducting degradable polymers have been reported to improve cell adhesion as well as proliferation and they could be used as scaffold materials for neural, cardiovascular, and bone tissue regeneration for which electroactivity is important.15
PAni has been exploited for electroactive hydrogels which are polymeric blends combining the responsive properties of electroactive polymers and highly hydrated hydrogels within an aqueous milieu that is hospitable to biological molecules such as peptide sequences, enzymes antibodies, and DNA.97 The combination of hydrogels and inherently conductive electroactive polymers allows both materials to retain their unique responsive properties. In addition, the electroconductive hydrogels engender a new class of devices with low interfacial impedances suitable for neural prosthetic devices such as deep brain stimulation electrodes, low voltage actuation for electrically stimulated drug release devices and potential for in vivo biocompatibility in implantable biosensors. In a study, a highly swelling grafted hydrogel composed of poly(acrylic acid) (PAA) and polyvinyl alcohol (PVA) containing PAni nanoparticles were prepared by in situ polymerization of aniline using ammonium persulphate as an oxidant.89 This study mainly focused on the synthesis and characterization of conductivity, swelling behaviour, biocompatibility, and microhardness. Impregnation of polyaniline into PVA-g-PAA resulted in a composite hydrogel which showed electroconductive and electroactive behaviour. The electrical conductivity varied with varying content of PAni in the composite and was found in the range of 0.04–0.06 S cm−1 for 5% PAni content. The native and PAni impregnated matrix not only showed a moderate biocompatibility and good mechanical strength but also exhibited good swelling properties in both distilled water and electrolyte solution. In another study by Yang et al. reported the synthesis of a bacterial cellulose (BC)/PAni nanofiber composite which is an electro-conductive hydrogel that may potentially be used for biosensors and tissue engineering applications.88 The hydrogel was synthesized in ammonium persulphate solution by in situ nano-assembly of BC nanofibers and PAni to enhance the electronic conductivity of BC nanofibers.88 The electrical conductivity of composite hydrogels was enhanced from 10−8 to 10−2 S cm−1.
Wallace and co-workers reported the synthesis of a single component CP hydrogel for potential applications as tissue engineering scaffolds.98 Poly(3-thiopheneacetic acid) hydrogels were fabricated by covalently crosslinking the polymer with 1,1′-carbonyldiimidazole. The hydrogels exhibited good swelling properties (with swelling ratios up to 850%) and the mechanical properties of the networks were found to be comparable to those of muscle tissue. Hydrogels were found to be electroactive and conductive at physiological pH. Fibroblast and myoblast cells cultured on the hydrogel substrates were shown to adhere and proliferate.
Hybrid composites comprising of a conducting polymer and silver, have also been shown to achieve high conductivity.83 These composites were produced mainly by the oxidation of aniline or pyrrole with silver ions.99–101 However, high electrical conductivity (>1000 S cm−1) of such composites is only achieved with high amount of silver (>60%, w/w) and seems to be controlled by percolation.83
Some relevant work on composites of CPs is summarised in Table 2.
| Composite/blend | Conductivity (S cm−1) | Properties | Suggested applications | Ref. |
|---|---|---|---|---|
| PPy/hyaluronic acid | 3.08 × 10−3 | Can support tissue growth and stimulate specific cell functions | Tissue engineering and wound-healing applications | 80 |
| PAni nanofibers/collagen | 0.27 | Well suited for cell culture | Scaffold material for biomedical applications | 86 |
| PPy/chitosan | 10−3–10−7 | Radical scavenger | Food packaging and biomedical applications | 84 |
| PEDOT/glycol | 1486 (maximum) | — | Implantable devices | 87 |
| PPy/cellulose acetate | 6.9 × 10−4 to 3.6 × 101 | — | — | 85 |
| PAni nanofibre/bacterial cellulose | 10−2 | Hydrogel | Biosensors, tissue engineering | 88 |
| PAni nanoparticles/poly(acrylic acid)/polyvinyl alcohol | 0.04–0.06 | Hydrogel, biocompatible, good mechanical strength and good swelling properties | — | 89 |
| Polythiophene derivative/PU | 2.23 × 10−5 | Suitable for supporting electrically stimulated cell growth | Tissue engineering | 90 |
| PEDOT:PSS/PU/ionic liquid | 8.8 × 10−5 | Mechanically flexible, stretchable | Actuating devices | 91 |
| PPy/poly(D,L-lactic acid) | 5.65 × 10−3 to 15.56 × 10−3 | Nerve tissue regeneration (in vivo rat), biocompatibility (PC12 cells) | Synthetic nerve conduits | 92 |
| PPy nanoparticles/PU | 2.3 × 10−6 (maximum) | Cytocompatible with C2C12 myoblast cells, elastomeric properties | Tissue engineering | 81 |
| PEDOT:PSS/PU (aqueous dispersion) | ∼120 | High pressure sensitivity | Electronic skin sensor | 93 |
| PEDOT/RG-O | 9.2 | Good thermal and environmental stability | — | 94 |
3.2 Composites of conductive nanoparticles/fillers with non-conjugated polymers
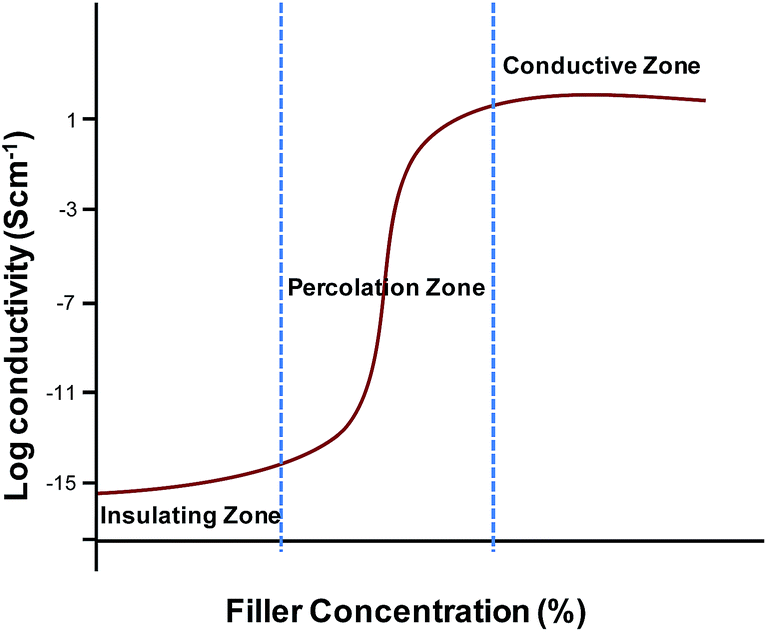 | ||
| Fig. 4 Conductivity of polymer composites as function of filler concentration. | ||
Graphene is a two dimensional monolayer of sp2-hybridized carbon arranged in honeycomb lattice and exhibits high mechanical strength, electrical conductivity and ultra high specific surface area.114 Graphene based polymer composites show superior mechanical, thermal and electrical properties as compared to the neat polymer.109,115 However, stable dispersions of graphene in polar solvents can only be obtained using suitable surfactants due to its hydrophobic nature.116,117
Graphene oxide (GO) is similar to graphene but has the oxygen containing polar functional groups which enhances its biocompatibility, compatibility with polar solvents or with a polymer matrix.107,120–123 Incorporation of hydrophilic graphene based fillers like GO also improves cell adhesion at the biomaterials surface.122 CNTs are also carbon based fillers which are used for making electrically conductive nanocomposites. CNTs exhibit very good electrical conductivity of >103 S cm−1, with a high aspect ratio reaching 100–1000 for μm long single-wall and multiwall CNTs.113,118,124 Apart from carbon based conductive fillers, metal nanoparticles have also been explored to impart conductivity to non-conjugated insulating polymers. Table 3 summarizes the various conductive fillers and their respective electrical conductivities.
One of the main challenges in the fabrication of carbon based conductive polymer composites is that the carbon fillers are usually difficult to be homogenously dispersed within polymer materials.125,126 Another challenge in fabrication of conductive composites for biomedical applications is to achieve both high conductivity and mechanical toughness at the same time. The limiting conductivity is as important as the percolation transition.102 The high conductivity is often achievable at the cost of mechanical strength. It also seems fairly clear that the direct use of nanosized materials does not provide a way to improve the making of conductive composite materials. However, if filler contact density can be reduced by sintering or using high-aspect ratio fillers, high conductivity can be achieved.102,127,128 The following section describes the methods commonly used for the preparation of conductive polymer composites.
3.2.2.1 Solution mixing. The most common method used for graphene and CNT based polymer composites is the solution mixing because it facilitates separation of graphene sheets or nanotube dispersion.113,123,129–131 By this method, a solution of polymer is prepared and the nanofiller is separately dispersed in a suitable solvent by sonication (Fig. 5).113 For CNT/polymer composites, this step requires the employment of surface-modified nanotubes (either covalent or non-covalent) to achieve a metastable dispersion. Once the filler is dispersed in the solvent, the polymer, which was previously dissolved in the same solvent, is added to the dispersion so that the polymer adsorbs on to the filler. The final step is removing the solvent by evaporation. Both organic solvents and water have been used to produce composites using this method.
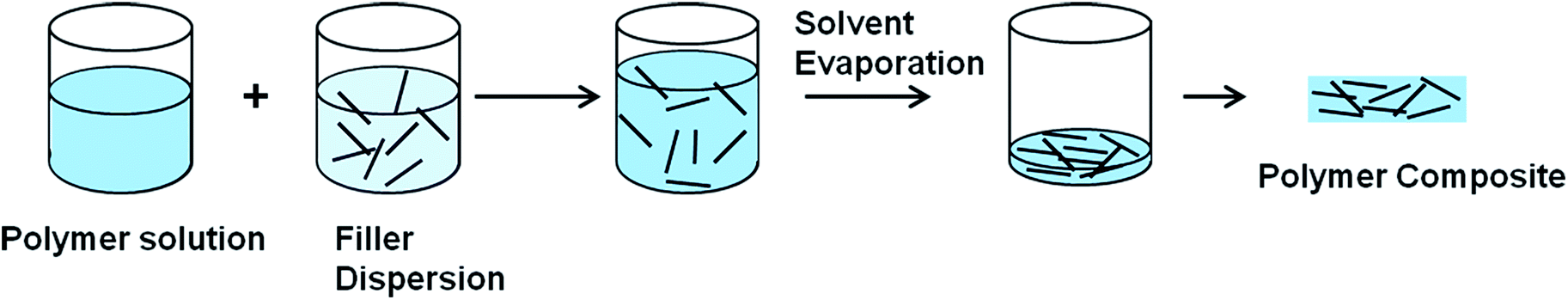 | ||
| Fig. 5 Preparation of conductive polymer composites using solvent mixing. | ||
3.2.2.2 In situ method. In the in situ polymerization method, the filler is first swollen in the liquid monomer. A suitable initiator is then diffused and polymerization is initiated either by heat or radiation.110In situ polymerization is extensively used for the preparation of polymer/CNT composites due to an advantage of formation of a covalent bond between the CNT and the matrix. The presence of polymeric chain onto the tubes' surface further facilitates their dispersion while providing a strong interface at the same time. This technique allows the preparation of composites with high nanotube loading, which can be diluted by other techniques.113
3.2.2.3 Melt processing. Melt blending or melt processing method has become attractive due to the advantage of being free of solvents. In this method, graphene or other nanofiller is mixed with the polymer matrix in molten state.126 The thermoplastic polymer is mixed mechanically with filler at elevated temperatures using conventional methods such as extrusion and injection molding.132,133 The polymer chains are then intercalated between the filler particles to form nanocomposites. The polymer chains experience a significant loss of conformational entropy during this process.113 Melt processing is preferred for industrial-scale processes, because of its speed and simplicity. This is also a preferred method for processing of polymers which are unsuitable for solution mixing or for in situ polymerization.110
3.2.2.4 Latex technology. Apart from the above methods, other methods have also been used by researchers to incorporate conductive fillers into a polymer matrix in order to obtain electrically conductive composites. Since the electrical conductivity arises from the formation of geometrical conduction pathway, integration of individual graphene nanosheets into well organized three dimensional assemblies and embedding them in polymer matrix is the key to achieve high conductivity.109,125 Latex technology is another method for making graphene and CNT based polymer composites and has advantages such as homogeneously dispersed fillers in the polymer matrix, easy processing and process up-scaling.109 In this method, any filler that can be dispersed to yield an aqueous colloidal dispersion can be used and similarly, any polymer that can be synthesized by emulsion polymerization or can artificially be brought into the form of a polymer latex is suitable.109 Latex technology facilitates direct incorporation of predominantly individual nanofillers into a highly viscous polymer matrix as well as it also allows the formation of three dimensional framework of filler particles in polymer matrix.109,134 Latex technology involves three main steps i.e. preparation of an aqueous colloidal dispersion of the nanofiller, mixing with a polymer latex to form a two-component colloidal mixture and drying (lyophilization) of the colloidal mixture in order to yield a composite.109 Latex technology has been successfully used to produce various CNT–polymer nanocomposites and graphene–polystyrene (PS) nanocomposites.109 The graphene–PS nanocomposites were prepared using latex technology with percolation thresholds as low as 0.8 wt% and maximum conductivity values of 0.15 S cm−1 for up to 2 wt% graphene loading.109 In this work, it was also demonstrated that controlled clustering of the graphene filler favours the lowering of the percolation threshold.
For biomedical applications such as cochlear implants, a polymer electrode with elastomeric mechanical properties and metal like conductivity can offer a solution to overcome the problems associated with electrical interfacing with neural tissue.145 The following sections present the current progress in developing conductive polymer composites of non-conjugated polymers with carbon and non-carbon conductive fillers such as graphene, CNTs and metal nanoparticles.
3.2.3.1 Carbon filler based conductive polymer composites. Ruoff and co-workers presented for the first time a general approach for the preparation of graphene–polymer composites via complete exfoliation of graphite and molecular-level dispersion of individual, chemically modified graphene sheets within polymer matrix (Fig. 6).115 A PS–graphene composite formed by this route exhibited a percolation threshold of 0.1 vol% for room temperature electrical conductivity. At only 1 vol%, this composite exhibited a conductivity of 0.001 S cm−1. Luo et al. reported the preparation of composite of PET resin and CNTs by melt compounding using a twin-screw extruder.135 The composites with 4 wt% loading of CNTs exhibited a volume electrical resistance of 103 Ω cm, 12 orders of magnitude lower than that of pure PET. Scanning electron microscopy (SEM) micrograph showed well dispersed CNTs in PET matrix although optical microscopy micrograph showed discontinuity of conductive phase existed in some segments of the composite fibre. The rheological behaviour of PET/CNTs composites showed that PET/CNTs composites containing high nanotube loadings exhibited a large decrease in viscosity with increasing shear frequency. A composite fiber was prepared using the conductive PET/CNTs composites and pure PET resin by a spinning process and a cloth was woven from the composite fiber and common terylene (composition 1
:3). The cloth showed good anti-static electricity property with a charge surface density of 0.25 μC m−2.
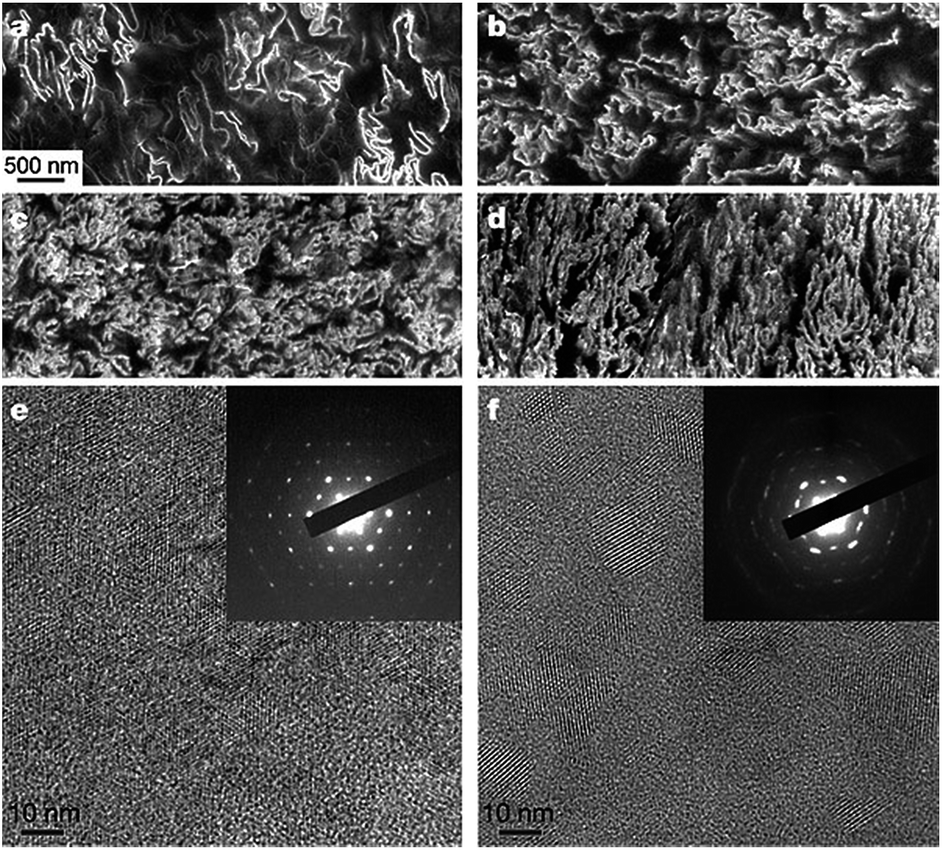 | ||
| Fig. 6 SEM and TEM images of graphene–polystyrene composite. (a–d) SEM images of the microtomed composites reveal different morphologies of the graphene sheets, including their packing, at different concentrations (vol%): (a) 0.24; (b) 0.96; (c) 1.44; and (d) 2.4. Scale bar, shown in (a) applies to (a–d). (e and f) High-resolution phase contrast images and SAED patterns (inset) of (e) cast film made from powder composite, and (f) microtomed composite sample. The SAED patterns show the six-fold rotational symmetry expected for diffraction with the beam incident along [0001]. Adapted from ref. 115. Reprinted by permission from Macmillan Publishers Ltd: Nature, 442(7100), 282–286, copyright 2006. | ||
In another study on PET, Yu and co-workers prepared PET/graphene nanocomposites by melt compounding which resulted in a sharp transition of PET from electrical insulator to semiconductor with a low percolation threshold of 0.47 vol%.133 Furthermore, the electrical conductivity of 2.11 × 10−2 S cm−1 was achieved with 3.0 vol% loading of graphene. The low percolation threshold and improved electrical conductivity were attributed to the high aspect ratio, large specific surface area and uniform dispersion of the graphene nanosheets in PET matrix.133 In a separate study, a nanocomposite paper was prepared from reduced graphene oxide sheets (rGO) and amine-modified nanofibrillated cellulose (A-NFC).146 Various rGO/A-NFC nanocomposites with varied content of graphene (0.1–10 wt%) were obtained. The rGO/A-NFC nanocomposites exhibited an electrical percolation threshold of 0.3 wt% with an electrical conductivity of 4.79 × 10−6 S cm−1 and a conductivity of 0.72 S cm−1 with 10% graphene loading. The composite showed improved tensile strength when compared to neat cellulose and graphene oxide paper, demonstrating an excellent reinforcement of graphene sheets.
PU-based composite films containing highly aligned graphene sheets were produced through an environmentally benign process developed by Kim and coworkers.138 An aqueous liquid crystalline dispersion of graphene oxide was reduced in situ in PU, hence producing a fine dispersion and a high degree of orientation of graphene sheets. The electrical conductivity of the composites was measured in the in-plane direction (surface conductivity) as well as in the through-the-thickness (or perpendicular) direction (volume conductivity). The conductivities of the composites containing 0.5 wt% graphene were of the same order of magnitude and almost identical in the in-plane and perpendicular directions, showcasing an isotropic behaviour and confirming homogeneous and random dispersions of GO. However, by increasing the graphene content to 2 and 5 wt%, there was several orders of magnitude of difference between the conductivities in two directions. The conductivity in the in-plane direction was significantly higher (10−3 S cm−1, 4 wt% filler loading) than that measured through the thickness (1.7 × 10−8 S cm−1). The significant anisotropy in electrical conductivity in composites with high graphene contents was attributed to alignment of graphene sheets so that conductive networks are preferentially formed along the plane direction whereas fewer conductive paths are present in the bulk. In another attempt by Cho et al.,139 highly flexible, conductive, and shape memory polyurethane nanocomposites were prepared for potential applications as materials for actuators, electronics and artificial muscles. Composites were prepared using both graphene and CNTs as conductive fillers and their effect on electrical and thermal conductivity of the composite was examined. CNTs and functionalized graphene sheets were incorporated as crosslinkers in the prepolymer. In comparison to pristine PU and CNT-crosslinked PU composite, the graphene-crosslinked PU composite exhibited better mechanical properties. The graphene-crosslinked PU composite also showed a higher electrical conductivity (1.67 × 10−3 S cm−1) than the CNT-crosslinked PU composite (2.30 × 10−4 S cm−1). The composites also exhibited good shape recovery, shape retention and fast electroactive shape recovery rate.
In a more recent study by Yang and coworkers, graphene incorporated polystyrene nanocomposites were prepared by integrating electrostatic self-assembly and latex technology.125 Positively charged polystyrene was synthesized first via disperse polymerization using a cationic co-monomer and then was directly co-assembled with graphene oxide. A honeycomb like graphene three dimensional framework was embedded in polystyrene matrix after in situ chemical reduction and hot compression molding. The resultant nanocomposites showed extremely low percolation threshold of 0.09 vol% and a conductivity of 0.252 S cm−1 at filler content of 1.22 vol%. This study demonstrated the use of integrating two methods to obtain composites with well-organised three dimensional microstructures and hence, better electrical conductivity.
In another very recent study, Jeong and coworkers examined the effect of extended thermal treatment to improve the conductivity of graphene loaded composites.136 Moderately functionalized graphenes were used to prepare electroconductive graphene/nylon 6 composites with a low percolation threshold of 0.39 wt% and an electrical conductivity of 6.84 × 10−4 S cm−1 for a low carbon incorporation of 0.54 wt%.136 The functionality of the graphenes was modulated by the thermal reduction time and the graphitic structure of graphene was strengthened by extended thermal treatment. It was observed that the main strengthening mechanism in the first 5 min was the generation of new sp2 domains followed by the growth of the domains during the next 5 min. This extended thermal treatment improved the conductivity of the graphene itself as well as the composite loaded with graphene. However, it led to a poor dispersion of graphene in the composites, reduced crystallization of nylon 6 and reduced reinforcement of nylon 6 by graphene.136
Furthermore, there are various reports found in literature on use of CNTs as conductive filler to make conductive polymer composites for various biomedical applications such as scaffolds for bone regeneration, tissue engineering and nerve regeneration.150–158
3.2.3.2 Non-carbon filler based conductive polymer composites. Metal nanoparticles have been also investigated as conductive fillers to prepare conductive polymer composites. Kotov and co-workers demonstrated the fabrication of stretchable conductors of PU containing spherical gold nanoparticles deposited by either layer-by-layer assembly or vacuum-assisted flocculation.149 High conductivity and stretchability were observed in both composites despite the minimal aspect ratio of the nanoparticles. These materials achieved electrical conductivities of up to 11
000 S cm−1 and also demonstrated the electronic tunability of mechanical properties resulting from dynamic self-organization of the nanoparticles under stress.
In another study, Pei et al. fabricated an elastomeric transparent composite electrode comprising a percolation network of copper nanowires (CuNWs) embedded in the surface layer of an elastomeric PU matrix.140 The composite electrode was fabricated by first forming a highly conductive CuNW network on glass, then overcoating with a layer of a liquid polyurethane precursor which was subsequently polymerized, and finally peeling off the resulting PU sheet. The composite retained the elastomeric stretchability of the polymer matrix. Pre-treatment of the CuNW network with 6-aminohexanoic acid enhanced the bonding between nanowires and PU matrix, and significantly improved the reversibility of the surface conductance of the composite electrode during repeated stretching at room temperature.
An overview of properties and applications of conductive composites of non conjugated polymers is provided in Table 4.
| Composite/blend | Loading of filler | Method of fabrication | Conductivity (S cm−1) | Properties | Applications | Ref. |
|---|---|---|---|---|---|---|
| Polystyrene/graphene | 2.5 vol% | Solution mixing | 1 | Good electrical conductivity | — | 115 |
| Polyethylene terephthalate/graphene | 3 vol% | Melt compounding | 2.11 × 10−2 | Enhanced electrical conductivity | Electromagnetic interference (EMI) shielding devices | 133 |
| Polyethylene terephthalate/CNT | 4 wt% | Melt compounding | 10−3 | Anti-static electrical property | — | 135 |
| Cellulose/graphene nanocomposite paper | 10 wt% | Solution mixing | 0.72 | Enhanced mechanical and electrical properties | Portable and bendable electronic equipment, EMI shielding devices and electromagnetic pulse protection | 146 |
| Nylon 6/graphene | 0.54 wt% | In situ polymerization | 6.84 × 10−4 | Enhanced electrical conductivity | — | 136 |
| Polystyrene/graphene nanocomposites | 1.22 vol% | Electrostatic assembly integrated latex technology | 0.252 | Enhanced electrical conductivity | — | 125 |
| PU/graphene | 2 wt% | In situ polymerization | >10−3 | Shape memory properties | — | 147 |
| PU/graphene | 4 wt% | In situ polymerization | 1.67 × 10−3 | Shape memory properties | Actuating devices, artificial muscles | 139 |
| PU/rGO (4%) | 4 wt% | Solution mixing | 10−3 | Enhanced electrical conductivity | — | 138 |
| Waterborne PU/acid treated CNT | 1.5 wt% | In situ polymerization | 1.1 × 10−3 | Enhanced thermal, conductive, and antistatic properties | Waterborne coatings | 148 |
| PU/CNT | 4 wt% | In situ polymerization | 2.30 × 10−4 | — | Actuating devices | 139 |
| PU/CuNW | — | In situ polymerization | — | Low sheet resistance of <102 Ω sq−1, elastomeric, transparent | Stretchable electrodes | 140 |
| PU/AuNP | 21.7 vol% | Layer by layer deposition and vacuum-assisted flocculation | Up to 1.1 × 104 | High electrical conductivity, flexible | Stretchable conductors in medical, optoelectronics, and energy storage devices | 149 |
In summary, conductive composites of CPs have been explored in order to overcome their insolubility, brittleness and low processability, while retaining their biological properties such as cell adhesion. Most of this work has focussed on biomedical applications and these studies have demonstrated that the electrical conductivity of CPs is usually compromised at the expense of mechanical properties. On the other hand, the work on conductive composites of non conjugated polymers is relatively recent and more focussed on understanding the fundamentals such as impact of different conductive fillers and their loadings. While the initial work on non conjugated polymer composites was based on conventional (non-biomedical polymers) such as PS, PET and nylon. In more recent work, conductive composites of polyurethane and biocompatible natural polymers have been investigated for biomedical applications. Amongst the organic fillers, graphene is a more popular choice as conductive filler due to its high conductivity and ease of incorporation. The ultimate goal is to achieve reasonable electrical conductivity with lowest possible amount of conductive filler, while retaining the properties of host polymer. The major challenges thus lie in selection of conductive filler, achieving low percolation threshold and retaining biocompatibility for biomedical applications.
4. Conclusions and future prospects
The review was focused on assessing the level of understanding of the potential of conducting polymers for biomedical applications based on many literature reported studies over the last decade. Most of the early studies have been focussed on evaluating the suitability of well known conducting polymers for biomedical applications. The limitations of conducting polymers, such as low processability, poor mechanical properties and biocompatibility, have prompted researchers to explore various chemical modification techniques, and blending with conducting nanoparticles and non-conducting polymers to overcome these limitations. The importance of the choice of conducting particles combined with appropriate blending techniques appear to be the key to develop useful composites that may find applications in biomedical implants. While this approach may help address processability, mechanical properties and biocompatibility, the improvement will come with some compromise in electrical conductivity, limiting the range of applications for these materials.In many of the reported studies, biocompatibility testing has been limited to in vitro screening and any further advancement of these materials require appropriate functional animal studies before they can be used in clinical applications. It is clear that CPs are promising materials to fulfil material requirements in medical implants, in particular implants used in neural stimulation and sensing. Tissue engineering is another area that these materials may find applications, mainly as substrates for regeneration of tissues where electrically conductivity can enhance cell growth. However, the area is full of unresolved technology challenges providing researchers with opportunities for further research and development work.
Acknowledgements
The authors would like to acknowledge the financial support provided by CSIRO Office of Chief Executive Postdoctoral Program.References
- A. K. Bakhshi and G. Bhalla, J. Sci. Ind. Res., 2004, 63, 715–728 CAS.
- W. Wei, H. Wang and Y. H. Hu, Int. J. Energy Res., 2014, 38, 1099–1111 CrossRef CAS PubMed.
- M. Ates, T. Karazehir and A. S. Sarac, Curr. Phys. Chem., 2012, 2, 224–240 CrossRef CAS.
- T. K. Das and S. Prusty, Polym.–Plast. Technol. Eng., 2012, 51, 1487–1500 CrossRef CAS PubMed.
- H. E. Katz and J. Huang, Annu. Rev. Mater. Res., 2009, 39, 71–92 CrossRef CAS.
- I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281–2305 CrossRef CAS PubMed.
- U. Lange, N. V. Roznyatovskaya and V. M. Mirsky, Anal. Chim. Acta, 2008, 614, 1–26 CrossRef CAS PubMed.
- A. Ramanavičius, A. Ramanavičiene and A. Malinauskas, Electrochim. Acta, 2006, 51, 6025–6037 CrossRef PubMed.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS PubMed.
- Z. Yin and Q. Zheng, Adv. Energy Mater., 2012, 2, 179–218 CrossRef CAS PubMed.
- J. F. Mike and J. L. Lutkenhaus, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 468–480 CrossRef CAS PubMed.
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2–18 CrossRef CAS PubMed.
- J. Jagur-Grodzinski, e-Polym., 2012, 12, 722–740 Search PubMed.
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS PubMed.
- B. Guo, L. Glavas and A. C. Albertsson, Prog. Polym. Sci., 2013, 38, 1263–1286 CrossRef CAS PubMed.
- R. Balint, N. J. Cassidy and S. H. Cartmell, Acta Biomater., 2014, 10, 2341–2353 CrossRef CAS PubMed.
- Z.-B. Huang, G.-F. Yin, X.-M. Liao and J.-W. Gu, Front. Mater. Sci., 2014, 8, 39–45 CrossRef.
- B. Bolto, R. McNeill and D. Weiss, Aust. J. Chem., 1963, 16, 1090–1103 CrossRef CAS.
- C. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis, S. C. Gau and A. G. MacDiarmid, Phys. Rev. Lett., 1977, 39, 1098–1101 CrossRef CAS.
- A. D. Bendrea, L. Cianga and I. Cianga, J. Biomater. Appl., 2011, 26, 3–84 CrossRef CAS PubMed.
- R. A. Green, N. H. Lovell, G. G. Wallace and L. A. Poole-Warren, Biomaterials, 2008, 29, 3393–3399 CrossRef CAS PubMed.
- A. J. Heeger, Angew. Chem., Int. Ed., 2001, 40, 2591–2611 CrossRef CAS.
- G. Wallace and G. Spinks, Soft Matter, 2007, 3, 665–671 RSC.
- M. J. Higgins and G. G. Wallace, Polym. Rev., 2013, 53, 506–526 CrossRef CAS PubMed.
- M. Ates, Mater. Sci. Eng., C, 2013, 33, 1853–1859 CrossRef CAS PubMed.
- M. Gerard, A. Chaubey and B. D. Malhotra, Biosens. Bioelectron., 2002, 17, 345–359 CrossRef CAS.
- R. Gracia and D. Mecerreyes, Polym. Chem., 2013, 4, 2206–2214 RSC.
- B. D. Malhotra, A. Chaubey and S. P. Singh, Anal. Chim. Acta, 2006, 578, 59–74 CrossRef CAS PubMed.
- S. C. Luo, Polym. Rev., 2013, 53, 303–310 CrossRef CAS PubMed.
- A. R. Harris, S. J. Morgan, J. Chen, R. M. I. Kapsa, G. G. Wallace and A. G. Paolini, J. Neural Eng., 2013, 10, 016004 CrossRef PubMed.
- R. Ravichandran, S. Sundarrajan, J. R. Venugopal, S. Mukherjee and S. Ramakrishna, J. R. Soc., Interface, 2010, 7, S559–S579 CrossRef CAS PubMed.
- A. F. Diaz, K. K. Kanazawa and G. P. Gardini, J. Chem. Soc., Chem. Commun., 1979, 635–636 RSC.
- K. Kaneto, K. Yoshino and Y. Inuishi, Solid State Commun., 1983, 46, 389–391 CrossRef CAS.
- J. L. Bredas and G. B. Street, Acc. Chem. Res., 1985, 18, 309–315 CrossRef CAS.
- L. Dai, in Intelligent Macromolecules for Smart Devices, Springer, London, 2004, pp. 41–80 Search PubMed.
- L. Dai, J. Macromol. Sci., Polym. Rev., 1999, 39, 273–387 CrossRef PubMed.
- H. Gustafsson, C. Kvarnström and A. Ivaska, Thin Solid Films, 2008, 517, 474–478 CrossRef CAS PubMed.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- J. Ouyang, C. W. Chu, F. C. Chen, Q. Xu and Y. Yang, Adv. Funct. Mater., 2005, 15, 203–208 CrossRef CAS PubMed.
- D. D. Ateh, H. A. Navsaria and P. Vadgama, J. R. Soc., Interface, 2006, 3, 741–752 CrossRef CAS PubMed.
- A. Gelmi, M. J. Higgins and G. G. Wallace, Biomaterials, 2010, 31, 1974–1983 CrossRef CAS PubMed.
- D. H. Kim, S. M. Richardson-Burns, J. L. Hendricks, C. Sequera and D. C. Martin, Adv. Funct. Mater., 2007, 17, 79–86 CrossRef CAS PubMed.
- R. T. Richardson, A. K. Wise, B. C. Thompson, B. O. Flynn, P. J. Atkinson, N. J. Fretwell, J. B. Fallon, G. G. Wallace, R. K. Shepherd, G. M. Clark and S. J. O'Leary, Biomaterials, 2009, 30, 2614–2624 CrossRef CAS PubMed.
- X. Cui, J. Wiler, M. Dzaman, R. A. Altschuler and D. C. Martin, Biomaterials, 2003, 24, 777–787 CrossRef CAS.
- S. Geetha, C. R. K. Rao, M. Vijayan and D. C. Trivedi, Anal. Chim. Acta, 2006, 568, 119–125 CrossRef CAS PubMed.
- I. S. Chronakis, S. Grapenson and A. Jakob, Polymer, 2006, 47, 1597–1603 CrossRef CAS PubMed.
- X. Cui, J. F. Hetke, J. A. Wiler, D. J. Anderson and D. C. Martin, Sens. Actuators, A, 2001, 93, 8–18 CrossRef CAS.
- D. H. Kim, M. Abidian and D. C. Martin, J. Biomed. Mater. Res., Part A, 2004, 71, 577–585 CrossRef PubMed.
- X. Zhang and S. K. Manohar, J. Am. Chem. Soc., 2004, 126, 12714–12715 CrossRef CAS PubMed.
- J. W. Lee, F. Serna, J. Nickels and C. E. Schmidt, Biomacromolecules, 2006, 7, 1692–1695 CrossRef CAS PubMed.
- A. Ramanaviciene, A. Kausaite, S. Tautkus and A. Ramanavicius, J. Pharm. Pharmacol., 2007, 59, 311–315 CrossRef CAS PubMed.
- K. Yang, H. Xu, L. Cheng, C. Sun, J. Wang and Z. Liu, Adv. Mater., 2012, 24, 5586–5592 CrossRef CAS PubMed.
- A. Vaitkuviene, V. Kaseta, J. Voronovic, G. Ramanauskaite, G. Biziuleviciene, A. Ramanaviciene and A. Ramanavicius, J. Hazard. Mater., 2013, 250–251, 167–174 CrossRef CAS PubMed.
- X. Wang, X. Gu, C. Yuan, S. Chen, P. Zhang, T. Zhang, J. Yao, F. Chen and G. Chen, J. Biomed. Mater. Res., Part A, 2004, 68, 411–422 CrossRef PubMed.
- D. K. Cullen, R. P. Ankur, F. D. John, H. S. Douglas and J. P. Bryan, J. Neural Eng., 2008, 5, 374 CrossRef PubMed.
- A. Borriello, V. Guarino, L. Schiavo, M. A. Alvarez-Perez and L. Ambrosio, J. Mater. Sci.: Mater. Med., 2011, 22, 1053–1062 CrossRef CAS PubMed.
- Y. Guo, M. Li, A. Mylonakis, J. Han, A. G. MacDiarmid, X. Chen, P. I. Lelkes and Y. Wei, Biomacromolecules, 2007, 8, 3025–3034 CrossRef CAS PubMed.
- S. Bhadra, D. Khastgir, N. K. Singha and J. H. Lee, Prog. Polym. Sci., 2009, 34, 783–810 CrossRef CAS PubMed.
- L. Ghasemi-Mobarakeh, M. P. Prabhakaran, M. Morshed, M. H. Nasr-Esfahani, H. Baharvand, S. Kiani, S. S. Al-Deyab and S. Ramakrishna, J. Tissue Eng. Regener. Med., 2011, 5, e17–e35 CrossRef CAS PubMed.
- S. Ameen, M. Shaheer Akhtar and M. Husain, Sci. Adv. Mater., 2010, 2, 441–462 CrossRef CAS PubMed.
- P. Humpolicek, V. Kasparkova, P. Saha and J. Stejskal, Synth. Met., 2012, 162, 722–727 CrossRef CAS PubMed.
- L. Huang, X. Zhuang, J. Hu, L. Lang, P. Zhang, Y. Wang, X. Chen, Y. Wei and X. Jing, Biomacromolecules, 2008, 9, 850–858 CrossRef CAS PubMed.
- R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- Y. Yagci and L. Toppare, Polym. Int., 2003, 52, 1573–1578 CrossRef CAS PubMed.
- M. Waugaman, B. Sannigrahi, P. McGeady and I. M. Khan, Eur. Polym. J., 2003, 39, 1405–1412 CrossRef CAS.
- G. Scarpa, A.-L. Idzko, S. Götz and S. Thalhammer, Macromol. Biosci., 2010, 10, 378–383 CrossRef CAS PubMed.
- P. Sista, K. Ghosh, J. S. Martinez and R. C. Rocha, J. Nanosci. Nanotechnol., 2014, 14, 250–272 CrossRef CAS PubMed.
- Y. Xiao, X. Cui and D. C. Martin, J. Electroanal. Chem., 2004, 573, 43–48 CAS.
- A. Peramo, M. G. Urbanchek, S. A. Spanninga, L. K. Povlich, P. Cederna and D. C. Martin, Tissue Eng., Part A, 2008, 14, 423–432 CrossRef CAS PubMed.
- C. A. Thomas, K. Zong, P. Schottland and J. R. Reynolds, Adv. Mater., 2000, 12, 222–225 CrossRef CAS.
- R. Starbird, C. A. García-González, I. Smirnova, W. H. Krautschneider and W. Bauhofer, Mater. Sci. Eng., C, 2014, 37, 177–183 CrossRef CAS PubMed.
- S. Venkatraman, J. Hendricks, Z. A. King, A. J. Sereno, S. Richardson-Burns, D. Martin and J. M. Carmena, IEEE Trans. Neural Syst. Rehabil. Eng., 2011, 19, 307–316 CrossRef PubMed.
- F. Greco, A. Zucca, S. Taccola, A. Menciassi, T. Fujie, H. Haniuda, S. Takeoka, P. Dario and V. Mattoli, Soft Matter, 2011, 7, 10642–10650 RSC.
- H. A. Lin, S. C. Luo, B. Zhu, C. Chen, Y. Yamashita and H. H. Yu, Adv. Funct. Mater., 2013, 23, 3212–3219 CrossRef CAS PubMed.
- L. Jin, T. Wang, Z. Q. Feng, M. K. Leach, J. Wu, S. Mo and Q. Jiang, J. Mater. Chem. B, 2013, 1, 1818–1825 RSC.
- F. Greco, V. Domenici, A. Desii, E. Sinibaldi, B. Zupančič, B. Zalar, B. Mazzolai and V. Mattoli, Soft Matter, 2013, 9, 11405–11416 RSC.
- G. Tarabella, A. G. Balducci, N. Coppedè, S. Marasso, P. D'Angelo, S. Barbieri, M. Cocuzza, P. Colombo, F. Sonvico, R. Mosca and S. Iannotta, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 4374–4380 CrossRef CAS PubMed.
- L. Sui, X. J. Song, J. Ren, W. J. Cai, L. H. Ju, Y. Wang, L. Y. Wang and M. Chen, J. Biomed. Mater. Res., Part A, 2014, 102, 1681–1696 CrossRef CAS PubMed.
- N. Gomez and C. E. Schmidt, J. Biomed. Mater. Res., Part A, 2007, 81, 135–149 CrossRef PubMed.
- J. H. Collier, J. P. Camp, T. W. Hudson and C. E. Schmidt, J. Biomed. Mater. Res., 2000, 50, 574–584 CrossRef CAS.
- C. R. Broda, J. Y. Lee, S. Sirivisoot, C. E. Schmidt and B. S. Harrison, J. Biomed. Mater. Res., Part A, 2011, 98, 509–516 CrossRef PubMed.
- J. Njuguna and K. Pielichowski, J. Mater. Sci., 2004, 39, 4081–4094 CrossRef CAS.
- J. Stejskal, Chem. Pap., 2013, 67, 814–848 CAS.
- R. J. Lee, R. Temmer, T. Tamm, A. Aabloo and R. Kiefer, React. Funct. Polym., 2013, 73, 1072–1077 CrossRef CAS PubMed.
- T. Takano, A. Mikazuki and T. Kobayashi, Polym. Eng. Sci., 2014, 54, 78–84 CAS.
- H.-S. Kim, H. L. Hobbs, L. Wang, M. J. Rutten and C. C. Wamser, Synth. Met., 2009, 159, 1313–1318 CrossRef CAS PubMed.
- E. M. Stewart, M. Fabretto, M. Mueller, P. J. Molino, H. J. Griesser, R. D. Short and G. G. Wallace, Biomater. Sci., 2013, 1, 368–378 RSC.
- Z. Shi, S. Zang, F. Jiang, L. Huang, D. Lu, Y. Ma and G. Yang, RSC Adv., 2012, 2, 1040–1046 RSC.
- A. K. Bajpai, J. Bajpai and S. N. Soni, J. Macromol. Sci., Part A: Pure Appl.Chem., 2009, 46, 774–782 CrossRef CAS PubMed.
- M. M. Perez-Madrigal, M. I. Giannotti, E. Armelin, F. Sanz and C. Aleman, Polym. Chem., 2014, 5, 1248–1257 RSC.
- H. Okuzaki, S. Takagi, F. Hishiki and R. Tanigawa, Sens. Actuators, B, 2014, 194, 59–63 CrossRef CAS PubMed.
- H. Xu, J. M. Holzwarth, Y. Yan, P. Xu, H. Zheng, Y. Yin, S. Li and P. X. Ma, Biomaterials, 2014, 35, 225–235 CrossRef CAS PubMed.
- C.-L. Choong, M.-B. Shim, B.-S. Lee, S. Jeon, D.-S. Ko, T.-H. Kang, J. Bae, S. H. Lee, K.-E. Byun, J. Im, Y. J. Jeong, C. E. Park, J.-J. Park and U. I. Chung, Adv. Mater., 2014, 26, 3451–3458 CrossRef CAS PubMed.
- L. K. H. Trang, T. Thanh Tung, T. Young Kim, W. S. Yang, H. Kim and K. S. Suh, Polym. Int., 2012, 61, 93–98 CrossRef CAS PubMed.
- N. Ferraz, M. Strømme, B. Fellström, S. Pradhan, L. Nyholm and A. Mihranyan, J. Biomed. Mater. Res., Part A, 2012, 100, 2128–2138 CrossRef PubMed.
- S. Meng, M. Rouabhia, G. Shi and Z. Zhang, J. Biomed. Mater. Res., Part A, 2008, 87, 332–344 CrossRef PubMed.
- A. Guiseppi-Elie, Biomaterials, 2010, 31, 2701–2716 CrossRef CAS PubMed.
- D. Mawad, E. Stewart, D. L. Officer, T. Romeo, P. Wagner, K. Wagner and G. G. Wallace, Adv. Funct. Mater., 2012, 22, 2692–2699 CrossRef CAS PubMed.
- F. Alam, S. A. Ansari, W. Khan, M. E. Khan and A. H. Naqvi, Funct. Mater. Lett., 2012, 5, 1250026 CrossRef.
- M. Atmeh and B. E. Alcock-Earley, J. Appl. Electrochem., 2011, 41, 1341–1347 CrossRef CAS.
- M. M. Ayad and E. Zaki, Appl. Surf. Sci., 2009, 256, 787–791 CrossRef CAS PubMed.
- D. Untereker, S. Lyu, J. Schley, G. Martinez and L. Lohstreter, ACS Appl. Mater. Interfaces, 2009, 1, 97–101 CAS.
- F. Du, R. C. Scogna, W. Zhou, S. Brand, J. E. Fischer and K. I. Winey, Macromolecules, 2004, 37, 9048–9055 CrossRef CAS.
- A. Nogales, G. Broza, Z. Roslaniec, K. Schulte, I. Šics, B. S. Hsiao, A. Sanz, M. C. García-Gutiérrez, D. R. Rueda, C. Domingo and T. A. Ezquerra, Macromolecules, 2004, 37, 7669–7672 CrossRef CAS.
- K. I. Winey, T. Kashiwagi and M. Mu, MRS Bull., 2007, 32, 348–353 CrossRef CAS.
- M. Antunes and J. I. Velasco, Prog. Polym. Sci., 2014, 39, 486–509 CrossRef CAS PubMed.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089–1115 CrossRef CAS PubMed.
- T. K. Das and S. Prusty, Polym.–Plast. Technol. Eng., 2013, 52, 319–331 CrossRef CAS PubMed.
- N. Grossiord, M.-C. Hermant and E. Tkalya, in Polymer-Graphene Nanocomposites, The Royal Society of Chemistry, 2012, pp. 66–85 Search PubMed.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS PubMed.
- X. Cheng, C. Li, L. Rao, H. Zhou, T. Li and Y. Duan, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2012, 27, 1053–1057 CrossRef CAS.
- P. J. Brigandi, J. M. Cogen and R. A. Pearson, Polym. Eng. Sci., 2014, 54, 1–16 CAS.
- T. Gurunathan, C. R. K. Rao, R. Narayan and K. V. S. N. Raju, J. Mater. Sci., 2013, 48, 67–80 CrossRef CAS.
- X. Sun, H. Sun, H. Li and H. Peng, Adv. Mater., 2013, 25, 5153–5176 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- M. J. Fernández-Merino, J. I. Paredes, S. Villar-Rodil, L. Guardia, P. Solís-Fernández, D. Salinas-Torres, D. Cazorla-Amorós, E. Morallón, A. Martínez-Alonso and J. M. D. Tascón, Carbon, 2012, 50, 3184–3194 CrossRef PubMed.
- A. M. Pinto, J. Martins, J. A. Moreira, A. M. Mendes and F. D. Magalhães, Polym. Int., 2013, 62, 928–935 CrossRef CAS PubMed.
- K. Kaneto, M. Tsuruta, G. Sakai, W. Y. Cho and Y. Ando, Synth. Met., 1999, 103, 2543–2546 CrossRef CAS.
- E. Dadrasnia, H. Lamela, M. B. Kuppam, F. Garet and J.-L. Coutaz, Adv. Condens. Matter Phys., 2014, 2014, 6 Search PubMed.
- J. Du and H. M. Cheng, Macromol. Chem. Phys., 2012, 213, 1060–1077 CrossRef CAS PubMed.
- H. Kim, A. A. Abdala and C. W. MacOsko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- A. M. Pinto, I. C. Gonçalves and F. D. Magalhães, Colloids Surf., B, 2013, 111, 188–202 CrossRef CAS PubMed.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS PubMed.
- S. Badaire, P. Poulin, M. Maugey and C. Zakri, Langmuir, 2004, 20, 10367–10370 CrossRef CAS PubMed.
- P. Zhao, Y. Luo, J. Yang, D. He, L. Kong, P. Zheng and Q. Yang, Mater. Lett., 2014, 121, 74–77 CrossRef CAS PubMed.
- E. Stefanescu, C. Daranga and C. Stefanescu, Materials, 2009, 2, 2095–2153 CrossRef CAS PubMed.
- H. Jiang, K. S. Moon, Y. Li and C. P. Wong, Chem. Mater., 2006, 18, 2969–2973 CrossRef CAS.
- S. Reich, S. Mazur, P. Avakian and F. C. Wilson, J. Appl. Phys., 1987, 62, 287–292 CrossRef CAS PubMed.
- J. Liang, Y. Xu, Y. Huang, L. Zhang, Y. Wang, Y. Ma, F. Li, T. Guo and Y. Chen, J. Phys. Chem. C, 2009, 113, 9921–9927 CAS.
- A. V. Raghu, Y. R. Lee, H. M. Jeong and C. M. Shin, Macromol. Chem. Phys., 2008, 209, 2487–2493 CrossRef CAS PubMed.
- A. Silvestri, P. M. Serafini, S. Sartori, P. Ferrando, F. Boccafoschi, S. Milione, L. Conzatti and G. Ciardelli, J. Appl. Polym. Sci., 2011, 122, 3661–3671 CrossRef CAS PubMed.
- C. Garzón and H. Palza, Compos. Sci. Technol., 2014, 99, 117–123 CrossRef PubMed.
- H.-B. Zhang, W.-G. Zheng, Q. Yan, Y. Yang, J.-W. Wang, Z.-H. Lu, G.-Y. Ji and Z.-Z. Yu, Polymer, 2010, 51, 1191–1196 CrossRef CAS PubMed.
- I. Jurewicz, P. Worajittiphon, A. A. K. King, P. J. Sellin, J. L. Keddie and A. B. Dalton, J. Phys. Chem. B, 2011, 115, 6395–6400 CrossRef CAS PubMed.
- Z. Li, G. Luo, F. Wei and Y. Huang, Compos. Sci. Technol., 2006, 66, 1022–1029 CrossRef CAS PubMed.
- C. I. Kim, S. M. Oh, K. M. Oh, E. Gansukh, H. I. Lee and H. M. Jeong, Polym. Int., 2014, 63, 1003–1010 CrossRef CAS PubMed.
- H. B. MacHado, R. N. Correia and J. A. Covas, J. Mater. Sci.: Mater. Med., 2010, 21, 2057–2066 CrossRef CAS PubMed.
- N. Yousefi, M. M. Gudarzi, Q. B. Zheng, S. H. Aboutalebi, F. Sharif and J. K. Kim, J. Mater. Chem., 2012, 22, 12709–12717 RSC.
- S. Rana, J. W. Cho and L. P. Tan, RSC Adv., 2013, 3, 13796–13803 RSC.
- W. L. Hu, R. R. Wang, Y. F. Lu and Q. B. Pei, J. Mater. Chem. C, 2014, 2, 1298–1305 RSC.
- S. Gogolewski, Colloid Polym. Sci., 1989, 267, 757–785 CAS.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110–4129 RSC.
- L. Xue and H. P. Greisler, J. Vasc. Surg., 2003, 37, 472–480 CrossRef PubMed.
- A. Rahimi and A. Mashak, Plast., Rubber Compos., 2013, 42, 223–230 CrossRef CAS PubMed.
- C. M. Williams, M. A. Nash and L. A. Poole-Warren, Progress in Biomedical Optics and Imaging, Proc. SPIE, 2005, 5651, 329–335 CrossRef CAS PubMed.
- N. D. Luong, N. Pahimanolis, U. Hippi, J. T. Korhonen, J. Ruokolainen, L.-S. Johansson, J.-D. Nam and J. Seppala, J. Mater. Chem., 2011, 21, 13991–13998 RSC.
- J. T. Choi, T. D. Dao, K. M. Oh, H. I. Lee, H. M. Jeong and B. K. Kim, Smart Mater. Struct., 2012, 21, 9 Search PubMed.
- J. Kwon and H. Kim, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3973–3985 CrossRef CAS PubMed.
- Y. Kim, J. Zhu, B. Yeom, M. Di Prima, X. Su, J.-G. Kim, S. J. Yoo, C. Uher and N. A. Kotov, Nature, 2013, 500, 59–63 CrossRef CAS PubMed.
- A. Abarrategi, M. C. Gutiérrez, C. Moreno-Vicente, M. J. Hortigüela, V. Ramos, J. L. López-Lacomba, M. L. Ferrer and F. del Monte, Biomaterials, 2008, 29, 94–102 CrossRef CAS PubMed.
- S. Shao, S. Zhou, L. Li, J. Li, C. Luo, J. Wang, X. Li and J. Weng, Biomaterials, 2011, 32, 2821–2833 CrossRef CAS PubMed.
- S. Nardecchia, M. C. Serrano, M. C. Gutierrez, M. L. Ferrer and F. D. Monte, J. Mater. Chem. B, 2013, 1, 3064–3072 RSC.
- S. L. Edwards, J. S. Church, J. A. Werkmeister and J. A. M. Ramshaw, Biomaterials, 2009, 30, 1725–1731 CrossRef CAS PubMed.
- K. D. McKeon-Fischer, D. H. Flagg and J. W. Freeman, J. Biomed. Mater. Res., Part A, 2011, 99, 493–499 CrossRef CAS PubMed.
- D. Y. Lewitus, J. Landers, J. R. Branch, K. L. Smith, G. Callegari, J. Kohn and A. V. Neimark, Adv. Funct. Mater., 2011, 21, 2624–2632 CrossRef CAS PubMed.
- M. Kabiri, M. Soleimani, I. Shabani, K. Futrega, N. Ghaemi, H. H. Ahvaz, E. Elahi and M. R. Doran, Biotechnol. Lett., 2012, 34, 1357–1365 CrossRef CAS PubMed.
- B. L. Behan, D. G. DeWitt, D. R. Bogdanowicz, A. N. Koppes, S. S. Bale and D. M. Thompson, J. Biomed. Mater. Res., Part A, 2011, 96, 46–57 CrossRef PubMed.
- M. C. Serrano, M. C. Gutiérrez and F. del Monte, Prog. Polym. Sci., 2014, 39, 1448–1471 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |