DOI:
10.1039/C5RA02055G
(Paper)
RSC Adv., 2015,
5, 46316-46324
Novel catalyst PTMA-PILC: structural properties and catalytic performance for the dehydration of bioethanol to ethylene†
Received
2nd February 2015
, Accepted 8th May 2015
First published on 8th May 2015
Abstract
Novel phosphomolybdic acid-pillared interlayer clay (PTMA-PILC) catalysts were prepared by three steps: acid treatment, ion-exchange and impregnation method. The resulting catalysts were initially characterized by a combination of XRD analysis, FT-IR, TG, NH3-TPD, pyridine-IR, TEM, and N2 adsorption–desorption. The characterization results indicated that the sandwich structure of the montmorillonite was delaminated on acid treatment, resulting in a reduction of the surface area to 335 m2 g−1 and the formation of plate-like particles. Surprisingly, after ion-exchange and impregnation, the stratified structure was restored. The strong and weak acidity contents also changed. The catalytic properties of this series of catalysts were tested in the dehydration of ethanol to ethylene. A significant improvement in catalytic activity was observed in the dehydration of ethanol (30 vol%) to ethylene on the synthesized PTMA-PILC catalyst, from which 93.2% conversion and 99.1% selectivity of ethylene were achieved at 300 °C. The improved activities were attributed to the layer structure and the balance of Lewis and Brønsted acid sites.
1 Introduction
Ethylene is one of the most important fine chemicals in the petrochemical industry, and it is mainly produced from the thermal cracking of petroleum or nature gas feedstock.1 With shortages of natural resources and energy and the soaring prices of crude oil, the production of petrochemicals from a non-petroleum, environmentally friendly feedstock has sparked a great deal of interest; the development of efficient ethylene production processes is a challenging research area.2,3 Compared with the traditional process, ethylene production from the dehydration of ethanol is economically feasible, with higher purity.4 Moreover, the utilization of ethanol from biomass fermentation instead of petroleum as a chemical feedstock may facilitate the reduction of CO2 emissions, which is a crucial problem in the chemical industry.5 In order to make ethanol dehydration more industry-friendly, scientists and researchers have developed different catalysts to enhance the yield of ethylene and lower the reaction temperature and alcohol concentration. The catalysts for acid-catalyzed alcohol dehydration can be classified in four categories:4 phosphoric acids,6 oxides,7 zeolites8,9 and heteropolyacids.10,11 With earlier γ-Al2O3 catalysts, ethanol dehydration required higher reaction temperatures (400–450 °C) and offered lower ethylene yields (80%).12,13 The obvious disadvantage was the high energy consumption required for the purification process of bioethanol; poor catalytic properties were observed when bioethanol solution, which contains large amounts of water, was used as the reactant over an Al2O3 catalyst.1 HZSM-5 zeolite was studied recently, and an ethanol conversion of 98% at lower temperature (300 °C) was reported.14,15 However, deactivation caused by coke deposition readily occurred on HZSM-5 zeolites during ethanol dehydration because of the smaller pore size and strong acidic properties of the zeolites.16 Arias et al.17 reported the dehydration of ethanol over SAPO-11 catalyst. The yield of ethylene achieved was about 90% at 320 °C. In order to improve the yield of ethylene, Zhang et al.13 chose metal doped-SAPO as a catalyst; the highest yield of ethylene was 92.3% for the dehydration of ethanol at 350 °C over Ni-substituted SAPO-34. Unfortunately, the yield only increased about 2% at the cost of increasing the temperature by 30 °C. Thus far, the dehydration reaction of bioethanol to ethylene has been studied extensively. Supported heteropoly acids (HPA) have become popular because of their redox natures, super acidic properties and Keggin structures. They function ubiquitously in homogeneous as well as heterogeneous catalytic reactions.18–23 However, HPA has several shortcomings, such as instability, small specific surface area and ease of aggregation. These problems can be overcome by loading the heteropolyacids on supports with high surface areas and stable structures.
Over the past decades, considerable attention has been paid to montmorillonites (MMT) as catalysts and catalyst supports because of their porosities, reactivities and thermal stabilities. Montmorillonite is a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 clay mineral with two tetrahedral silica layers of Mg or Al that form an octahedral metal oxide structure. The layers are separated from each other by an interlayer or a gallery containing cations (Na+, K+, Ca2+, etc.), which balance the excess negative charges created by the natural substitution of some of the atoms forming the crystal.24 These cations are exchangeable, which is the reason for the high adsorption capacity of MMT for cations (cation exchange capacity, CEC). MMT in its natural form does not show much catalytic activity. To improve its porosity and stability, a pillaring method is often employed, which involves intercalating large organic or inorganic molecules25 between the silicate layers of clays. Pillared interlayer clays (PILCs) can be used as supports for conventional metal catalysts; Al-metal pillared clays are the most commonly used. PILCs are also considered to be important members of the porous solids family.26,27 In the traditional method, Al137+ ([AlO4Al12(OH)24(OH2)12]7+) units are inserted into the untreated MMT. The polymer Al137+ has a Keggin structure. It consists of 12 octahedral Al atoms arranged in four groups of three octahedra around a central (AlO4) tetrahedron.28 However, Al137+ cannot be distributed completely, since the hydroxyl polymer cannot be replaced by other cations, as chemical bonds can be formed between the H+ ions around the OH groups of [Al(OH)6] and the O2− ions of the MMT silicate layers.
:1 clay mineral with two tetrahedral silica layers of Mg or Al that form an octahedral metal oxide structure. The layers are separated from each other by an interlayer or a gallery containing cations (Na+, K+, Ca2+, etc.), which balance the excess negative charges created by the natural substitution of some of the atoms forming the crystal.24 These cations are exchangeable, which is the reason for the high adsorption capacity of MMT for cations (cation exchange capacity, CEC). MMT in its natural form does not show much catalytic activity. To improve its porosity and stability, a pillaring method is often employed, which involves intercalating large organic or inorganic molecules25 between the silicate layers of clays. Pillared interlayer clays (PILCs) can be used as supports for conventional metal catalysts; Al-metal pillared clays are the most commonly used. PILCs are also considered to be important members of the porous solids family.26,27 In the traditional method, Al137+ ([AlO4Al12(OH)24(OH2)12]7+) units are inserted into the untreated MMT. The polymer Al137+ has a Keggin structure. It consists of 12 octahedral Al atoms arranged in four groups of three octahedra around a central (AlO4) tetrahedron.28 However, Al137+ cannot be distributed completely, since the hydroxyl polymer cannot be replaced by other cations, as chemical bonds can be formed between the H+ ions around the OH groups of [Al(OH)6] and the O2− ions of the MMT silicate layers.
In this study, Na-montmorillonite (Na-MMT) precursors were first modified using sulfuric acid. The acid treatment enhanced the unique properties of MMT. The skeleton crystal lattice dehisced and the interlayer space of the MMT underwent an obvious increase. MMT was pillared by polymer Al137+ units. The structure of MMT was restored because of its “memory effect”. We further improved the catalytic properties by supporting phosphomolybdic acid (PTMA) on the modified MMT, which has a large surface area. These modified MMTs served as robust catalysts in the simulated dehydration of bioethanol (30 vol%) to ethylene with enhanced performance.
2 Experimental
2.1 Chemicals
Sodium montmorillonite (Na-MMT) was supplied by Zhejiang Sanding Technology Co., Ltd., China. The chemical composition of Na-MMT is shown in Table S1.† All chemicals used in this study, including NaOH, H2SO4 (98%), AlCl3·6H2O (Shanghai Jinshan Chemical Plant) and phosphomolybdic acid (Tianjin Kaida plant), were of analytical reagent grade.
2.2 Catalyst preparation
Acid treatment of MMT: purified Na-MMT (10 g) was dispersed in 40 mL H2SO4 with 25 percent mass and refluxed at 70 °C for 5 h. Then the slurry was cooled, filtered and washed thoroughly with distilled water. The MMT was dried at 80 °C for 6 h and named AT-MMT (acid treatment-montmorillonite).
Preparation of the pillared montmorillonite: the pillaring agent was prepared from the hydrolysis of AlCl3 solution with 0.2 mol L−1 NaOH. Sodium hydroxide was added to the aluminium chloride solution under vigorous stirring at room temperature. The titration was stopped when the OH−/Al3+ = 2.4, followed by aging at 70 °C for 24 h. A suspension of MMT with 10 g of AT-MMT and 400 mL of distilled water was first prepared and stirred for 1 h. A certain amount of the pillaring agents which were obtained by ion-exchange was added to the clay slurry; then the reaction mixture was vigorously stirred for 2 h at room temperature. After aging for 24 h at 60 °C, the pillared clay was washed until the chloride ions were totally eliminated; the clay was then dried at 70 °C for 24 h (named PILC (a), pillared interlayer clay (a)),and finally calcined at 400 °C for 3 h (named PILC, pillared interlayer clay).
Preparation of PTMA-PILC: 10 g of PILC and 0.5 g of PTMA (phosphomolybdic acid) were weighed separately. The PTMA was dissolved in 10 mL distilled water. The PTMA solution was added slowly to the suspension liquid of PILC with constant stirring. The sample was further dried in an oven at 80 °C in order to remove water completely, and the dried catalyst sample was maintained in a sealed bottle. Prior to use, the sample was dried in an oven to remove moisture.
The catalyst characterizations and catalytic performance tests are listed in the ESI.†
3 Results and discussion
3.1 XRD
Fig. 1 shows the XRD patterns of the Na-MMT, AT-MMT, PILC (a), PILC and PTMA-PILC catalysts. The lines at 7.08° and 19.77° of Na-MMT correspond to the support 001 and 100 reflections, and the lines at 2θ of 26.67° are attributed to quartz impurities. The obtained XRD pattern for Na-MMT exhibits a basal distance (d001) of 1.26 nm, typical of Na-MMT. However, the pattern for the 001 and 100 reflections of AT-MMT disappeared. It was deduced that the acid treatment dramatically changed the structure of the MMT and that the lamellar structure had been delaminated. After Al137+ was inserted into the interlayers of MMT, rather to our surprise, the structure and elementary properties of MMT were restored and the interlayer spacing d001 of PILC (a) increased from 1.26 nm to 1.70 nm, indicating a great expansion of the 001 interlayer distance. However, the height of the Al137+ layer was 0.9 nm,29 and the layer thickness obtained for AT-MMT was 0.8 nm (Fig. S2†), which is slightly lower than that found in previous research.30 The reduction of the MMT layer thickness may be due to the dealumination of the aluminium–oxygen octahedron structure. After calcination at 400 °C, a large number of hydroxyl groups were removed. The thermal treatment caused the transformation of the interlayer compounds from polycations to oxides. These results were in agreement with the chemical composition measured by XRF, shown in Table S2,† in which the amount of Al2O3 and the Al/Si molar ratio in PILC were higher than in AT-MMT. The interlayer spacing (d001) of PILC was reduced from 1.70 nm to 1.40 nm, possibly due to the reduction of the MMT layer thickness. The two-step pillaring process, wherein the firm molecular skeleton of MMT was first stripped by acid treatment and then recombined with the aid of Al137+, enabled the uniform dispersement of the active centre in the MMT carrier. There is no strong variation in the patterns and peaks attributed to other crystalline impurities for the PTMA-PILC catalyst, indicating that the PTMA was loaded onto the surface and in the interlamination of the PILC. The schematic model of the two-step method pillaring process of PTMA-PILC and the traditional methods of PILC (b), pillared interlayer clay (b), are shown in Scheme 1.
 |
| | Fig. 1 XRD patterns of different catalysts. | |
 |
| | Scheme 1 Schematic of the synthetic process of PTMA-PILC. | |
3.2 FT-IR
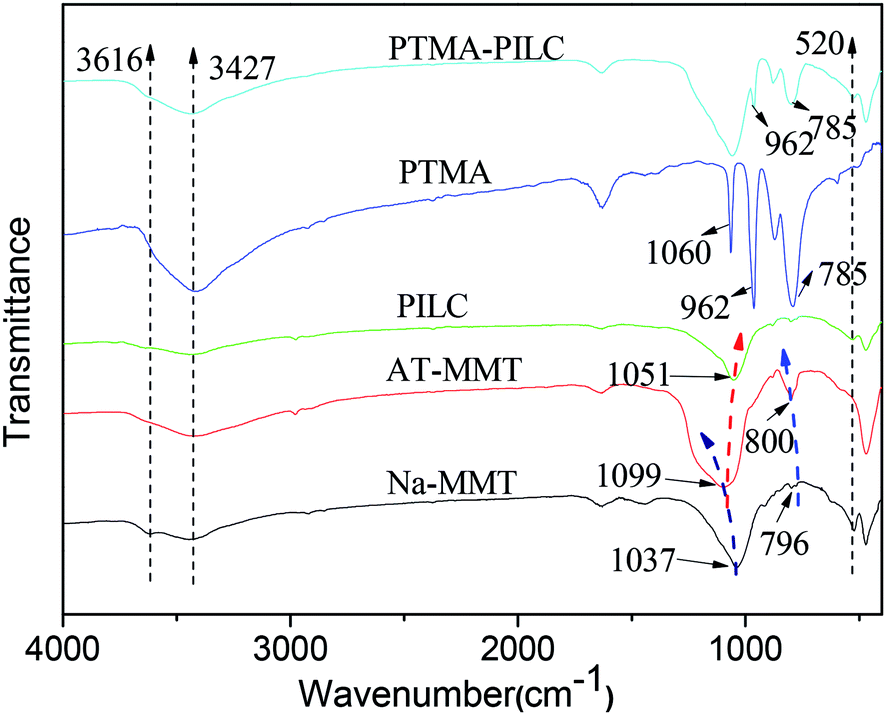
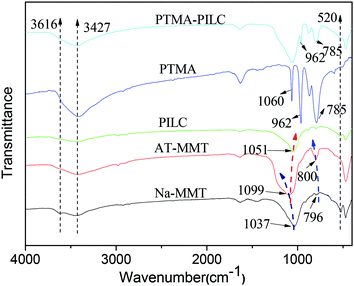
In the FT-IR spectra of the Na-MMT samples, the peaks at 1037 and 520 cm−1 were attributed to the Si–O in-plane stretching and Si–O bending vibrations respectively. The band at 796 cm−1 is attributed to the presence of cristobalite. The broad band at 3427 cm−1 belongs to the stretching vibration of the hydroxyl groups of water molecules. The band at 3616 cm−1 was assigned to the OH–Al bonds of octahedral cations.31 In the FT-IR spectra of AT-MMT, the position and shape of the band changed slightly. The band at 1037 cm−1 was shifted to a higher wavenumber (1099 cm−1) and the peak at 520 cm−1 disappeared. This was in agreement with the results of the XRD analysis, which showed that acid treatment had dramatically changed the structure of the MMT. The band at 796 cm−1 was shifted to 800 cm−1 and its intensity increased significantly after acid treatment. After pillaring, the band at 1099 cm−1 was shifted to a lower wavenumber (1051 cm−1), and the band at 3427 cm−1 was broadened, which was interpreted as the effect of pillaring.20 The increase in the intensity of the band at 3616 cm−1 was attributed to the presence of aluminum oxocations in the samples. This finding confirmed that the lamellar structure was recombined and the aluminium oxide was intercalated into the interlayers of the MMT. For pure PTMA, the characteristic absorption bands of Mo–O (785 cm−1), P–O (962 cm−1 and 1060 cm−1) were clearly observed. For the PTMA-PILC, the main characteristic peaks of PTMA were visible, proving that the novel supported catalyst had been synthesized (Fig. 2).
 |
| | Fig. 2 FT-IR spectras of different catalysts. | |
3.3 N2 adsorption–desorption isotherms
Nitrogen adsorption–desorption isotherms and the pore size distribution (inset) of the different catalysts are shown in Fig. 3. The size and shape of the hysteresis loops changed under acid treatment. In the case of Na-MMT, a typical H4 hysteresis loop can be expected because the branches of adsorption and desorption are nearly horizontal and parallel in a wide range of P/P0, which is indicative of narrow slit-like pores.32 At low values of P/P0, the nitrogen adsorption–desorption isotherms of Na-MMT shifted to x-offset, indicating the presence of small intercrystalline micropores. However, AT-MMT exhibited a type I isotherm, indicating a strong interaction with nitrogen. This is because the Na+ ions leached out and were replaced by hydrogen ions, and the edges of the platelets became more open; the particles were more dense due to the delaminating of the MMT laminar structure. Generally, loosely coherent plate-like particles are indicated by an H3 hysteresis loop.33 At high values of P/P0, after activation by sulfuric acid, a gradual change of the isotherm from an H4 to an H3 hysteresis loop is observed. In consequence, the delamination of the MMT laminar structure and an increase in the number of layers per stack were proved to have occurred. This explanation is confirmed by the BJH pore size distribution (inset). As is shown, Na-MMT shows very similar pore size distributions and the peak pore size is centered at about 3.7 nm. Contrastingly, AT-MMT shows a very broad pore size distribution and a clear shift toward a larger pore size of 9.1 nm. More mesopores were created by the acid treatment and the pore size distribution became wider, which is due to the partial destruction of the layered MMT structure. After the pillaring reaction and the loading process, the isotherms recovered their H4 shapes and the pore size shifted to the original size (3.7 nm), showing that the sandwich structure of MMT was restored. These processes fit well with the XRD and FT-IR analysis. The calculated textural data of the different catalysts are summarized in Table S2.† In the case of Na-MMT, its surface area is about 21 m2 g−1. However, unbelievably, after activation by sulfuric acid, the BET surface area of Na-MMT increased to about 335 m2 g−1. This is certainly due to the large number of micropores and stacking pores. Moreover, the surface area of PILC was reduced to 301 m2 g−1. This is because Al137+ accessed the layers of MMT and was transformed into non-crystalline oxides after calcination. There was no other significant change for PMA-PILC, except that the surface area decreased to 253 m2 g−1. This is merely because the PTMA was loaded on the surface of the PILC catalyst.
 |
| | Fig. 3 N2 adsorption–desorption isotherms and pore size distributions (inset) of different catalysts. | |
3.4 TG-DTG analysis
The TG-DTG curves (Fig. S3†) revealed that weight loss occurred for all the samples at temperatures below 100 °C, corresponding to the loss of hydration water adsorbed on the surface or in the MMT layers. For Na-MMT, a mass loss of 5.12% was observed at 78 °C. However, the mass loss percentages of AT-MMT, PILC and PTMA-PILC were 3.66%, 3.46%, and 2.80%, respectively. The weight loss of hydration water decreased obviously and the weightlessness peak was shifted from 78 °C to 90 °C for the modified MMT. The presence of large quantities of hydrated cations was attributed to the gallery containing cations (Na+, Ca2+ etc.). The lamellae were delaminated and the bonding forces between the interlayers weakened after acid treatment; therefore, the hydrated cation adsorption was reduced. The negative charges were further balanced by Al137+, and Na-MMT was converted from a hydrophilic to a hydrophobic substance. The weight loss between 570 °C and 628 °C was ascribed to the dehydroxylation of structural OH groups located in the tetrahedral sheets of the 2:1 MMT layers.34 Comparing the Na-MMT and modified MMT results, this peak shifted to a lower temperature and the mass loss percentages also decreased from 6.49% to 5.83% to 2.5% to 1.88%. The skeleton crystal lattices of Na-MMT were dehisced in the acid treatment process; thus, the OH groups could be easily exposed and removed. After pillaring by an acid solution of Al137+, the hydroxyl groups were removed more readily because chemical bonds formed between the H+ ions around the OH groups of Al137+ and the O2− ions of the MMT silicate layers. On the PILC catalyst, a weight loss was observed at 449 °C, which is due to the dehydroxylation of the polyhydroxy aluminium cations.35 For the PTMA-PILC catalyst, the first weight loss occurred near 90 °C and was attributed to the adsorbed water; the second mass loss step, occuring at 255 °C, was attributed to the bound water on the phosphomolybdic acid. As the temperature was increased to 450 °C, the acidic protons were lost and the Keggin structure of PTMA began to decompose. When the temperature was increased to 690 °C, P2O5 and MoO3 were formed. The thermal decomposition process of the phosphomolybdic acid is shown in eqn (1).| |
 | (1) |
3.5 NH3-TPD
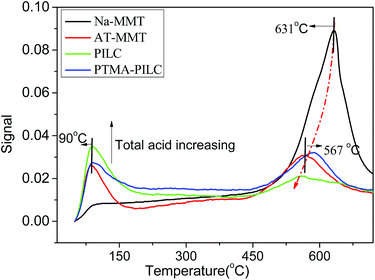
NH3-TPD was used to characterize the acidic properties of the samples. The desorption temperature indicates the acid strength of sample. The higher desorption the temperature, the stronger the acid strength. The acid amounts corresponding to the amounts of adsorbed NH3 were estimated from the areas underlying the NH3-TPD curves. As shown in Fig. 4, there are two ammonia desorption peaks for all the catalysts. The low temperature desorption peaks centered near 90 °C were attributed to weak acid sites present as surface hydroxyl groups and other acids. As can be seen from the first peak in Fig. 4, the number of weak acid sites of the samples increased from 0.086 to 0.112 mmol g−1 (Table S3†) after acid treatment, but the acid strength did not change. This increase was greater for the PILC, as the polycations transformed into non-crystalline aluminium oxides. The number of weak acid sites increased further to 0.178 mmol g−1. For the second peak, however, the dehydroxylation of structural OH groups located in the MMT framework took place between 570 °C and 628 °C. It is important to note that the changes in the TG and DTG curves for the modified MMT were completely consistent with the changes in the NH3 desorption peaks. Therefore, the desorption peak appeared at a high temperature on account of the desorption of water from the condensation of the MMT framework.
 |
| | Fig. 4 NH3-TPD profiles of different catalysts. | |
3.6 Pyridine-IR
IR studies of pyridine can confirm the Brønsted and Lewis acidity of a catalyst. Fig. S4† shows the IR spectra of pyridine adsorbed on the different catalysts. The peaks at 1450 and 1548 cm−1 represent the Lewis acid sites and Brønsted acid sites, respectively. The band at 1493 cm−1 was attributed to pyridine bonded to both the Lewis and Brønsted acid sites (L + B).36 Na-MMT presents weak Brønsted acid centres and Lewis acid sites. The Brønsted acidity has two sources: (1) the compensating cations, which may have a strong polarising effect on coordinated water molecules. (2) Specific sites at the layer edges; layer termination results in broken bonds which may be compensated by the formation of OH groups.28 Coordinatively unsaturated Al and Mg are readily formed at the edges and should behave as Lewis acidic centers.28 AT-MMT presents more Brønsted acid centres and Lewis acid sites. The Brønsted acidity of AT-MMT was enhanced, possibly due to two reasons. Firstly, the basal planes of MMT exposed the bridging oxides of the tetrahedral sheet, which have basic properties; they also exhibited pseudo hexagonal holes within which the structural OH groups of the octahedral sheets were accessible. These OH groups were very weak Brønsted acids in most clays.28 However, after modification by sulfuric acid, the skeleton crystal lattices were dehisced and the lamellae were delaminated; therefore, pseudo hexagonal holes arose and the OH groups were exposed. Secondly, the interlayer cations were exchanged with H+, and these protons may have also enhanced the Brønsted acidity. The dealumination of the framework was obvious during the acid treatment process, and could be observed by the changes in the compositions of the different catalysts (Table S2†). The increase in unsaturated Al benefited the Lewis acid sites. For PILC, the pillars are a major factor that may induce an increase in the number of Lewis acid sites; the Al137+ would lose water molecules after calcination. The Al–O–Al structure was created in the interlayers of MMT and the Al–O–Si structure was bridged with the lamellar structures. The Lewis acid sites and the (L + B) sites increased. Phosphomolybdic acid is a typical strong Brønsted acids and can offer strong option. There is no doubt that the Brønsted acidity of PTMA-PILC increased.
3.7 TEM
Transmission electron microscopy was performed in order to elucidate the structure of the materials. Fig. 5 shows micrographs of the Na-MMT, AT-MMT, PILC and PTMA-PILC samples; they are greatly different. The typical TEM image of Na-MMT shows the lamellar structure, which is compactly arranged and contains numerous metal Na+ ions. After acid treatment, the ion impurities leached out. At the same time, the layered structure disappeared, the skeleton of the MMT was disrupted, and a large number of mesopores appeared. However, in the image of the PILC, the lamellar structure of MMT can be clearly observed again. The polymerized hydroxyl ions transformed the aluminium oxide and were dispersed adequately and uniformly in the MMT. This layer structure was able to disassemble and then reassemble in the process of acid treatment and the pillaring reaction, indicating that MMT has a memory function. These changes were exactly consistent with the XRD, FT-IR and N2 adsorption–desorption isotherm results. The TEM image of PTMA-PILC showed that the mesoporous layer changed little, confirming that PTMA impregnation did not disrupt the structure of the catalyst. The PTMA particles on the surface of the catalyst were homogeneously dispersed on the catalyst surface.
 |
| | Fig. 5 TEM images of (A) Na-MMT, (B) AT-MMT, (C) PILC and (D) PTMA-PILC samples. | |
3.8 Catalytic performance
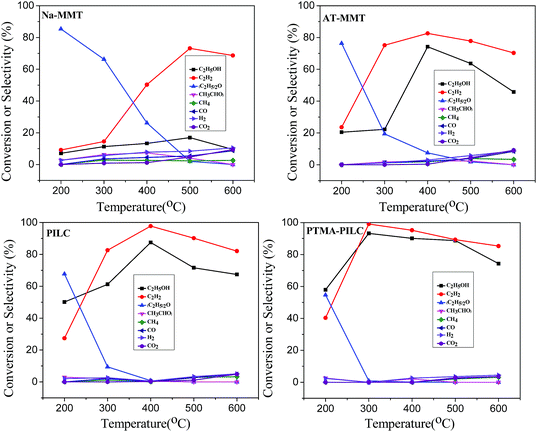
The ethanol conversions and the selectivities of different products over various catalysts at different temperatures are shown in Fig. 6. To evaluate the reactivity of the different catalysts, ethanol dehydration was investigated from 200 °C to 600 °C. The reactant used was simulated bioethanol with a volume fraction of only 30%. In the presence of an acid catalyst, the ethanol was dehydrated into ethylene and water. It is known that the presence of water in the reaction can have a detrimental effect on the active sites of the solid acid catalysts. As shown in Fig. 6, at lower temperatures, the ethanol conversion was lower. With increasing temperature, both the conversion of ethanol and the selectivity towards ethylene increased for all the catalysts but decreased at different temperatures. At 500 °C, the conversion of ethanol was 16.9% for the Na-MMT sample. At 400 °C, the conversion of ethanol was 74.2% for AT-MMT and 87.5% for PILC. In contrast, the PTMA-PILC sample displayed the highest conversion of ethanol, up to 93.2% at a lower temperature of 300 °C. With all four catalysts, the selectivity for ethylene showed the same change tendency. The selectivity for ethylene increased with increasing temperature up to a certain point, after which it decreased. At 200 °C, the selectivity for ethylene was lower due to the higher production of diethyl ether. For all the catalysts, diethyl ether formation showed a significant decline with increasing temperature and disappeared at high temperatures. In addition, other by-products such as CH4, CO and H2 appeared at this stage. As previous reports observed,37 two reactions can occur in parallel during the catalytic dehydration of ethanol: (eqn (2) and (3))| | |
2C2H5OH → C2H5OC2H5 + H2O
| (3) |
 |
| | Fig. 6 The conversion of ethanol and the selectivity for different products vs. temperature over different catalysts. | |
The main reaction and the side reaction are endothermic and exothermic, respectively. At lower temperatures, diethyl ether is produced in significant quantities, while at higher temperatures ethylene is the major product.38 Methane may be formed during ethanol steam reforming by ethanol decomposition (eqn (4)), and this reaction is thermodynamically favored at low temperatures.39 For the Na-MMT catalyst, the ethanol dehydrogenation (eqn (5)) also took place in the low temperature range of 200–400 °C, which was due to the multiple mixtures of metal oxides (Table S1†) of Na-MMT.40,41 However, it can be seen that the acetaldehyde products decreased during the process over the AT-MMT catalyst. That is because the metal oxides were dissolved by the acid treatment.
| | |
C2H5OH → CH4 + CO + H2
| (4) |
| | |
C2H5OH + 3H2O → 6H2 + 2CO2
| (7) |
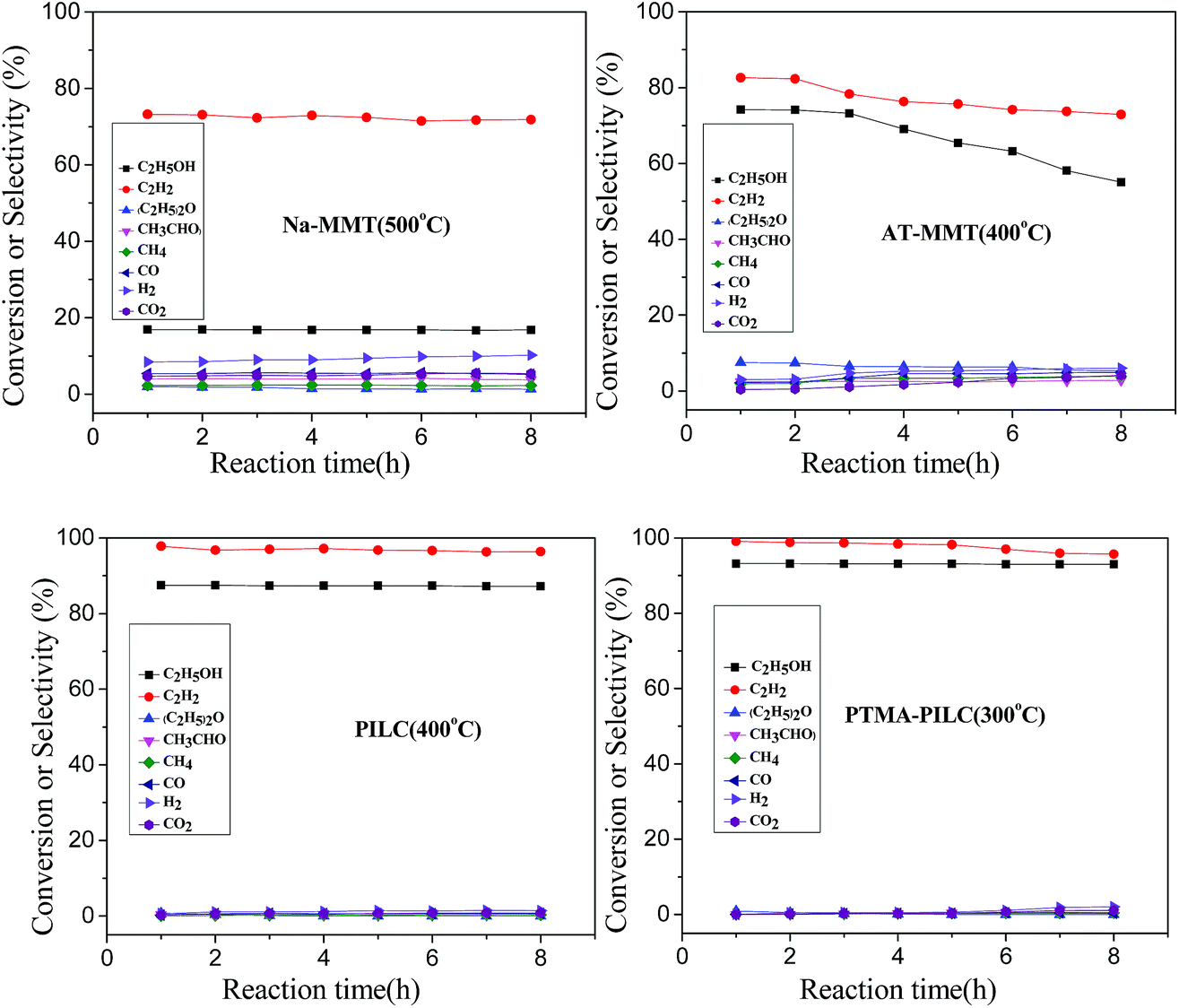
The ethylene formation increased distinctly as the temperature increased. At higher temperatures (over 400 °C), the selectivity for ethylene began to decrease, not only due to the formation of outgrowth produced by ethanol dehydrogenation (eqn (5)) and acetaldehyde decomposition (eqn (6)), but also as a consequence of the steam reforming reaction of ethanol, which occurred due to the presence of water in the feed (eqn (7)). With Na-MMT, AT-MMT, PILC and PTMA-PILC, the selectivity for ethylene was maximal (73.2%, 82.6%, 97.8%, 99.1% at 500 °C, 400 °C, 400 °C, and 300 °C, respectively). PTMA-PILC exhibited greater selectivity to ethylene than any of the other catalysts at lower temperatures. In summary, the conversion of ethanol and the selectivity for ethylene decreased in the order PTMA-PILC > PILC > AT-MMT > Na-MMT. In order to reveal the reason why PTMA-PILC exhibited remarkably enhanced ethylene yield in the reaction, the relationships between the catalytic performance of ethanol dehydration and the structural properties of the catalysts were carefully considered.
Firstly, the effect of the layer structure was correlated. Generally, a mesoporous/microporous structure may benefit catalytic reactions, which is mainly attributed to the enhanced diffusion features of such structures.42 In our work, no direct relation was found between the BET surface area and the ethylene yield. For example, AT-MMT possesses a larger specific surface area than the PTMA-PILC catalyst, but showed low ethylene selectivity and ethanol conversion at low temperature (300 °C). This was in agreement with the analysis by Hongchuan Xin.43 The delamination of the MMT skeleton and recombination assisted by Al137+ benefited the uniform dispersion of the active centers in the MMT. PTMA was distributed homogeneously on the PILC. The PTMA-PILC catalyst and PILC possessed abundant crystal lattice defects despite the fact that the pillaring reaction restored the lamellar structure of the MMT, which provided a large number of active centers for the catalytic reaction. This may have contributed to the ethanol conversion at low temperature. Furthermore, the Al2O3 phase, having an amorphous state and existing steadily in the MMT, could maintain a high activity at a low concentration for the dehydration of ethanol to ethylene.
Secondly, the effect of acidity was studied. The exact chemical nature of the acid sites is still a matter of debate.44,45 However, at a low temperature, the relationship demonstrated that the weak acid centers most likely promoted the formation of ethylene.44 It is generally accepted that the primary reactions involved in the conversion of alcohol to lower hydrocarbons can occur on weak acid sites, but the subsequent reactions (e.g., oligomerization, dehydrocyclization and hydrogenation) occur on sites with comparatively higher acid strength.46–48 In addition, the Lewis and Brønsted acid sites should play an important role in the reaction. Topchieva et al.49 suggested that the dehydration of ethanol occurs essentially with the aid of Brønsted acid sites via the formation of ethoxide (C2H5O−) surface species. However, according to Hussein et al.,50 the different mechanistic pathways for the dehydration of ethanol to diethyl ether and ethene depended largely on the surface hydroxylation. The mechanisms are listed in the ESI.† In a number of papers, it was shown that the dehydration of ethanol over solid catalysts takes place predominantly on the strong acid sites. However, the Na-MMT contains a few weak Lewis acid sites. After modification, the most obvious change in the MMT was the change in the ratio of Lewis and Brønsted acid sites. These mechanisms also indicated that the performance of the catalyst in ethanol dehydration benefited from a balance of Lewis and Brønsted acid sites.
Due to coking of the catalytic material and other reasons, the catalytic activity in a reaction may decrease with reaction time. In this study, the stabilities of the Na-MMT, AT-MMT, PILC and PTMA-PILC catalysts were studied per hour under optimal conditions. As can be seen in Fig. 7, Na-MMT exhibited less catalytic activity but considerable stability; it remained unchanged for 8 h. In contrast, AT-MMT exhibited higher catalytic activity but less stability; the selectivity for ethylene decreased slowly over 2 h. Compared to the Na-MMT and AT-MMT catalysts, PILC and PTMA-PILC showed higher activities. The PTMA-PILC catalyst, which showed the highest initial activity (93.2% conversion of ethanol and 99.1% selectivity of ethylene), presented a slight decrease after 6 h of reaction and remained stable after that. Since the reactants were simulated bioethanol, the presence of water may have resulted in a reduction in the Brønsted acidity of the PTMA-PILC catalyst. In addition, coke formation from ethylene polymerization was also considered as a reason for the deactivation of the catalyst. Even so, this novel catalyst, exhibiting considerably high activity for the dehydration of bioethanol at low temperature, can build a foundation for further research into catalysts for the dehydration of bioethanol.
 |
| | Fig. 7 The stabilities of different catalysts. | |
4 Conclusions
Na-MMT catalysts modified by acidic treatment, ion-exchange and impregnation displayed efficient performance for the dehydration of ethanol to ethylene. The XRD patterns of the samples showed that the acid treatment dramatically changed the structure of the minerals, resulting in delamination of the lamellar structure. The structures of surface Si\O\Si and Si\O\Al in the framework of MMT were slightly destroyed by acidic treatment. Additionally, the specific surface area and pore size significantly increased. The pore size had a clear shift toward a larger pore size of 9.1 nm, which was beneficial to the pillaring reaction. The structural and elementary properties of MMT were restored after pillaring by Al137+, due to the “memory effect”. The active center was uniformly dispersed and remained stable in the MMT, and the Lewis and Brønsted acid sites were further balanced by PTMA. These characteristics benefited the performance of the catalyst and enabled the achievement of greater catalytic activity for the dehydration of ethanol to ethylene; the highest catalytic activity, with 93.2% conversion of ethanol and 99.1% selectivity of ethylene, was obtained at a low temperature of 300 °C.
Acknowledgements
The project was supported by the National Natural Science Foundation of China (50872086), Special/Youth Foundation of Taiyuan University of Technology (2012L022), Scientific and technological project of Shanxi Province (02010145). Scientific and technological project of Shanxi Province (2013021008-4).
Notes and references
- Y. Chen, Y. L. Wu, L. Tao, B. Dai, M. D. Yang, Z. Chen and X. Y. Zhu, J. Ind. Eng. Chem., 2010, 16, 717–722 CrossRef CAS PubMed.
- C. J. Pereira, Science, 1999, 285, 670–671 CrossRef CAS.
- Y. Gucbilmez, T. Dogu and S. Balci, Ind. Eng. Chem. Res., 2006, 45, 3496–3502 CrossRef CAS.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514 CrossRef CAS.
- V. V. Bokade and G. D. Yadav, Appl. Clay Sci., 2001, 53, 263–271 CrossRef PubMed.
- J. Bedia, R. Barrionuevo, J. Rodríguez-Mirasol and T. Cordero, Appl. Catal., B, 2011, 103, 302–310 CrossRef CAS PubMed.
- S. Golay, R. Doepper and A. Renken, Appl. Catal., A, 1998, 172, 97–106 CrossRef CAS.
- J. B. Bi, X. W. Guo, M. Liu and X. S. Wang, Catal. Today, 2010, 149, 143–147 CrossRef CAS PubMed.
- A. M. Zheng, L. Chen, J. Yang, M. J. Zhang, Y. C. Su, Y. Yue, C. H. Ye and F. Deng, J. Phys. Chem. B, 2005, 109, 24273–24279 CrossRef CAS PubMed.
- L. D. Chen, X. S. Wang, X. W. Guo, H. C. Guo, H. O. Liu and Y. Y. Chen, Chem. Eng. Sci., 2007, 62, 4469–4478 CrossRef CAS PubMed.
- N. D. Feng, A. M. Zheng, S. J. Huang, H. L. Zhang, N. Y. Yu, C. Y. Yang, S. B. Liu and F. Deng, J. Phys. Chem. C, 2010, 114, 15464–15472 CAS.
- O. Winter and M. T. Eng, Hydrocarbon Process., 1976, 55, 125–133 Search PubMed.
- X. Zhang, R. J. Wang, X. X. Yang and F. B. Zhang, Microporous Mesoporous Mater., 2008, 116, 210–215 CrossRef CAS PubMed.
- C. B. Phillips and R. Datta, Ind. Eng. Chem. Res., 1997, 36, 4466–4475 CrossRef CAS.
- T. M. Nguyen and R. Levanmao, Appl. Catal., 1990, 58, 119–129 CrossRef CAS.
- Q. Sheng, K. C. Ling, Z. R. Li and L. F. Zhao, Fuel Process. Technol., 2013, 110, 73–78 CrossRef CAS PubMed.
- D. Arias, A. Colmenares, M. L. Cubeiro, J. Goldwasser, C. M. Lopez, F. J. Machado and V. Sazo, Catal. Lett., 1997, 45, 51–58 CrossRef CAS.
- A. C. Garade, V. R. Mate and C. V. Rode, Appl. Clay Sci., 2009, 43, 113–117 CrossRef CAS PubMed.
- A. C. Garade, V. S. Kshirsagar, R. B. Mane, A. A. Ghalwadkar, U. D. Joshi and C. V. Rode, Appl. Clay Sci., 2010, 48, 164–170 CrossRef CAS PubMed.
- M. Misono, Catal. Rev., 1987, 29, 269–321 CAS.
- M. Nehate and V. V. Bokade, Appl. Clay Sci., 2009, 44, 255–258 CrossRef CAS PubMed.
- L. Zhang, Q. Jin, L. Shan, Y. Liu, X. Wang and J. Huang, Appl. Clay Sci., 2010, 47, 229–234 CrossRef CAS PubMed.
- C. H. Zhou, Appl. Clay Sci., 2010, 48, 1–4 CrossRef CAS PubMed.
- A. D. Mali, N. G. Shimpi and S. Mishra, Soc. Chem. Ind., 2014, 63, 338–346 CAS.
- T. Pratyaporn and S. Punnama, Mater. Res. Bull., 2013, 48, 4856–4866 CrossRef PubMed.
- J. A. Zazo, J. Bedia, C. M. Fierro, G. Pliego, J. A. Casas and J. J. Rodriguez, Catal. Today, 2012, 187, 115–121 CrossRef CAS PubMed.
- L. A. Galeano, A. Gil and M. A. Vicente, Appl. Catal., B, 2010, 100, 271–281 CrossRef CAS PubMed.
- J. F. Lambert and G. Poncelet, Top. Catal., 1997, 4, 43–56 CrossRef.
- Q. Lu, X. R. Lei, Z. D. Tang and H. F. Liu, Geol. Sci. Technol. Inf., 2001, 1, 91–98 Search PubMed.
- Y. H. Zhao, Q. Q. Hao, Y. H. Song, W. B. Fan, Z. T. Liu and Z. W. Liu, Energy Fuel, 2013, 27, 6362–6371 CrossRef CAS.
- A. Paula, A. B. Carolina, Q. Raúl, A. Pilar and R. H. Eduardo, Appl. Catal., A, 2013, 453, 142–150 CrossRef PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- M. R. Mostafa, A. M. Youssef and S. M. Hassan, Mater. Lett., 1991, 12, 207–213 CrossRef CAS.
- C. B. Hedley, G. Yuan and B. K. G. Theng, Appl. Clay Sci., 2007, 35, 180–188 CrossRef CAS PubMed.
- F. Bergaya, A. Aouad and T. Mandalia, Dev. Clay Sci., 2006, 1, 393–421 CAS.
- F. Kooli, Microporous Mesoporous Mater., 2014, 184, 184–192 CrossRef CAS PubMed.
- G. W. Chen, S. L. Li, F. J. Jiao and Y. Quan, Catal. Today, 2007, 125, 111–119 CrossRef CAS PubMed.
- X. Zhang, R. Wang, X. Yang and F. Zhang, Microporous Mesoporous Mater., 2008, 116, 210–215 CrossRef CAS PubMed.
- M. G. Vanesa, L. Eduardo, S. Maria and L. Jordi, J. Power Sources, 2009, 192, 208–215 CrossRef PubMed.
- V. García, E. López, M. Serra and J. Llorca, J. Power Sources, 2009, 192, 208–215 CrossRef PubMed.
- V. García, E. López, M. Serra, J. Llorca and J. Riera, Int. J. Hydrogen Energy, 2010, 35, 9768–9775 CrossRef PubMed.
- J. Perez-Ramirez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC.
- H. H. Xin, X. P. Li, Y. Fang, X. F. Yi, W. H. Hu, Y. Y. Chu, F. Zhang, A. M. Zheng, H. P. Zhang and X. B. Li, J. Catal., 2014, 312, 204–215 CrossRef CAS PubMed.
- W. Xia, A. Takahashi, I. Nakamura, H. Shimada and T. Fujitani, J. Mol. Catal. A: Chem., 2010, 328, 114–118 CrossRef CAS PubMed.
- J. P. Marques, I. Gener, P. Ayrault, J. C. Bordado, J. M. Lopes, F. R. Ribeiro and M. Guisnet, Microporous Mesoporous Mater., 2003, 60, 251–262 CrossRef CAS.
- K. K. Ramasamy and Y. Wang, J. Energy Chem., 2013, 22, 65–71 CrossRef CAS.
- V. S. Nayak and V. R. Choudhary, Appl. Catal., 1984, 9, 251–261 CrossRef CAS.
- S. N. Chaudhuri, C. Halik and J. A. Lercher, J. Mol. Catal., 1990, 62, 289–295 CrossRef CAS.
- K. V. Topchieva, K. Yun-Pin and I. V. Smirnova, Adv. Catal., 1957, 9, 799–806 Search PubMed.
- G. A. M. Hussein, N. Sheppard, M. I. Zaki and R. B. Fahim, J. Chem. Soc., Faraday Trans., 1991, 87, 2661–2668 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra02055g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Baoru Li,
Xu Wu and
Xia An
,
Baoru Li,
Xu Wu and
Xia An