
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient organo-photocatalysis system of an n-type perylene derivative/p-type cobalt phthalocyanine bilayer for the production of molecular hydrogen from hydrazine†
Toshiyuki
Abe
*a,
Yoshinori
Tanno
a,
Naohiro
Taira
a and
Keiji
Nagai
b
aDepartment of Frontier Materials Chemistry, Graduate School of Science and Technology, Hirosaki University, 3 Bunkyo-cho, Hirosaki 036-8561, Japan. E-mail: tabe@hirosaki-u.ac.jp
bChemical Resources Laboratory, Tokyo Institute of Technology, Suzukake-dai, Midori-ku, Yokohama 226-8503, Japan
First published on 8th May 2015
Abstract
The stoichiometric decomposition of hydrazine (N2H4) into N2 and H2 was observed to occur efficiently in a photocatalysis system of an organic p–n bilayer. The primary feature of the present system is that the entire visible-light energy spectrum can be utilised for N2H4 decomposition. Furthermore, this paper presents the first demonstration of H2 formation by the reducing power photogenerated at the n-type perylene derivative in an organic bilayer.
Introduction
Solar hydrogen has attracted attention as a potential clean energy technology for the establishment of a sustainable society and is actively being investigated in the areas of photoelectrochemistry and photocatalysis.1,2 Ever since Honda and Fujishima reported a photoelectrochemical water-splitting system with UV-responsive TiO2,3 photocatalysis researchers have aimed to extend the visible-light response of catalysts to longer-wavelength regions, particularly to enhance their photocatalytic activity towards the water-splitting reaction. However, the literature contains only a few examples of photocatalysts that are capable of H2/O2 evolution, even in the near-infrared region;4–7i.e. the conventional photocatalysts of band-engineered semiconductors usually exhibit diminished activity because of band-gap reduction, which is a serious issue that needs to be addressed.We studied and reported photocatalysis systems featuring an organic p–n bilayer in terms of H2 evolution from H+,8,9 as well as the decomposition of trimethylamine, alcohol and thiol into CO2,10,11 thereby demonstrating novel approaches to photocatalysis. These organo-photocatalysis systems can utilise the entire visible-light energy spectrum, wherein a series of photophysical events (i.e. exciton formation, charge separation at a heterojunction and subsequent hole and electron conduction within p-type and n-type layers, respectively) can be induced in a manner similar to that in the corresponding photovoltaic systems.12,13 Thus, a photocatalytic reaction can be achieved by the oxidising and reducing power generated at the surface of p-type and n-type conductors, respectively.
In this study, a photocatalysis system of a 3,4,9,10-perylenetetracarboxylic-bis-benzimidazole (PTCBI, n-type)/cobalt(II) phthalocyanine (CoPc, p-type) bilayer was applied to the decomposition of a carbon-free hydrogen storage material, i.e. hydrazine (N2H4). The literature contains only a few examples of UV-responsive TiO2 photocatalysts that are capable of photocatalysing the decomposition of N2H4 into N2 and H2 through a four-electron transfer (vide infra).14 We recently reported a novel instance of a bilayer of n-type C60 and p-type Zn phthalocyanine (ZnPc) photocatalytically inducing the stoichiometric decomposition of N2H4, particularly under visible-light irradiation;8 however, the photocatalytic activity of the present system is twice that of our previous system (based on a comparison of the optimised photocatalysis systems). Herein, we present and discuss the details of the photocatalytic decomposition of N2H4 by a PTCBI/CoPc bilayer.
Experimental
The PTCBI/CoPc bilayer was prepared by vapour deposition (pressure, <1.0 × 10−3 Pa; deposition speed, ca. 0.03 nm s−1), and an indium–tin oxide (ITO)-coated glass plate was used as the base material. The organic bilayer comprised a PTCBI coating on ITO and CoPc coating on top of the PTCBI layer (denoted as ITO/PTCBI/CoPc). Furthermore, a Nafion membrane (denoted as Nf) was used in combination with the bilayer, wherein (i) an alcoholic solution of Nf was cast onto the CoPc surface, followed by solvent evaporation under air, which resulted in (ii) the loading of a 1 μm thick Nf film onto that surface (the resulting photocatalytic device is abbreviated as ITO/PTCBI/CoPc/Nf).A twin compartment cell that was separated by a salt bridge was utilised for the photocatalytic experiments (see Scheme 1). Other experimental details are provided in the ESI.†
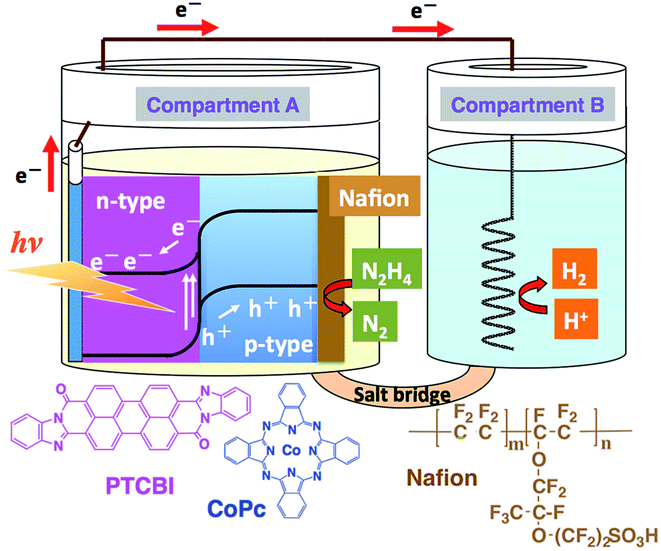 | ||
| Scheme 1 Twin-compartment cell employed for photocatalysis experiments and the structures of the chemicals used in this study. | ||
Results and discussion
ITO/PTCBI/CoPc and ITO/PTCBI/CoPc/Nf were applied to photoanodes in an aqueous phase containing N2H4; this setup was used for the initial evaluation of these devices (see Fig. S1 in the ESI†). The resulting voltammograms exhibited photoanodic characteristics due to N2H4 oxidation (in each case, almost no electrochemical response was recorded in the absence of light). These results are consistent with those reported in our previous study, wherein the p-type material/water interface in an organic p–n bilayer induced oxidation under irradiation.15 The aforementioned voltammetric characteristics also reveal that ITO/PTCBI/CoPc/Nf is superior to ITO/PTCBI/CoPc. This result indicates that Nf functions as an absorbent material for N2H4, thus producing a high surface concentration of the reactant, which ensures its efficient oxidation. Details of the function and role of Nf in the oxidation of N2H4 are provided in the ESI.†8Photoelectrochemical decomposition of N2H4 at the organo-photoelectrodes was conducted under a potentiostatic condition of +0.3 V (vs. Ag/AgCl); a twin-compartment cell was used in this study (see Scheme S1 in the ESI†). Photoelectrolysis data related to the decomposition of N2H4 are presented in Table 1. In the cases of both ITO/PTCBI/CoPc/Nf (entry 1) and ITO/PTCBI/CoPc (entry 2), the stoichiometric decomposition of N2H4 into N2 (oxidation product) and H2 (reduction product) was confirmed to occur according to the following equations (eqn (1)–(3)):
| N2H4 → N2 + 2H2 (overall reaction) | (1) |
| N2H4 → N2 + 4H+ + 4e− (oxidation) | (2) |
| 4H+ + 4e− → 2H2 (reduction) | (3) |
| System | N2 evolved/μL (compartment A) | H2 evolved/μL (compartment B) | Note |
|---|---|---|---|
| a ITO/PTCBI/CoPc/Nf was used as the photoanode, except in the case of entry 2. b Film thickness: PTCBI = 200 nm, CoPc = 150 nm, Nf = 1 μm; effective area (i.e. geometrical area) of the photoelectrode: 1 cm2; electrolyte solution in compartment A, 5 mM N2H4 (pH = 11); electrolyte solution in compartment B, H3PO4 (pH = 2); applied potential, +0.3 V vs. Ag/AgCl (satd.); light intensity, ∼70 mW cm−2; irradiation direction, back side of the ITO-coated face; electrolysis time, 1 h. c Each potentiostatic electrolysis experiment was performed under conditions similar to those used for ITO/PTCBI/CoPc/Nf (i.e. entry 1). d Irradiation of ITO/PTCBI/CoPc/Nf in compartment A was conducted in an open circuit. | |||
| Entryb 1 | 53.1 | 115.8 | Full conditions |
| Entryc 2 | 38.7 | 80.2 | Without Nf |
| Entryc 3 | 1.72 | 0 | Without applied potentiald |
| Entryc 4 | 0 | 0 | Without irradiation |
| Entryc 5 | 0 | 0 | In the absence of N2H4 |
When the Faradaic efficiency (F.E.) of N2 formation from N2H4 was estimated under the assumption of a four-electron transfer oxidation (i.e.eqn (2)), the F.E. values for entries 1 and 2 were calculated to be >90% (see the ESI† for the F.E. calculation procedure). The N2H2 intermediate may be formed through a two-electron transfer oxidation of N2H4 (i.e. N2H4 → N2H2 + 2H+ + 2e−), followed by the further spontaneous decomposition of N2H2 (i.e. N2H2 → N2 + H2).16 However, such processes can be neglected, i.e. in the case of N2 formation via N2H2, the F.E.value should be only ca. 50%; in addition, particularly in compartment A (see Scheme S1†), no detection of H2 from N2H2 was confirmed in the present study. As supported by the aforementioned voltammograms (see Fig. S1†), the results in Table 1 also indicate that ITO/PTCBI/CoPc/Nf is the more efficient photoanode for N2H4 decomposition than ITO/PTCBI/CoPc. Furthermore, the results of the control experiments (i.e. entries 3, 4 and 5) indicate that the efficient decomposition of N2H4 can occur, particularly when employing the complete components of entry 1.
The photocatalytic decomposition of N2H4 was conducted in a system containing ITO/PTCBI/CoPc/Nf (oxidation site) and Pt wire (reduction site) (see Scheme 1). The results are listed in Table 2, which includes the results for the control experiments. Stoichiometric N2H4 decomposition was observed to occur in the constructed photocatalysis system (i.e. entry 1). In the control experiment, the photocatalytic decomposition of N2H4 was conducted in the presence of O2 (electron acceptor), wherein only compartment B was left under aerobic conditions (i.e. entry 2). A comparison of the results for entry 2 with those for entry 1 revealed that the amount of N2 evolved is almost invariable. The similarity of these results indicates that the rate-limiting oxidation of N2H4 can proceed at ITO/PTCBI/CoPc/Nf. The Nf absorbent resulted in an increase in the surface concentration of N2H4 (vide supra); thus, its use can lead to an enhancement of the rate-determining reaction. Furthermore, the lack of components in entry 1 in Table 2 for the experimental results of the negative control experiments (i.e. entries 3, 4 and 5) indicates inactivity towards N2H4 decomposition.
| System | N2 evolved/μL (compartment A) | H2 evolved/μL (compartment B) | Note |
|---|---|---|---|
| a These experiments were conducted under a greater chemical bias between the compartments compared to those listed in Table 1. b Film thickness: PTCBI = 200 nm, CoPc = 150 nm, Nf = 1 μm; effective area (i.e. geometrical area) of the photocatalytic device, 1 cm2; electrolyte solution in compartment A, 5 mM N2H4 (pH = 11); electrolyte solution in compartment B, H3PO4 (pH = 0); light intensity, ∼70 mW cm−2; irradiation direction, back side of the ITO-coated face; irradiation time, 1 h. c Controlled experiments were performed under conditions similar to those for entry 1. d The experimental conditions were the same as those listed for entry 1 in this table, except that a photocatalyst device of 200 nm C60, 150 nm ZnPc and 1 μm Nf was used. | |||
| Entryb 1 | 20.5 | 41.1 | Full conditions |
| Entryc 2 | 18.6 | — | Aerobic atmosphere in compartment B |
| Entryc 3 | 1.72 | 0 | No connection between ITO/PTCBI/CoPc/Nf and Pt wire |
| Entryc 4 | 0 | 0 | Without irradiation |
| Entryc 5 | 0 | 0 | In the absence of N2H4 |
| Entryd 6 | 10.7 | 20.4 | Data for N2H4 decomposition in C60/ZnPc photocatalysis system (ref. 8) |
The factors affecting the photocatalysis of N2H4 decomposition, specifically, the thickness of the film employed (Fig. S2 in the ESI†), the light intensity (Fig. S3 in the ESI†) and the N2H4 concentration in compartment A (Fig. S4 in the ESI†), were examined. These results demonstrate that the conditions listed in entry 1 in Table 2 are the most appropriate conditions for the operation of the present photocatalysis system.
ITO/PTCBI/CoPc/Nf was also used in a prolonged photocatalysis study. This study was conducted under the aforementioned optimum conditions, except that the concentration of N2H4 was 10 mM. The amounts of N2 and H2 evolved after 12 h of irradiation (N2 amount, 265.7 μL; H2 amount, 548.4 μL) were almost proportional to those after 1 h of irradiation (N2 amount, 22.3 μL; H2 amount, 45.8 μL), indicating stable performance of the present photocatalysis system.
External quantum efficiency (EQE) was estimated on the basis of the amount of H2 evolved (see the ESI† for the EQE calculation procedure), and the decomposition of N2H4 photocatalysed by ITO/PTCBI/CoPc/Nf under irradiation by monochromatic light was studied. Fig. 1 shows the relation between the EQE values and radiation wavelength; the resulting EQE values are consistent with the absorption spectrum of the PTCBI monolayer (Fig. 1) rather than with the spectrum of the PTCBI/CoPc bilayer (Fig. S5 in the ESI†). Moreover, N2H4 decomposition was induced over the entire visible-light wavelength region of λ < 750 nm. Such action spectral characteristics, which originate from the sole absorption of PTCBI in an organic p–n bilayer, have been reported by authors.15,17,18 These details are discussed in the following paragraph, along with the mechanism of N2H4 decomposition.
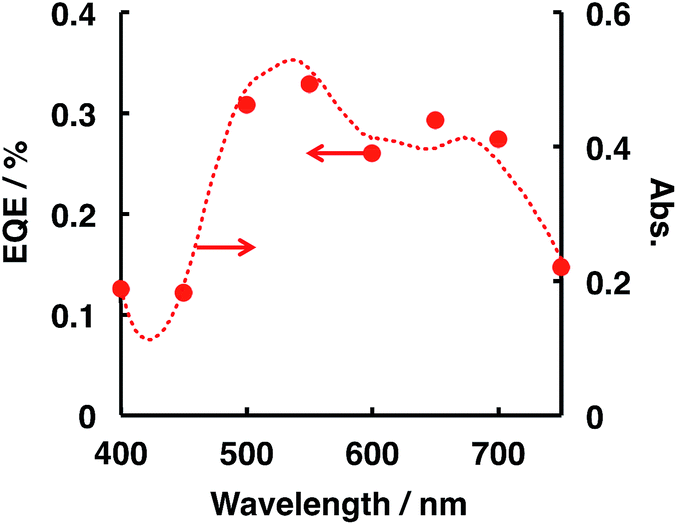 | ||
| Fig. 1 Dependence of the EQE value (closed circles) on the incident wavelength in the ITO/PTCBI/CoPc/Nf and Pt wire photocatalysis system and the absorption spectrum (dashed line) of the PTCBI single layer. The conditions for entry 1 in Table 2 were used for the EQE measurements, except that monochromatic light was used. The irradiated intensity for the photocatalytic device was 0.83 mW cm−2. | ||
The photocatalysis system of ITO/PTCBI/CoPc/Nf is discussed with respect to the N2H4 decomposition mechanism. A schematic for the present N2H4 decomposition is presented in Scheme 2. The photocatalytic function of the PTCBI/CoPc bilayer originates in the absorption of visible light by PTCBI (see Fig. 1 and S1†), thus resulting in the generation of oxidising power at the CoPc surface via a series of photophysical events within the p–n bilayer (see Introduction). An exciton, which is photogenerated within the PTCBI layer, can consequently undergo charge separation into electrons and holes at the heterojunction, followed by carrier conduction through the PTCBI and CoPc layers. In other words, the CoPc layer needs to transport hole carriers towards the N2H4 oxidation occurring at the phthalocyanine surface when the visible-light absorption of CoPc cannot participate in carrier generation (refer to a supporting result in Fig. S6 in the ESI†). The formal potential of CoPc+/CoPc (estimated to be +0.68 V vs. Ag/AgCl from ref. 19 and 20) is more positive than that of N2/N2H4 (−1.18 V (at pH = 11) vs. Ag/AgCl);21 thus, in compartment A, the photocatalytic oxidation of N2H4 into N2 is reasonably assumed to occur at the CoPc surface. The reducing power photogenerated at PTCBI is transferred to the Pt wire (i.e. co-catalyst for H2 evolution) in compartment B where the reduction of H+ into H2 can occur; notably, however, the formal potential of PTCBI/PTCBI− (i.e. −0.15 V vs. Ag/AgCl, as estimated on the basis of the data from ref. 22–24) is very similar to that of H+/H2 (i.e. −0.20 V (at pH = 0) vs. Ag/AgCl). To gain insight into the process of H2 formation induced by PTCBI−, we conducted a separate experiment to examine the relation between the amount of H2 evolved and pH in compartment B. As shown in Fig. S7 in the ESI,† decreasing amounts of H2 were formed with increasing pH. This result may indicate that the PTCBI/PTCBI− potential is independent of pH, thus leading to a shortage of reducing power for H2 evolution at a high pH (i.e. the difference between the PTCBI/PTCBI− and H+/H2 potentials is enhanced by an increase in pH level). The magnitude of the steady concentration of electrons at the Pt wire is associated with the magnitude of reducing power for H2 formation; i.e. efficient H2 evolution at pH = 0 is attributed to an effective gain in reducing power, which is based on the locally concentrated electrons.
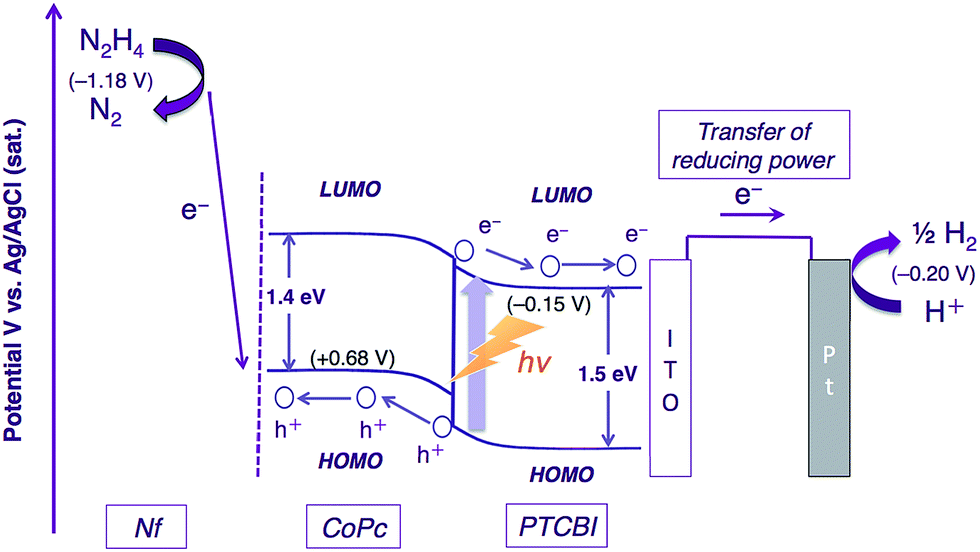 | ||
| Scheme 2 Decomposition of N2H4 in the photocatalysis system of ITO/PTCBI/CoPc/Nf and a Pt wire. | ||
Conclusions
In the present study, we demonstrated a novel photocatalysis system featuring a PTCBI/CoPc bilayer, which led to the stoichiometric decomposition of N2H4 into N2 and H2 over the complete visible-light wavelength range (λ < 750 nm). Furthermore, this study included the first instance of H2 evolution that originates from the reducing power of PTCBI (i.e. PTCBI−). We recently reported an optimised photocatalysis system of an n-type C60/p-type ZnPc bilayer for N2H4 decomposition;8 however, the photocatalytic activity of the present system is double that of the C60/ZnPc system (see entry 6 in Table 2). A large built-in potential at the heterojunction can lead to a high carrier concentration, which is critical to the rate-limiting step in the photocatalytic reaction. However, the built-in potentials in both systems were constant (according to a previous procedure,18 the potential in each system was estimated to be ca. 400 mV). Although the details are currently unclear, the more efficient activity in the PTCBI/CoPc system is speculated to originate from the higher steady concentration of holes for the rate-limiting N2H4 oxidation. We first realised and clarified the instance of C60 (n-type) participating in H2 formation,25 and in the current study, revealed that PTCBI is also an n-type organic material that is capable of H2 generation. Organophotocatalysts have an advanced characteristic in the entire visible-light spectrum available for photocatalyzed reactions. The development of such organophotocatalysts is expected to open a path to the large-scale production of solar hydrogen. Towards this end, materials and active structures with p–n bilayers based on the abundant varieties of organic semiconductors must be developed in order to realise efficient photocatalysis.Acknowledgements
This work was partly supported by a grant from Hirosaki University Institutional Research and a Grant-in-Aid for Scientific Research on innovative areas (no. 25107505) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (T.A.).References
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2013, 135, 10238 CrossRef CAS PubMed.
- Y. P. Xie, Z. B. Yu, G. Liu, X. L. Ma and H.-M. Cheng, Energy Environ. Sci., 2014, 7, 1895 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- Y. Zhao, B. Li, Q. Wang, W. Gao, C. J. Wang, M. Wei, D. G. Evans, X. Duan and D. O'Hare, Chem. Sci., 2014, 5, 951 RSC.
- R. Abe, K. Shinmei, N. Koumura, K. Hara and B. Ohtani, J. Am. Chem. Soc., 2013, 135, 16872 CrossRef CAS PubMed.
- H. Kaga, K. Saito and A. Kudo, Chem. Commun., 2010, 46, 3779 RSC.
- O. Game, U. Singh, A. A. Gupta, A. Suryawanshi, A. Banpurkar and S. Ogale, J. Mater. Chem., 2012, 22, 17302 RSC.
- T. Abe, N. Taira, Y. Tanno, Y. Kikuchi and K. Nagai, Chem. Commun., 2014, 50, 1950 RSC.
- T. Abe, J. Chiba, M. Ishidoya and K. Nagai, RSC Adv., 2012, 2, 7992 RSC.
- K. Nagai, T. Abe, Y. Kaneyasu, Y. Yasuda, I. Kimishima, T. Iyoda and H. Imaya, ChemSusChem, 2011, 4, 727 CrossRef CAS PubMed.
- S. Zhang, A. Prabhakarn, T. Abe, T. Iyoda and K. Nagai, J. Photochem. Photobiol., A, 2012, 244, 18 CrossRef CAS PubMed.
- P. Peumans, S. Uchida and S. R. Forrest, Nature, 2003, 425, 158 CrossRef CAS PubMed.
- K. Suemori, T. Miyata, M. Yokoyama and M. Hiramoto, Appl. Phys. Lett., 2005, 86, 063509 CrossRef PubMed.
- Y. Oosawa, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 1507 RSC.
- T. Abe, K. Nagai, S. Kabutomori, M. Kaneko, A. Tajiri and T. Norimatsu, Angew. Chem., Int. Ed., 2006, 45, 2778 CrossRef CAS PubMed.
- H. Yuzawa, T. Mori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 4126 CAS.
- T. Abe, K. Nagai, M. Kaneko, T. Okubo, K. Sekimoto, A. Tajiri and T. Norimatsu, ChemPhysChem, 2004, 5, 716 CrossRef CAS PubMed.
- T. Abe, S. Miyakushi, K. Nagai and T. Norimatsu, Phys. Chem. Chem. Phys., 2008, 10, 1562 RSC.
- J. H. Zagal, M. A. Gulppi, C. Depretz and D. Leliévre, J. Porphyrins Phthalocyanines, 1999, 3, 355 CrossRef CAS.
- D. Geraldo, C. Linares, Y.-Y. Chen, S. Ureta-Zañartu and J. H. Zagal, Electrochem. Commun., 2002, 4, 182 CrossRef CAS.
- J. Sanabria-Chinchilla, K. Asazawa, T. Sakamoto, K. Yamada, H. Tanaka and P. Strasser, J. Am. Chem. Soc., 2011, 133, 5425 CrossRef CAS PubMed.
- T. Matsushima, H. Matsuo, T. Yamamoto, A. Nakao and H. Murata, Sol. Energy Mater. Sol. Cells, 2014, 123, 81 CrossRef CAS PubMed.
- R. O. Loutfy and Y. C. Cheng, J. Chem. Phys., 1980, 73, 2902 CrossRef CAS PubMed.
- B. Białek, I. G. Kim and J. I. Lee, Thin Solid Films, 2006, 513, 110 CrossRef PubMed.
- T. Abe, S. Tobinai, N. Taira, J. Chiba, T. Itoh and K. Nagai, J. Phys. Chem. C, 2011, 115, 7701 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, function and role of Nf as absorbent, illustration of twin-compartment cell used for photoelectrolysis experiments, cyclic voltammograms, action spectrum for photocurrents, absorption spectra of PTCBI/CoPc bilayer and single-layered CoPc, photocatalysis data for N2H4 decomposition with respect to film thickness of photocatalyst device of PTCBI/CoPc, light intensity, hydrazine concentration and pHs employed in compartment B. See DOI: 10.1039/c5ra03842a |
| This journal is © The Royal Society of Chemistry 2015 |