
Exceptional proton affinities of push–pull nitriles substituted by the guanidino and phosphazeno groups†‡
Ewa D. Raczyńska*a,
Jean-François Galb and
Pierre-Charles Mariab
aDepartment of Chemistry, Warsaw University of Life Sciences (SGGW), ul. Nowoursynowska 159c, 02-776 Warszawa, Poland. E-mail: ewa_raczynska@sggw.pl; Fax: +48 22 5937635; Tel: +48 22 5937623
bInstitut de Chimie de Nice (ICN) – UMR CNRS 7272, University Nice Sophia Antipolis, Parc Valrose, 06108 Nice Cedex 2, France. E-mail: gal@unice.fr; pcmaria@unice.fr; Tel: +33 492076361
First published on 27th February 2015
Abstract
Effects of the pushing groups (electron donors) for nitriles increase as follows: H2N < H2N–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N < H2N–CHCH < H2N–CHN < (H2N)2CCH < (H2N)2CN < (H2N)3PN. The G2(MP2)-calculated PA(N-cyano) for (H2N)2CN–C
N < H2N–CHCH < H2N–CHN < (H2N)2CCH < (H2N)2CN < (H2N)3PN. The G2(MP2)-calculated PA(N-cyano) for (H2N)2CN–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and (H2N)3PN–CN are larger than that of HCN by 186 and 250 kJ mol−1, respectively. The hypothesis of protonation in the gas phase at the N-imino and N-amino atoms, corresponding respectively to PAs weaker by 30 and 70 kJ mol−1 than that of the N-cyano site, can be rejected.
N and (H2N)3PN–CN are larger than that of HCN by 186 and 250 kJ mol−1, respectively. The hypothesis of protonation in the gas phase at the N-imino and N-amino atoms, corresponding respectively to PAs weaker by 30 and 70 kJ mol−1 than that of the N-cyano site, can be rejected.
Thermodynamic parameters of protonation and deprotonation reactions are frequently used for characterizing the basicity of organic and inorganic compounds in the gas phase. One essential parameter is the proton affinity (PA). For a neutral base (B), it is defined as the enthalpy change (ΔH°) associated with the deprotonation reaction (1) of the conjugated acid (BH+),1 and is therefore equal to the negative of the enthalpy change of the reverse reaction, protonation of B. The PA value of B can be determined by various experimental and/or quantum-chemical methods.1,2 For theoretical estimations, eqn (2) can be applied. For H+, only the translational energy term is not equal to zero, and H298(H+) = Htransl(H+) = 3/2RT = 3.7 kJ mol−1 at 298.15 K. The PA values can be calculated for each possible site of protonation for polyfunctional bases. A larger PA value corresponds to a site of stronger basicity.
| BH+ → B + H+ | (1) |
| PA(B) = H298(B) + H298(H+) − H298(BH+) | (2) |
For the family of organic N-bases, monofunctional nitriles exhibit weaker basicity in the gas phase than the corresponding imines and amines.1 Their PA order is as follows: RCN < RCHNH < RCH2NH2 for the same substituent R, alkyl or aryl group. The substituent R affects the basicity of the N-site in the gas phase by its polarizability, field/inductive, and/or resonance effect.3 The electron-donating group (donor, D) increases the PA value, whereas that possessing the electron-accepting character (acceptor, A) decreases it. When a donor is directly linked with an acceptor (D–A) or separated by a π-electron fragment [e.g., Dn–(XY)i–Ak, i, k, n = 1, 2, 3, etc.], such conjugated systems are called “push–pull” and display very attractive properties.4 For example, the order of basicity is reversed for push–pull N-bases.1 Many push–pull imines are stronger bases than amines and belong to the family of superbases in the gas phase (PA > 1000 kJ mol−1). A large number of their experimental data have been included by Hunter and Lias in their last compilation published in 1998 and in the database maintained by the NIST.1 This experimental scale has been now extended for superbasic guanidines and phosphazenes.5
However, nitriles with PA values close to 1000 kJ mol−1 have not yet been discovered. Only a few PA values <900 kJ mol−1 for push–pull derivatives have been reported.1,6–9 Looking for more basic nitriles (with PA > 900 kJ mol−1), we considered derivatives containing the strong electron-donating guanidino [(H2N)2CN] or phosphazeno groups [(H2N)3PN] (Fig. 1). These groups may possess stronger pushing effects than others, more common D-substituents such as the amino (H2N), vinamino (H2N–CHCH), and formamidino (H2N–CN) groups.6–9a
 | ||
| Fig. 1 Nitriles with the guanidino (a) and phosphazeno (b) group. | ||
Since guanidines and phosphazenes themselves possess superbasic properties in the gas phase, it is not evident which site, the N-imino in the pushing group or the N-cyano in the pulling one, will be preferentially protonated for (H2N)2CN–CN and (H2N)3PN–CN. For this reason, we applied the G2 and G2(MP2) theories10 to determine the favored sites of protonation and to predict their proton affinities. For comparison of internal effects in push–pull nitriles, we also included in the series the cyano derivatives with the H2N, H2N–NN, H2N–CHN, H2N–CHCH, and (H2N)2CCH groups, and we performed similar quantum-chemical calculations. For the simplest push–pull nitrile, cyanamide (H2N–CN), the G3 and G3B3 theories11 were also applied. It is well recognized that the Gn theories lead to basicity data corresponding to a “chemical” accuracy (≈5 kJ mol−1) equivalent to that of most of the experimental PA values (or even better).12,13
For H2N–XY–CN, where X, Y = CH or N, two isomers (Z and E) could be taken into account. However, it has been already shown that the E-isomer has lower energy than the other one for the vinamine and formamidine derivatives.6c,8,9a Hence, solely E-configurations were considered for the neutral and monocationic forms. For all calculations, the Gaussian-03 programs14 containing the Gn procedures were used.
G2 and G2(MP2) calculations, performed for the neutral, N-cyano, N-imino, and N-amino protonated forms of push–pull nitriles, showed without any doubt that the N-cyano atom is the favored site of protonation in the gas phase for all investigated compounds, even for the guanidino and phosphazeno derivatives. The energies of the N-imino and N-amino protonated forms are larger than those protonated at the favored site by more than 30 and 70 kJ mol−1, respectively. Hence, they can be neglected for the monoprotonation reaction in the gas phase. All derivatives can be classified in the family of push–pull nitriles.
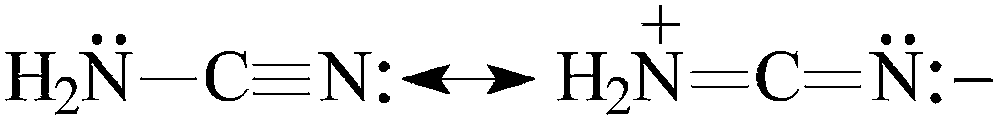
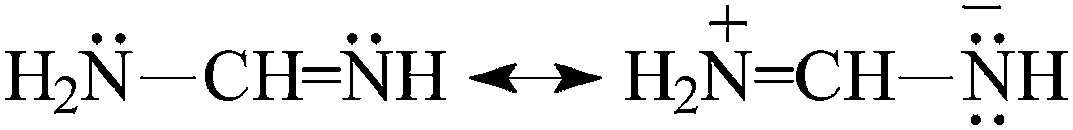
For H2N–CN, the Gn-calculated PA(N-cyano) values are close to the experimental one (Fig. 2). Differences between these PA values are not larger than 4 kJ mol−1. It is noteworthy that the calculated PA(N-amino) value is lower than that of PA(N-cyano) by more than 100 kJ mol−1. Our calculations are in good agreement with the previous ones.7 Direct link of the H2N and CN groups in cyanamide strongly reduces the basicity of the pushing H2N group and augments that of the pulling CN one in comparison to the monofunctional bases, NH3 and HCN, respectively. These opposite basicity effects originate mainly from the n–π conjugation of the amino and cyano groups [eqn (3)]. The electron-donating resonance effect of the H2N group is considerably stronger than its relatively weak electron-accepting field/inductive and polarizability effects.3 Consequently, the experimental PA value of cyanamide (Fig. 2) increases for the favored N-cyano site by 93 kJ mol−1 when compared to that of HCN.1 A similar effect is found at the G2 (89 kJ mol−1) and G2(MP2) levels (88 kJ mol−1). On the other hand, when going from NH3 to cyanamide, the G2- and G2(MP2)-calculated PA(N-amino) decrease by 160 and 159 kJ mol−1, respectively. The PA data for HCN and NH3 were taken from ref. 12. The n–π conjugation effect for H2N–CN is slightly stronger than that for H2N–CHNH [eqn (4)], for which the N-imino atom is the favored site of protonation. Its PA value exceeds that of H2CNH by 81 kJ mol−1 at the G2 level (Table 1).15
 | (3) |
 | (4) |
 | ||
| Fig. 2 Experimental and Gn-calculated PA values for the two potential protonation N-sites in cyanamide (in kJ mol−1). | ||
| Compound | G2 | G2(MP2) | ||||
|---|---|---|---|---|---|---|
| N-Cyano | N-Imino | N-Amino | N-Cyano | N-Imino | N-Amino | |
| a In kJ mol−1 at 298.15 K.b Calculated values taken from ref. 12.c According to ref. 1, experimental PA 712.9 kJ mol−1.d According to ref. 1, experimental PA value is equal to the G2-calculated one.e Taken from ref. 15.f Calculated values taken from ref. 18.g According to ref. 1, experimental PA 986.3 kJ mol−1.h PA for the N-imino linked to the cyano group (N–CN).i PA for the N-imino linked to the amino group (H2N–N).j Experimental gas-phase basicity9a is not very different from that of ammonia.k PA for the Nsyn-amino.l PA for the Nanti-amino. |
||||||
| H–CNb,c |
712.0 | 713.8 | ||||
| H2N–Hb,d | 853.6 | 853.5 | ||||
| H2CN–He |
862.3 | |||||
| H2N–CHN–He |
943.1 | |||||
| (H2N)2CN–Hf,g |
986.6 | 986.2 | ||||
| H2N–NN–CN |
822.6 | 754.0h, 705.0i | 719.1 | 823.4 | 754.6h, 706.3i | 720.1 |
| H2N–CHN–CN |
870.6 | 821.0 | 728.6 | 871.4 | 821.6 | 729.6 |
| H2N–CHCH–CNj |
860.0 | 784.7 | 860.5 | 785.5 | ||
| (H2N)2CCH–CN |
892.9 | 802.9k, 790.4l | 893.5 | 803.7k, 791.4l | ||
| (H2N)2CN–CN |
898.3 | 864.9 | 748.7k, 757.2l | 899.4 | 865.4 | 749.7k, 758.3l |
| (H2N)3PN–CN |
962.5 | 906.7 | 806.5 | 963.5 | 906.8 | 806.8 |
The other push–pull nitriles of general formula (H2N)nXY–CN considered in this communication [H2N–NN–CN, H2N–CHN–CN, H2N–CHCH–CN, (H2N)2CCH–CN, (H2N)2CN–CN, and (H2N)3PN–CN], include a XY fragment separating the pushing H2N and pulling CN groups. Except for aminoacrylonitriles, this fragment possesses N-imino atom(s), which could be also protonated in the gas phase.1,2,5,6c,8 However, this atom cannot be treated as the favored site for the monoprotonation reaction, because the G2-calculated PA(N-imino) values are lower than those of PA(N-cyano) by more than 30 kJ mol−1 (Table 1). Unfortunately, there are no experimental data for (H2N)nXN–CN for comparison. An exception is H2N–CHCH–CN. Our quantum-chemical calculations are in good agreement with those (G3B3) reported previously.9a The experimental estimations of the gas-phase basicity are close to the theoretical ones. HO–CHCH–CN and HS–CHCH–CN display similar gas-phase basicities to their amino analogue.9b
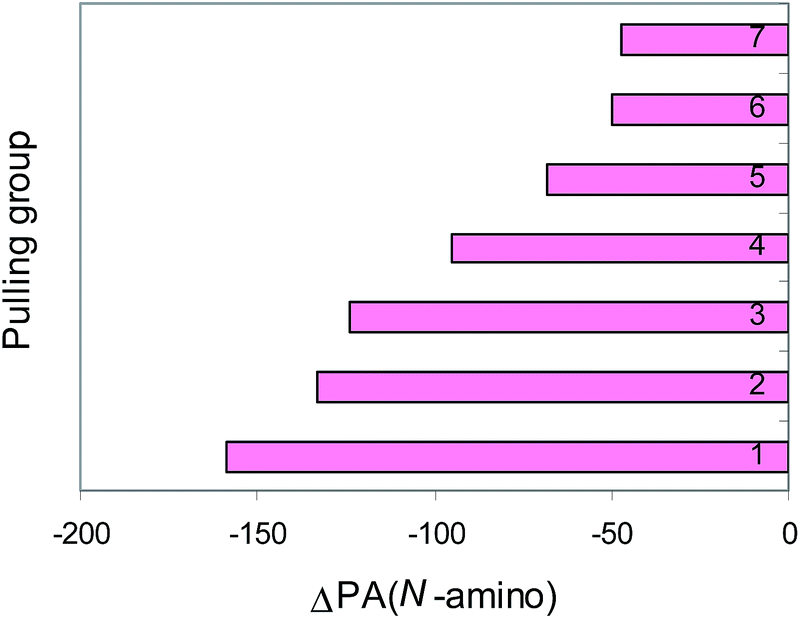
The XY transmitter group modifies the PA values of the pushing H2N and pulling CN groups by its polarizability, field/inductive, and resonance effects (Fig. 3 and 4). The weakest effects are exerted by the NN group in H2N–NN–CN. In this case, the polarizability and resonance effects of the NN group are strongly reduced by its opposite field-inductive effect. When proceeding from H2N–CN to the E-isomer of H2N–NN–CN, the G2- and G2(MP2)-calculated PA(N-cyano) values increase by ca. 20 kJ mol−1. A larger PA(N-cyano) increase (by ca. 60–70 kJ mol−1) occurs for the E-isomer of H2N–CHN–CN containing the CHN group and for the E-isomer of H2N–CHCH–CN with the CHCH transmitter. The second amino group for (H2N)2CN–CN and (H2N)2CCH–CN augments the PA(N-cyano) values by ca. 30 kJ mol−1 when compared to H2N–CHN–CN and H2N–CHCH–CN, respectively.
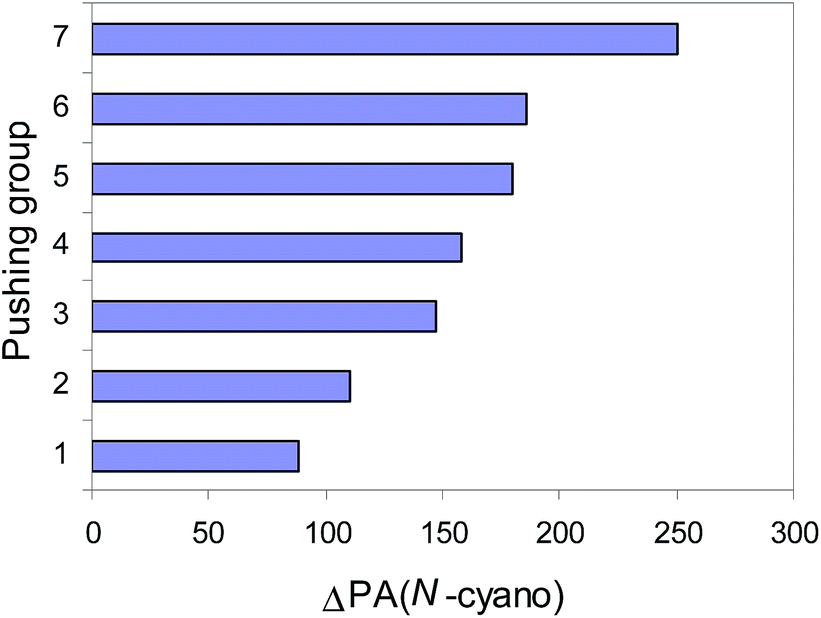 | ||
| Fig. 3 Pushing effects estimated at the G2(MP2) level [ΔPA(N-cyano) = PA(N-cyano in nitrile) − PA(N-cyano in HCN), in kJ mol−1] for the following groups: H2N– (1), H2N–NN– (2), H2N–CHCH– (3), H2N–CHN– (4), (H2N)2CHCH– (5), (H2N)2CN– (6), and (H2N)3PN– (7). | ||
 | ||
| Fig. 4 Pulling effects estimated at the G2(MP2) level [ΔPA(N-amino) = PA(N-amino in nitrile) − PA(N-amino in NH3), in kJ mol−1] for the following groups: –CN (1), –NN–CN (2), –CHCH–CN (3), –(H2N)CN–CN (4), –CHN–CN (5), –(H2N)CHCH–CN (6), and –(H2N)2PN–CN (7). | ||
It should be also noted here that among the two NH2 groups in (H2N)2CX–CN, the NH2 group at the synperiplanar position to the CN group possesses a larger PA value for aminoacrylonitrile than that at the antiperiplanar one. For the guanidino derivative, a reverse situation takes place. The NH2 group at the antiperiplanar position to the CN group possesses a larger PA value than that at the synperiplanar one. This is attributed to differences in intramolecular interactions between the H2NsynH+ group and π-electrons of the CN group for (H2N)2CCH–CN and between the H2NantiH+ group and n-electrons of the N-imino atom for (H2N)2CN–CN (Chart 1).
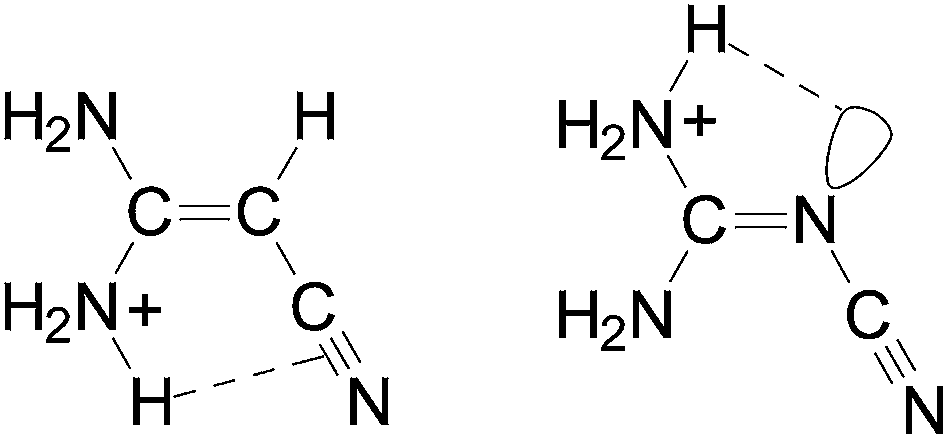 | ||
| Chart 1 Favored structures for the N-amino protonated forms of (H2N)2CX–CN. | ||
The presence of π-electrons in the XY fragment does not interrupt the simple n–π conjugation between the N-amino and N-cyano atoms in H2N–XY–CN [eqn (5)]. The N-imino atom in H2N–XN–CN is also n–π conjugated with the pushing H2N group [eqn (6)], but solely the strong electron-accepting field/inductive and resonance effects of the CN group can explain why the N-imino atom is less basic than the N-cyano one. At the G2 level, the PA(N-imino)-decreasing effect for H2N–CHN–CN is equal to 122 kJ mol−1 when the PA(N-imino) in H2N–CHNH and H2N–CHN–CN are compared, whereas the PA(N-cyano)-increasing effect in H2N–CHN–CN is equal to 159 kJ mol−1 when going from H–CN to H2N–CHN–CN (Table 1). In the case of H2N–NN–CN, the second N-imino atom linked with the N-amino atom does not participate in the resonance conjugation, and its basicity depends solely on the polarizability and field/inductive effects of the amino and cyano groups. Its PA value in H2N–NN–CN is lower than that of HNNH16 by 69 kJ mol−1 at the G2 level.
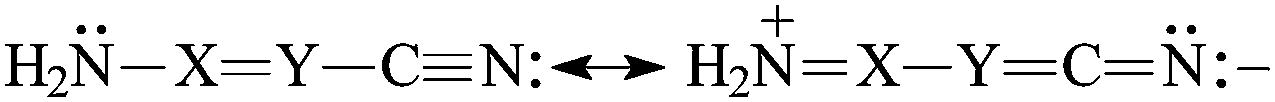 | (5) |
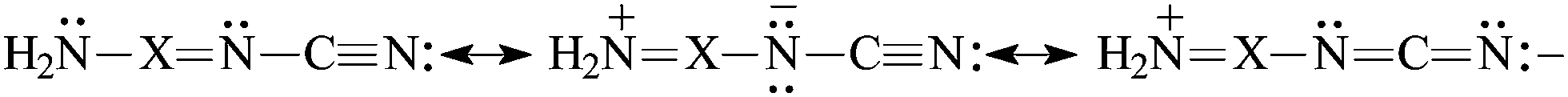 | (6) |
Guanidine itself [(H2N)2CNH], possessing the two amino pushing groups directly linked with the imino pulling one, is a strong base in the gas phase. Its experimental PA value is larger than those of 1,8-diaminonaphthalene and triethylamine.1,17,18 Various theories and explanations were proposed in the literature to answer the question, “why is guanidine a strong base?” Some of them were focused on the Y-aromaticity (Y-delocalization, also called Y-conjugation) of the system.18
For (H2N)2CN–CN, the guanidino group transmits its Y-conjugation to the pulling cyano group [eqn (7)] and increases its basicity. At the same time, the basicity of the N-imino atom decreases due to the strong electron-accepting effect of the cyano group. The PA(N-imino)-decreasing effect, equal to 121 kJ mol−1 at the G2(MP2) level when proceeding from (H2N)2CNH to (H2N)2CN–CN, is similar to that for H2N–CHN–CN. The PA(N-cyano) value increases by 186 kJ mol−1 when going from H–CN to (H2N)2CN–CN. This effect is stronger than that for H2N–CHN–CN. The resonance electron-accepting effect of CN is stronger than that of CN,3 and the N-cyano site is preferred for the monoprotonation reaction of (H2N)2CN–CN. The PA(N-cyano) value is larger than that of PA(N-imino) by 34 kJ mol−1 at the G2(MP2) level. The protonation of the N-amino atoms leads to PA values lower than that for the favored site by 140–150 kJ mol−1, respectively.
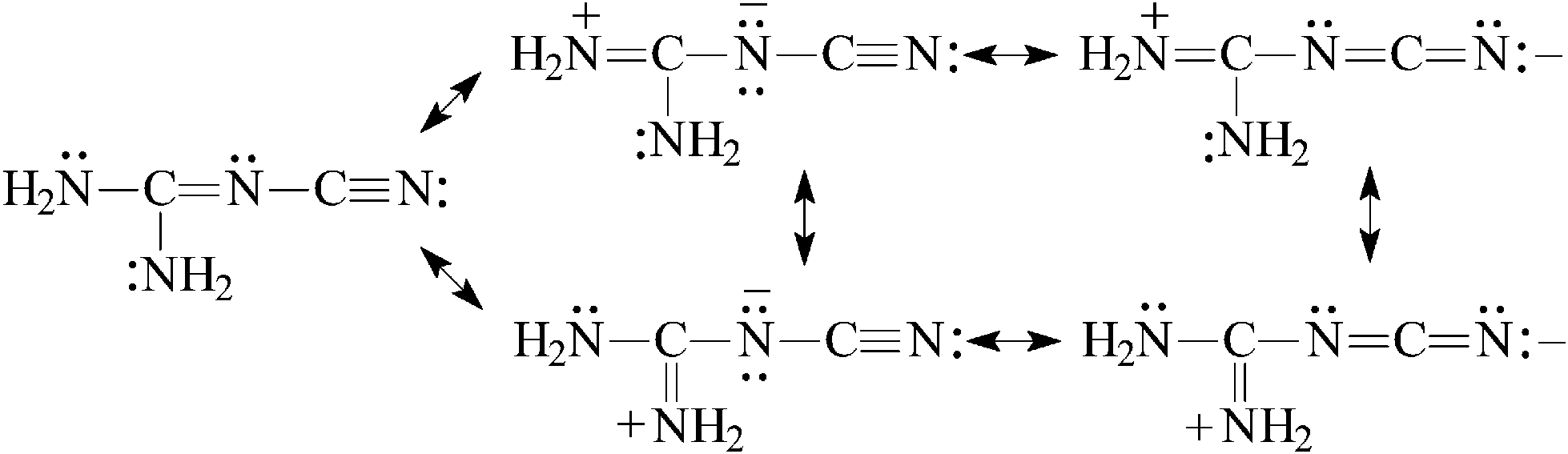 | (7) |
Phosphazene [(H2N)3PNH] possesses three NH2 groups linked with the PN group, and can be regarded as a push–pull imine, by analogy with formamidine and guanidine. The n–π conjugation of the pushing and pulling groups, which can be called cross-conjugation (T-delocalization), explains its stronger basicity than that of guanidine. For example, the experimental PA value of (Me2N)3PNH is larger than that of (Me2N)2CNH by ca. 50 kJ mol−1.5a Experiments for phosphazene were based on the extended gas-phase basicity scale derived from the known basicity of (Me2N)2CNH.1
In the case of (H2N)3PN–CN, the phosphazeno group transmits its cross-conjugation to the cyano group [eqn (8)] and increases its basicity. The push–pull effect seems to be stronger than that for (H2N)2CN–CN. At the G2(MP2) level the difference between the PA(N-cyano) and PA(N-imino) values for (H2N)3PN–CN is equal to 57 kJ mol−1 and that between the PA(N-cyano) and PA(N-amino) values is larger than 150 kJ mol−1. (H2N)3PN–CN possesses the largest PA value in the series of push–pull nitriles (Fig. 5). When compared to H–CN, its PA(N-cyano) value is larger by 250 kJ mol−1.
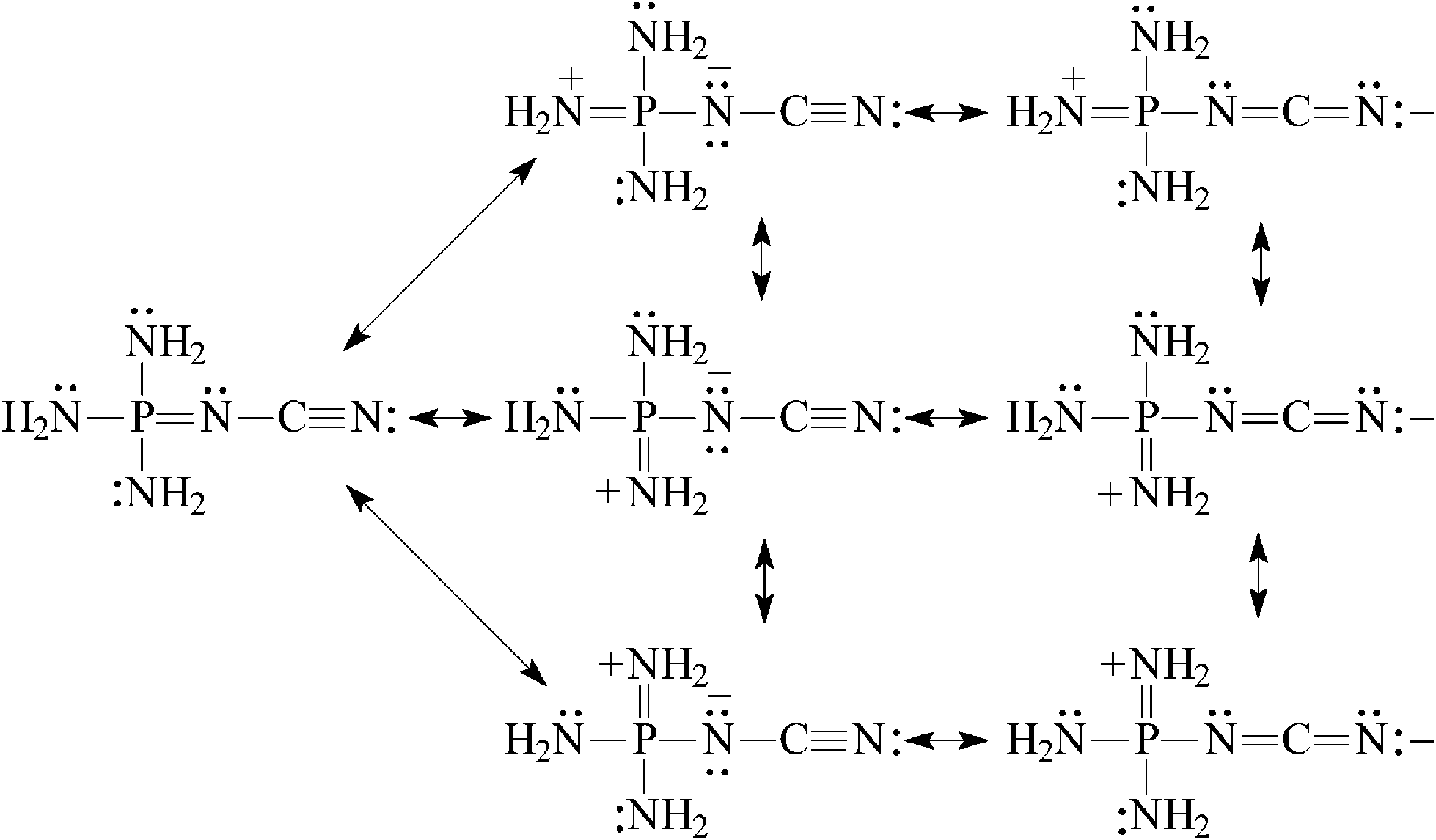 | (8) |
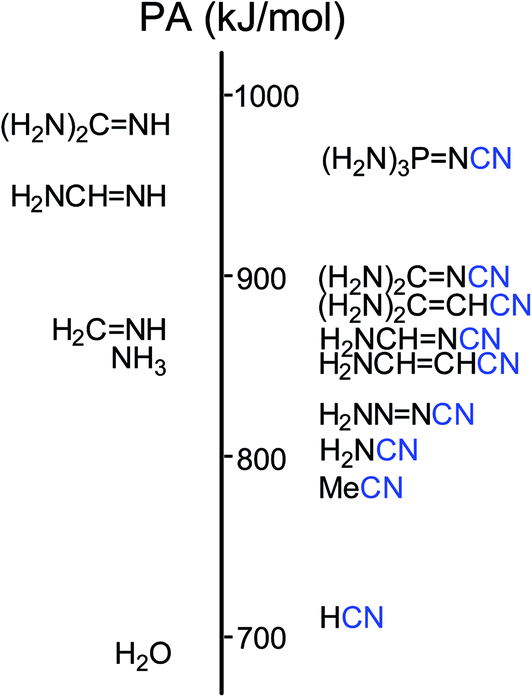 | ||
| Fig. 5 PA scale for push–pull nitriles estimated at the G2(MP2) level. | ||
Conclusions
The push–pull effect clearly explains the preference of unsubstituted nitriles (H2N)n(XY)iCN (n = 1, 2, or 3, i = 0 or 1) to be always protonated at the N-cyano site. It also explains the high PAs of push–pull nitriles when compared to the other members of the family of alkylnitriles.1 In comparison with the calculated PA value of H–CN,12 the increasingly pushing effect of the H2N, H2N–NN, H2N–CHCH, H2N–CHN, (H2N)2CCH, (H2N)2CN, and (H2N)3PN groups linked directly with the pulling CN group augments the proton affinity of the N-cyano site by 80–250 kJ mol−1 (Fig. 3). Cyanamide (H2N–CN) displays the weakest basicity in the family of push–pull nitriles. When proceeding from H2N–CN to H2N–XY–CN, the transmitter XY group (NN, CHN, and CHCH) increases the PA values of the N-cyano site by 20–70 kJ mol−1 (Fig. 5). The Y-conjugation possible for (H2N)2CCH–CN and (H2N)2CN–CN causes a stronger effect (90–100 kJ mol−1) on the PA(N-cyano) than the simple n–π conjugation for H2N–CHCH–CN and H2N–CHN–CN. The strongest push–pull effect (250 kJ mol−1) is found for the cross-conjugated (H2N)3PN–CN. Its calculated PA is close to that of guanidine. This suggests that the alkyl derivatives (R2N)3PN–CN may exhibit stronger basic properties than guanidine and may be classified in the family of superbases in the gas phase (PA > 1000 kJ mol−1). The polarizability effect of the alkyl groups and the push–pull effects on the PA(N-amino) and PA(N-cyano), along with the favored site of protonation will be investigated in the near future for alkyl derivatives.
References
- (a) E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS PubMed; (b) E. P. L. Hunter and S. G. Lias, in NIST Chemistry WebBook, NIST Standard Reference Database No. 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, 2014 Search PubMed.
- J.-F. Gal, P.-C. Maria and E. D. Raczyńska, J. Mass Spectrom., 2001, 36, 699–716 CrossRef CAS PubMed.
- (a) R. W. Taft and R. D. Topsom, Prog. Phys. Org. Chem., 1987, 16, 1–83 CrossRef; (b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- (a) S. J. Lord, N. R. Conley, H. D. Lee, S. Y. Nishimura, A. K. Pomerantz, K. A. Willets, Z. Lu, H. Wang, N. Liu, R. Samuel, R. Weber, A. Semyonov, M. He, R. J. Twieg and W. E. Moerner, ChemPhysChem, 2009, 10, 55–65 CrossRef CAS PubMed; (b) M. E. Belowich and J. Fraser Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC; (c) F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC.
- (a) E. D. Raczyńska, M. Decouzon, J.-F. Gal, P.-C. Maria, G. Gelbard and F. Vielfaure-Joly, J. Phys. Org. Chem., 2001, 14, 25–34 CrossRef; (b) I. Kaljurand, I. A. Koppel, A. Kütt, E.-I. Rõõm, T. Rodima, I. Koppel, M. Mishima and I. Leito, J. Phys. Chem. A, 2007, 111, 1245–1250 CrossRef CAS PubMed; (c) Z. Glasovac, F. Pavošević, V. Štrukil, M. Eckert-Maksić, M. Schlangen and R. Kretschmer, Int. J. Mass Spectrom., 2013, 354–355, 113–122 CrossRef CAS PubMed.
- (a) S. Marriott, R. D. Topsom, C. B. Lebrilla, I. Koppel, M. Mishima and R. W. Taft, THEOCHEM, 1986, 137, 133–141 CrossRef; (b) P.-C. Maria, J.-F. Gal and R. W. Taft, New J. Chem., 1987, 11, 617–621 CAS; (c) M. Berthelot, M. Helbert, C. Laurence, J.-Y. Le Questel, F. Anvia and R. W. Taft, J. Chem. Soc., Perkin Trans. 2, 1993, 625–627 RSC.
- F. Cacace, G. de Petris, F. Grandinetti and G. Occhiucci, J. Phys. Chem., 1993, 97, 4239–4245 CrossRef CAS.
- M. Makowski, E. D. Raczyńska and L. Chmurzyński, J. Phys. Chem. A, 2001, 105, 869–874 CrossRef CAS.
- (a) A. Luna, O. Mó, M. Yáñez, J.-F. Gal, P.-C. Maria and J.-C. Guillemin, Chem.–Eur. J., 2006, 12, 9254–9261 CrossRef CAS PubMed; (b) A. Luna, O. Mó, M. Yáñez, J.-C. Guillemin, J.-F. Gal and P.-C. Maria, Int. J. Mass Spectrom., 2007, 267, 125–133 CrossRef CAS PubMed.
- (a) L. A. Curtiss, K. Raghavachari, G. W. Trucks and J. A. Pople, J. Chem. Phys., 1991, 94, 7221–7230 CrossRef CAS PubMed; (b) L. A. Curtiss, K. Raghavachari and J. A. Pople, J. Chem. Phys., 1993, 98, 1293–1298 CrossRef CAS PubMed.
- (a) L. A. Curtiss, K. Raghavachari, P. C. Redfem, V. Rassolov and J. A. Pople, J. Chem. Phys., 1998, 109, 7764–7776 CrossRef CAS PubMed; (b) A. G. Baboul, L. A. Curtiss, P. C. Redfem and K. Raghavachari, J. Chem. Phys., 1999, 110, 7650–7657 CrossRef CAS PubMed.
- (a) B. J. Smith and L. Radom, J. Am. Chem. Soc., 1993, 115, 4885–4888 CrossRef CAS; (b) B. J. Smith and L. Radom, J. Phys. Chem., 1995, 99, 6468–6471 CrossRef CAS.
- S. Hammerum, Chem. Phys. Lett., 1999, 300, 529–532 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- J. Tortajada, E. Leon, A. Luna, O. Mó and M. Yáñez, J. Phys. Chem., 1994, 98, 12919–12926 CrossRef CAS.
- E. D. Raczyńska, K. Woźniak, E. Dolecka and M. Darowska, J. Phys. Org. Chem., 2002, 15, 706–711 CrossRef.
- B. Amekraz, J. Tortajada, J.-P. Morizur, A. I. Gonzalez, O. Mó, M. Yáñez, I. Leito, P.-C. Maria and J.-F. Gal, New J. Chem., 1996, 20, 1011–1021 CAS.
- E. D. Raczyńska, M. K. Cyrański, M. Gutowski, J. Rak, J.-F. Gal, P.-C. Maria, M. Darowska and K. Duczmal, J. Phys. Org. Chem., 2003, 16, 91–106 CrossRef.
Footnotes |
| † Dedicated to the memory of Professor Nicolaas Martinus Maria Nibbering who supported as referee our first paper on superbases in the gas phase. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra02716k |
| This journal is © The Royal Society of Chemistry 2015 |