
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrospinning of ultrafine metal oxide/carbon and metal carbide/carbon nanocomposite fibers†
Jennifer S. Atchison‡
a,
Marco Zeiger‡ab,
Aura Tolosa‡ab,
Lena M. Funkeab,
Nicolas Jäckelab and
Volker Presser*ab
aINM – Leibniz Institute for New Materials, Campus D2 2, 66123 Saarbrücken, Germany. E-mail: volker.presser@inm-gmbh.de
bSaarland University, Campus D2 2, 66123 Saarbrücken, Germany
First published on 27th March 2015
Abstract
Electrospinning has emerged as a facile technology for the synthesis of ultrafine fibers and even nanofibers of various materials. While carbon nanofibers have been extensively investigated, there have also been studies reported on metal oxide and metal carbide fibers. Yet, comparative studies, especially following the same general synthesis approach, are lacking. In our comprehensive study, we use a sol gel process by which a carrier polymer (cellulose acetate or polyvinylpyrrolidone) is mixed with titanium butoxide, zirconium(IV) acetylacetonate, or niobium n-butoxide to yield nanotextured titania/carbon, zirconia/carbon, or niobia/carbon nonwoven textiles. Carbothermal reduction between 1300 °C and 1700 °C effectively transforms the metal oxide/carbon fibers to metal carbide/carbon nanocomposite while preserving the fiber integrity. As a beneficial effect, the fiber diameter decreases compared to the as-spun state and we obtained ultrafine fibers: 294 ± 108 nm for ZrC/C, 122 ± 28 nm for TiC/C, and 65 ± 36 nm for NbC/C. The highly disordered and porous nature of the carbon matrix engulfing the metal carbide nanocrystals enables a high specific surface area of up to 450 m2 g−1 (TiC/C) after carbothermal reduction.
1. Introduction
Over the last decades, electrospinning has emerged as a facile technology to produce ultrafine carbon fibers (i.e., <0.7 denier; ca. <10 μm) and nanofibers (<100 nm) from polymer solutions.1 Especially the synthesis of carbon nanofibers via electrospinning of polyacrylonitrile (PAN) has been extensively studied for applications in the field of energy conversion and storage, capacitive deionization, catalysis, adsorption/separation, and in the field of biomedicine.2,3 To meet the complex requirements of advanced applications, synthesis routes to produce porous carbon nanofibers with high surface area and tunable pore size distribution have been developed by electrospinning polymer/sol–gel systems4 which yield composites of metal oxides and carbon (MOC).5 The latter can be carbothermally reduced at elevated temperatures of typically above 1200 °C to yield metal carbide/carbon (MCC) nanocomposite fibers.6 Recently, we have shown that MCC nanocomposites can easily be transformed into porous carbon by selectively etching metal ions7 and leaving behind so called carbide-derived carbon (CDC).8 This represents an attractive synthesis route to obtain binder-free, high-performance electrodes for electrochemical energy storage (supercapacitors) devices.9 So far, CDCs have only been reported from Si(O)C or TiC/C fiber mats and it is highly desirable to have an array of metal carbide/carbon nanocomposite fiber compositions available. Thus, the scope of this work is to establish in addition to optimized TiC/C fibers also ZrC/C and NbC/C as freestanding nonwoven textiles following a sol gel approach.The best investigated electrospun metal carbide fiber system to date is silicon carbide, mostly derived from polycarbosilane10 or polycarbomethylsilane.11 Commonly, such fibers are in the micrometer range,11,12 although fiber diameters as small as ca. 500 nm have been reported recently when using polymethylphenylsilsesquioxane and polyvinylpyrrolidone as carrier polymer.13 The low annealing temperatures at or below 1000 °C result in a silicon carbide that lacks the formation of an important feature of MCCs, namely carbide nanocrystals engulfed in a matrix of highly porous, partially graphitic carbon.7
TiO2/C and TiC/C ultrafine fibers have been electrospun from preformed TiO2 nanoparticles (e.g., derived from titanium isopropoxide or TiCl4) and a carrier polymer.14 Alternatively, nanofibers have been successfully electrospun from solutions containing a sol and a carrier polymer, growing in situ a metal oxide network that was later carbothermally reduced to metal carbide. The smooth ultrafine TiO2/C fibers and nanofibers have also been reported by electrospinning a mixture containing polyvinylpyrrolidone (PVP) with a sol using titanium isopropoxide as the metal precursor, with the addition of acetic acid or acetylacetonate as stabilizers.4,15,16 The obtained fibers presented diameters between 50–250 nm, but had a low specific surface area of 68–70 m2 g−1.14,17 In addition, TiC/C composite and TiC–CDC with non-uniform morphology including beaded fibers and average diameters around 100–120 nm have been produced using a phenolic resin and TiCl4, as carbon and metal sources, respectively, exhibiting a high surface area of 523 m2 g−1 for the MCC.18 Also, ultrafine fibers and ribbons with 300 nm width and 40 nm thickness have been obtained by electrospinning titanium(IV) n-butoxide and furfuryl alcohol as titanium and carbon sources, respectively.6 Based on this approach, in our previous study smooth TiC ultrafine fibers (131 nm) and TiC–CDC were obtained with the same fiber diameter.7
Zirconia fibers with diameters between 50–1000 nm have been produced by electrospinning mixtures containing a carrier polymer and a metal oxide precursor followed by calcination between 540 °C and 930 °C.19 Several routes have been proposed, using zirconium(IV) oxychloride octahydrate,19,20 zirconium(IV)isopropoxide,21 or zirconium(IV)acetylacetonate (ZrAcAc)22 as metal oxide sources. Composite ZrC/C ultrafine fibers have been only obtained by electrospinning sol gel systems containing ZrAcAc with a carbon precursor which also acts as a carrier polymer, mixed for C/Zr ratios higher than 3 to produce stoichiometric ZrC.23,24 The thinnest ZrC fibers obtained so far measured 730 nm in diameter after annealing at 1600 °C,23 using ZrAcAc, cellulose acetate as carbon source, and acetic acid to stabilize the sol. Instead of using a sol gel approach, recently Li et al. reported thinner fibers by electrospinning polyzirconoxane with a primary carbon source (PAN or sucrose).25 After annealing at 1400 °C, fiber diameters around 1–2 μm were reported for PAN and 500–600 nm for a mixture of PAN and sucrose. Thus, 500 nm diameter remains as a limit for the current state-of-the-art of ZrC/C fibers.
Mixing pre-existing niobium oxide nanoparticles with poly(vinyl alcohol) (PVA) with niobia, niobium carbide fibers have been produced by electrospinning.26 A thermal treatment at 1500 °C produces NbC fibers of about 110 nm diameter and a low surface area (4 m2 g−1) by annealing at 800 °C was observed. The synthesis of ultrafine Nb2O5 fibers by sol gel and electrospinning has been reported by mixing niobium n-butoxide27 or niobium ethoxide28 with a dissolved carrier polymer and an acid catalyst. As reported by Macias et al. when using HCl as the acid catalyst, ultrafine fibers (400–600 nm) were obtained;27 however, the fiber mats lost structural integrity upon calcination. Viswanathamurthi et al. reported ultrafine fibers (500 nm) by adding acetic acid, which acted as a catalyst for the hydrolysis and condensation reaction; the latter also formed a complex with niobium ethoxide which retarded the reaction and effectively prevented precipitation.28 This way, electrospinning was possible, but only during such a small time window that no suitable mats were obtained. Thus, until now, there has been no report of a sol–gel-approach of forming a niobium carbide phase without already starting with admixed niobia nanoparticles.
In this study, we present data for TiC/C, NbC/C, and ZrC/C nanocomposites fibers with partially unprecedented small diameters compared to the state-of-the-art. While we build on the previously reported synthesis of TiC/C via carbothermal reduction of TiO2/C nanofibers, we present a novel chemistry system to electrospin NbC/C and ZrC/C nanocomposites for a non-woven textile solely employing sol gel synthesis as a versatile technology platform. Thus, we abstain from adding metal oxide nanoparticles. Instead, metal oxide nanodomains within the electrospun fibers are formed in an in situ synthesis approach. Presenting these three related synthesis routes side-by-side, we provide a comprehensive technology platform to build on for further technological exploitation of these unique nanocomposite fibers, for example, as electrodes for supercapacitors, capacitive deionization, or other electrochemical applications.
2. Experimental
2.1. Materials
All chemicals were used as received. Zirconium(IV)acetylacetonate (Zr(IV)AcAc) and titanium butoxide (Ti(IV)BO) were purchased from Sigma-Aldrich. Niobium n-butoxide (NbBO) was purchased from Alfa Aesar. Carrier polymers cellulose acetate (CA) and polyvinylpyrrolidone (PVP) were purchased from Sigma-Aldrich as well the following solvents: acetylacetone (AcAc), anhydrous dimetylformamide 99.8% purity (DMF), acetic acid (AA), acetic anhydride (Ac2O), and furfuryl alcohol (FA). Air-sensitive materials (NbBO, AA, and Ac2O) were stored in an inert gas atmosphere under nitrogen in a glove box.2.2. Fiber synthesis
Fibers mats were electrospun using a MECC Co. NF-103V nanofiber system. Each sol–polymer system used slightly different spinning parameters due to variations in solution properties. All samples were spun on a stationary grounded target wrapped with aluminum foil. The spinneret head moved with respect to the collector along the x-axis parallel to the collector (travel distance). In addition, the head moved along the z-axis, perpendicular to the collector, which adjusts the distance from the spinneret tip to the collector or the distance the fiber flies (fly distance). The velocity of the head traveling in the x-axis can be defined as the travel velocity and is reported in mm s−1. A variable pump rate (mL h−1) is used to supply the spin dope to the spinneret. Finally, the temperature and relative humidity were monitored by a portable thermometer–hygrometer. More specific parameters for each system are summarized in Table 1 and shortly outlined below.| Parameter | TiBO + PVP | NbBO + PVP | ZrAcAc + cellulose acetate |
|---|---|---|---|
| Applied voltage (kV) | 15 | 23 | 20 |
| Fly distance (mm) | 250 | 165 | 250 |
| Pump rate (mL h−1) | 0.2 | 0.5 | 0.5 |
| Travel speed (mm s−1) | 50 | 0 | 30 |
| Travel distance (mm) | 100 | 0 | 200 |
| Temperature (°C) | 23 | 25 | 26 |
| Relative humidity | 21–32 | 11–15 | 21–25 |
The TiBO sol precursor was prepared adopting the method established by Zhang et al.6 In short, the sol was prepared in ambient conditions by mixing the liquids TiBO, FA, AA, and DMF in a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.25:6.75; then, 1 g of PVP is added to this mixture to make a 10 mass% solution. The mixture is aged while stirring for 48 h at room temperature. The final solution is translucent amber color with a viscosity of 0.27 Pa s at a shear rate of 500 s−1. Fibers were spun under the influence of a 15 kV potential. The collected fibers were allowed to age an additional 72 h exposed to ambient relative humidity (32 to 45%) to encourage hydrolysis and the formation of an extended metal–oxygen–metal network. Carbothermal reduction to convert metal oxides to carbides was accomplished by a two-step heat treatment profile under 99.5% argon in a Thermal Technologies furnace. The TiBO complex fibers were heated to 325 °C at 5 °C min−1 and annealed at that temperature for 3 h. The second step of the temperature profile is a 5 °C min−1 ramping to 1400 °C. At this temperature, TiC is thermodynamically favored over Ti–O in the presence of excess carbon from the pyrolyzed PVP and FA.29 The reaction is completed in 5 h and the carbide/carbon nanocomposite nonwoven textile is cooled to room temperature.
:1:0.25:6.75; then, 1 g of PVP is added to this mixture to make a 10 mass% solution. The mixture is aged while stirring for 48 h at room temperature. The final solution is translucent amber color with a viscosity of 0.27 Pa s at a shear rate of 500 s−1. Fibers were spun under the influence of a 15 kV potential. The collected fibers were allowed to age an additional 72 h exposed to ambient relative humidity (32 to 45%) to encourage hydrolysis and the formation of an extended metal–oxygen–metal network. Carbothermal reduction to convert metal oxides to carbides was accomplished by a two-step heat treatment profile under 99.5% argon in a Thermal Technologies furnace. The TiBO complex fibers were heated to 325 °C at 5 °C min−1 and annealed at that temperature for 3 h. The second step of the temperature profile is a 5 °C min−1 ramping to 1400 °C. At this temperature, TiC is thermodynamically favored over Ti–O in the presence of excess carbon from the pyrolyzed PVP and FA.29 The reaction is completed in 5 h and the carbide/carbon nanocomposite nonwoven textile is cooled to room temperature.
Zirconium carbide/carbon nanocomposite textiles were produced from a precursor of cellulose acetate in 10 mL of 1:1 by volume AA to AcAc (16 mass% polymer) allowed to solvate overnight until the solution was optically transparent. To this 5 g of Zr(IV)AcAc was added and mixed for 24 h until the solution was optically transparent and the viscosity reached 0.93 Pa s at a shear rate of 500 s−1. The solution was electrospun at 20 kV and subjected to a 2-step heat treatment regime. The Zr complex textiles were heated at 5 °C min−1 to 350 °C and held at temperature for 3 h to stabilize the fibers then heated up at 5 °C min−1 to 1500 °C and held at temperature for 2 h then cooled to room temperature.23
The precursor sol for niobium oxide was prepared under nitrogen to prevent premature gelation. In this preparation, NbBO was mixed with Ac2O (2.47 mL:0.55 mL), and stirred for 60 s. To this, 25 mL of DMF and 3 g of PVP were added and mixed for 48 h until the solution was translucent while exhibiting a viscosity of 0.032 Pa s at a shear rate of 500 s−1. The solution was electrospun under an applied potential of 23 kV. The niobium complex fibers were allowed to age in ambient humidity to promote condensation of the metal oxide domains for 72 h prior to heat treatment. The same furnace was used for the two-step temperature profile as was used for the other syntheses using an annealing temperature of 400 °C for 3 h with a 5 °C min−1 heating rate. For the carbothermal reduction heat treatment study, the pyrolyzed fibers were subjected to a high temperature annealing at 1100, 1300, 1500, or 1700 °C for 4 h with a 5 °C min−1 heating rate. One carbon/niobia sample was processed under each temperature profile for a total of 4 samples. In addition, one sample was heat treated at 400 °C for 3 h for characterization of the stabilized oxide fibers.
2.3. Characterization methods
The fibers were imaged using a FEI ESEM Quanta 400 FEG scanning electron microscope (SEM) under variable pressure and a JEOL JSM 7500F field emission scanning electron microscope (FESEM). Non-conductive samples were coated with a nanoscopically thin layer of Au or Pt/Au. ImageJ software was used to process the SEM images and diameters of at least 150 fibers were measured.30 Cross sectional focused ion beam cutting was performed with a Helios Nanolab 600 (FEI) using a working distance of 4 mm, a dwell time of 1 μs, and a cutting depth of 1 μm. Before cutting the sample was sputter coated with platinum for stabilization. High-resolution transmission electron (TEM) micrographs of the fibers were recorded after dispersing fibers in ethanol via sonication for 5 min and placing the solution over a copper grid with a lacey carbon film and imaging with a JEOL 2100F TEM operated at 200 kV.Infrared vibrational spectroscopy was performed with an attenuated total internal reflectance crystal (ATR) attachment on a Bruker Tensor 27 FTIR (Fourier-transform infrared) and spectra were integrated over 28 scans. Raman spectra were measured using a Renishaw inVia Raman system equipped with an Nd-YAG laser and an excitation wavelength of 532 nm with power of 0.2 mW at the focal point focused on a spot of ca. 2 μm diameter. A grating with 2400 lines per mm and a 50× objective (numeric aperture: 0.9) was used, yielding a spectral resolution of 1.2 cm−1. Peak fitting was accomplished assuming two Lorentzian peaks, one for the D-mode and one for the G-mode, respectively.
The free carbon content was measured using a VG Scienta furnace system, applying 700 °C for 1 h with a heating rate of 20 °C min−1 in synthetic air. The mass before and after thermal treatment was measured to calculate the initial carbon content.
A Bruker AXS D8 Advance diffractometer was used to collect X-ray diffractogramms using Cu-Kα radiation (0.15418 nm) with a step size of 0.02° and a measurement time of 1 s per step. The system was calibrated with a LaB6 single crystal prior to each measurement. Rietveld analysis was employed to determine lattice parameters and average coherence length values (corresponding roughly with the crystal domain size) using TOPAS software from Bruker.
Nitrogen gas sorption measurements at −196 °C were carried out with an Autosorb iQ system (Quantachrome). The samples were outgassed at 150 °C for 10 h under vacuum conditions (100 Pa) to remove adsorbed water and other surface functionalities. Nitrogen gas sorption was performed in liquid nitrogen in the relative pressure range from 5 × 10−7 to 1.0 in 68 steps. The specific surface area (SSA) was calculated with the ASiQwin-software using the Brunauer–Emmett–Teller (BET) equation in the linear relative pressure range 0.01–0.20.
Sol viscosities were measured using a modular compact Physica rheometer from Anton Paar with a 49.94 mm 1° cone and plate configuration. The sample chamber was left open to ambient humidity and viscosities were measured at constant shear rate of 500 s−1.
3. Results and discussion
3.1. Titanium carbide/carbon nanocomposite fibers
As-spun ultrafine fibers obtained from the TiBO precursor shown in Fig. 1A exhibit a smooth surface morphology and show an average diameter of 264 ± 53 nm (Fig. 2A). The fibers maintained their morphology after pyrolysis in argon at 325 °C for 3 h (Fig. 1B), but the fiber diameters were reduced by 18% to 217 ± 49 nm (Fig. 2A). The low temperature heat treatment stabilized the fiber morphology by promoting the FA to polymerize, evolved off water and organic residuals, and drove condensation and formation of an oxydic network.29 Subsequent carbothermal reduction in argon at 1400 °C for 5 h produced fibers with irregular oblate morphologies as seen in Fig. 1C and D with mean fiber diameters of 122 ± 28 nm (Fig. 2A) and this value aligns very well with our previous study (i.e., 131 ± 52 nm).7 As an important feature, we note that the open architecture afforded by the nonwoven fibers is preserved throughout the heat treatment process and the final textile is mechanically stabilized because the fiber crossovers are sintered together forming an isotropic percolated network of titanium carbide nanocrystals and carbon (Fig. 1E). This way, free-standing fiber mats are obtained. | ||
| Fig. 1 Scanning (A–E) and transmission (F) electron micrographs of final TiC/C nanocomposite fibers. (A) As spun. (B) After heat treatment in argon at 325 °C for 3 h. (C–F) After heat treatment in argon at 1400 °C for 5 h. | ||
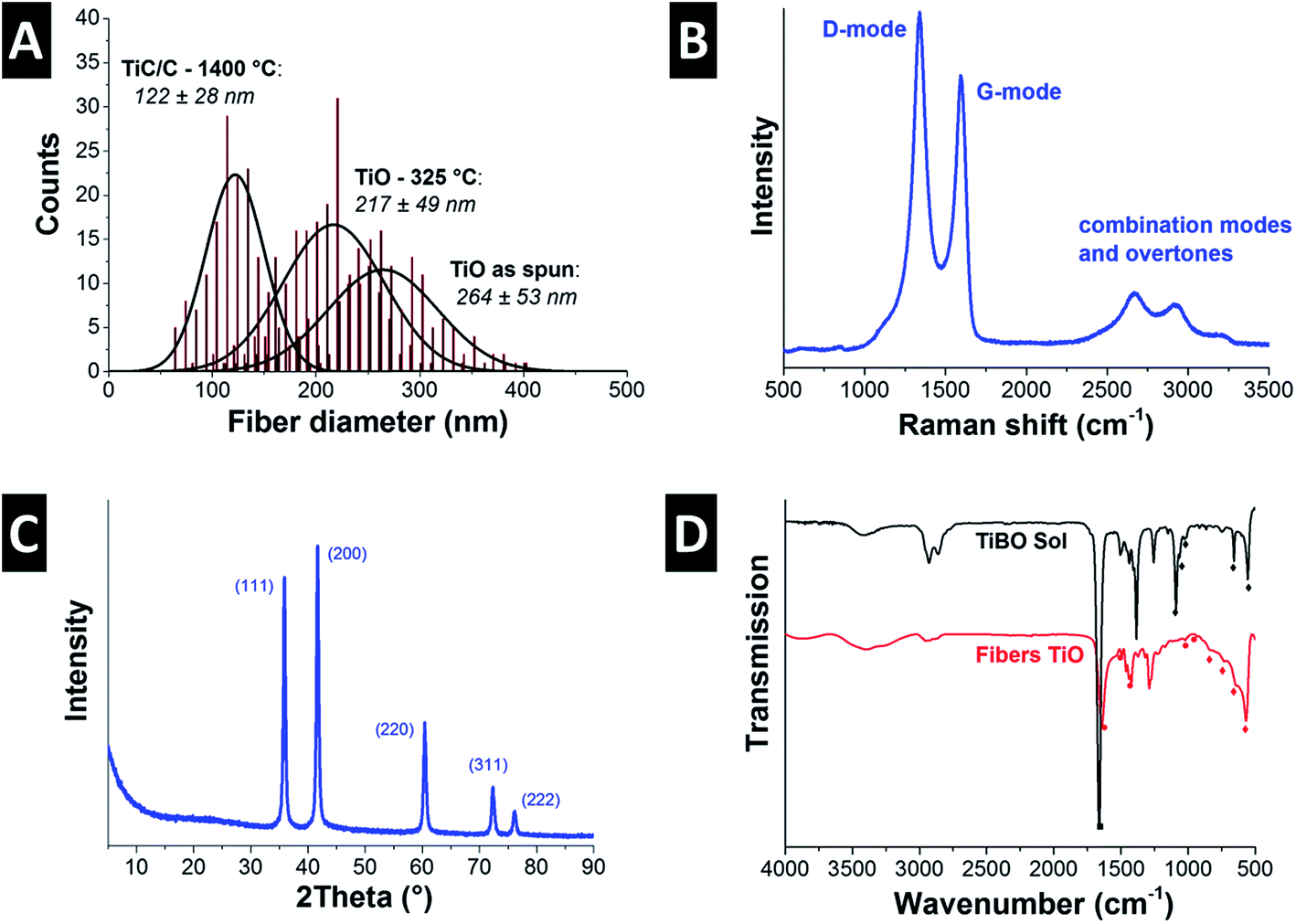 | ||
| Fig. 2 (A) Fiber diameter distributions of the as spun TiO fibers and heat treated fibers at 325 °C and 1400 °C. (B) Raman spectrum of TiC/C, and (C) X-ray diffractogram of the TiC/C nanocomposite fibers after heat treatment in argon at 1400 °C for 5 h, and (D) FTIR spectra of the TiBO sol and the as spun fibers. | ||
TEM investigation shows that the fibers were nanocomposites composed of TiC nanocrystals embedded in a continuous network of disordered carbon in a brick-and-mortar-like fashion (Fig. 1F). The rough fiber morphology is a result of the nanoscopic character of the carbide nanocrystals with a size of ca. 20 nm which are engulfed by 1–3 layers of disordered carbon (Fig. 1F). As determined from high resolution TEM, the corresponding lattice spacing of 0.25 nm is characteristic for the (111) d-spacing of TiC. We also note that the fibers do not show any tendency for core–shell formation but instead exhibit a very homogenous texture throughout the fiber (Fig. 1F). The isotherm measured using gas sorption analysis shows a typical combination between type I and II characteristic for micro- and macroporous materials (ESI; Fig. S1A†). The combination of fully dense carbide nanocrystals and disordered porous carbon yields a rather high specific surface area (SSA) of 450 m2 g−1 (Table 2). This value is slightly higher than what we have reported before for this system (+10%; i.e., 414 m2 g−1; ref. 7) and somewhat smaller than what has recently been reported by the Kaskel group (namely, 523 m2 g−1; ref. 18). For comparison, we note that a fully dense TiC nanofiber of the same average diameter would only show a SSA of 10 ± 3 m2 g−1 which illustrates the large pore volume maintained within the carbon phase. In our system the free carbon content was measured to 32 mass% (Table 2). Considering the non-porous nature of TiC nanocrystals, we can estimate a porosity of the amorphous carbon phase of more than 1400 m2 g−1.
| Sample | Free carbon mass% | BET SSA (m2 g−1) | ID/IG-ratio | FWHM (cm−1) | |
|---|---|---|---|---|---|
| D-mode | G-mode | ||||
| TiC/C – 1400 °C | 32 | 450 | 2.1 ± 0.1 | 98 ± 0.9 | 58 ± 0.4 |
| ZrC/C – 1500 °C | 15 | 224 | 1.7 ± 0.0 | 66 ± 3.6 | 63 ± 2.2 |
| NbC/C – 1100 °C | 15 | 151 | 2.6 ± 0.0 | 148 ± 1.6 | 60 ± 0.3 |
| NbC/C – 1300 °C | 19 | 147 | 1.9 ± 0.3 | 94 ± 1.0 | 64 ± 6.5 |
| NbC/C – 1500 °C | 20 | 121 | 2.0 ± 1.0 | 67 ± 0.7 | 60 ± 10.7 |
| NbC/C – 1700 °C | 29 | 39 | 1.2 ± 0.2 | 53 ± 0.4 | 57 ± 4.8 |
Raman spectroscopy confirmed the presence of free carbon in the fibers, that is, carbon that is not bound in form of the metal carbide (Fig. 2B, Table 2). Free carbon is in a highly disordered state and stems from FA, AA, and BO residuals forming a cohesive matrix around TiC nanocrystals, as seen in Fig. 1F. The two dominant features in the Raman spectrum, namely the band at 1341 ± 0.1 cm−1 (FWHM: 98 ± 0.9 cm−1) and the band at 1598 ± 0.5 cm−1 (FWHM: 58 ± 0.4 cm−1) are assigned to the D-mode and G-mode of carbon, respectively.31 The integral intensity ratio, ID/IG, is a metric of carbon ordering and the high value of 2.1 ± 0.1 is indicative of incompletely graphitized carbon32 and typical for nanoporous carbon materials.33 In contrast to carbon, the other component of the ultrafine fibers, titanium carbide, is present in a highly crystalline phase. The TiC nanocrystals can be assigned to the space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m with a lattice spacing of 0.431 nm (cf. PDF-32-1383: 0.433 nm).34 In more detail, X-ray diffraction (Fig. 2C) confirms the nanocrystalline character of titanium carbide. Rietveld analysis for samples after carbothermal reduction for 5 h in argon yields a value of 22 nm, which is in perfect agreement with the TEM observations (Fig. 1F).
m with a lattice spacing of 0.431 nm (cf. PDF-32-1383: 0.433 nm).34 In more detail, X-ray diffraction (Fig. 2C) confirms the nanocrystalline character of titanium carbide. Rietveld analysis for samples after carbothermal reduction for 5 h in argon yields a value of 22 nm, which is in perfect agreement with the TEM observations (Fig. 1F).
To better understand the evolution of the oxide network formation, FTIR spectroscopy was carried out on the as-spun fibers and the precursor (Fig. 2D). The sol was investigated without PVP and DMF and the main interaction in the sol is between TiBO and acetic acid. The acid slows down the condensation by coordinating with titanium ions. FTIR spectra of the AA-TiBO-FA show bands near 1665 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) from the AA and absorption bands between 1150–900 cm−1 assigned to the C–O terminal vibration arising from the Ti–O–C4H9 mixed with bridging Ti–C–O vibrations at 1023 cm−1 and 912 cm−1. Other peaks can be assigned to the formation of M–O below 800 cm−1. In particular, the combination of peaks at 3410 cm−1 (O–H) and 950 cm−1 are evidence of butanol formed as a reaction product.35,36 During electrospinning, the DMF and acetic acid evaporate from the fibers leaving behind PVP and a nascent titanium oxide network (Fig. 2D). In addition, the peaks between 1500 cm−1 and 1400 cm−1 are assigned to the CH2 scissoring vibrations on the PVP rings, while the small peak at 1016 cm−1 corresponds to the C–C polymer backbone, and the band at 930 cm−1 is the breathing mode of the ring.37 The very broad peaks below 800 cm−1 are related to the O–Ti–O network.35 This amorphous oxygen–metal–oxygen network evolves at higher temperatures to the TiC nanocrystals via carbothermal reduction.
O stretch) from the AA and absorption bands between 1150–900 cm−1 assigned to the C–O terminal vibration arising from the Ti–O–C4H9 mixed with bridging Ti–C–O vibrations at 1023 cm−1 and 912 cm−1. Other peaks can be assigned to the formation of M–O below 800 cm−1. In particular, the combination of peaks at 3410 cm−1 (O–H) and 950 cm−1 are evidence of butanol formed as a reaction product.35,36 During electrospinning, the DMF and acetic acid evaporate from the fibers leaving behind PVP and a nascent titanium oxide network (Fig. 2D). In addition, the peaks between 1500 cm−1 and 1400 cm−1 are assigned to the CH2 scissoring vibrations on the PVP rings, while the small peak at 1016 cm−1 corresponds to the C–C polymer backbone, and the band at 930 cm−1 is the breathing mode of the ring.37 The very broad peaks below 800 cm−1 are related to the O–Ti–O network.35 This amorphous oxygen–metal–oxygen network evolves at higher temperatures to the TiC nanocrystals via carbothermal reduction.
3.2. Zirconium carbide/carbon nanocomposite fibers
The cylindrical fibers electrospun from Zr(IV)AcAc and CA in a solution of AA/AcAc show a very smooth morphology without beads or other visible defects (Fig. 3A). Unlike the fibers spun from the TiBO precursor, the Zr(IV)AcAc precursor yielded significantly higher fiber diameters, namely 715 ± 395 nm. The larger diameter is a result of the higher polymer content necessary to produce uniform fibers. Lower polymer content solutions resulted in extensive beading and low quality nonwoven textiles were obtained. However, the large standard deviation in fiber diameters is reflective of a broad size distribution, ranging from a population of very fine fibers smaller than 200 nm up to larger sized fibers with more than 2 μm (Fig. 4A). | ||
| Fig. 3 Scanning (A–D) and transmission (E and F) electron micrographs of final ZrC/C nanocomposite fibers. (A) As spun. (B) After heat treatment in argon at 350 °C for 3 h. (C–F) After heat treatment in argon at 1500 °C for 2 h. | ||
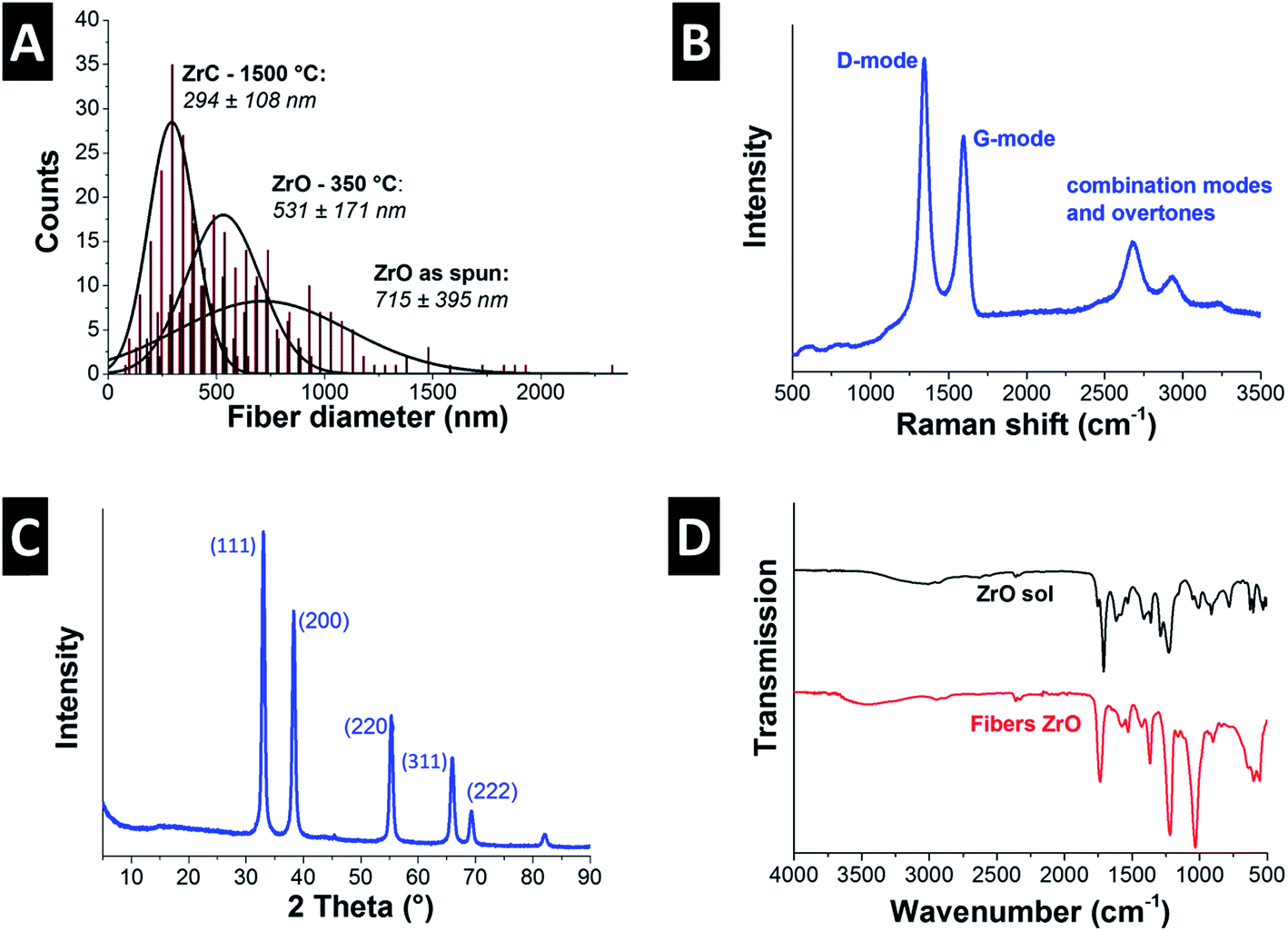 | ||
| Fig. 4 (A) Fiber diameter distributions of the as spun fibers and heat treated fibers at 350 °C and 1500 °C. (B) Raman spectrum and (C) X-ray diffractogram of the ZrC/C nanocomposite fibers after heat treatment in argon at 1500 °C for 2 h. (D) FTIR spectra of the ZrO sol and the as spun fibers. | ||
Pyrolysis was accomplished via heat treatment at 350 °C for 3 h under argon resulting in a 26% reduction of the mean fiber diameter (Fig. 3B and 4A). During the heat treatment, some of the smaller fibers were consumed and the remaining population has a mean diameter of 531 ± 171 nm. Carbothermal reduction at 1500 °C for 2 h produced fibers with very rough surfaces and an average size of 294 ± 108 nm corresponding to a 59% reduction compared to the as spun fibers (Fig. 3C & D and 4A). This value is significantly smaller than what was recently reported for ZrC/C fibers by Li et al. (i.e., 500 nm to 1.5 μm) using polyzirconoxane and PAN.25 The homogenous fibers after carbothermal reduction did not exhibit any indication of a core–shell geometry and are composed of carbide nanocrystals embedded in a matrix of porous carbon (Fig. 3E). The free carbon content was measured to be 15 mass% (Table 2) and we see a similar coating of graphene-like carbon layers around the metal carbide domains (Fig. 3F). The isotherm measured via gas sorption analysis shows a similar shape than for TiC/C (ESI, Fig. S1A†) and confirms the highly porous nature of the fibers and a surface area of 224 m2 g−1 (Table 2). This value is lower compared to the TiC/C-system; yet, we have to consider the higher density of zirconium carbide (6.7 g cm−3) compared to TiC (4.9 g cm−3) and the lower free carbon content of 15 mass% compared to 32 mass% with a high porosity.
Unlike the PVP in the TiBO system, CA and ACAC both participate in the condensation reaction as can be seen when comparing the FTIR spectra of the Zr(IV)AcAc sol and ZrO fibers (Fig. 4D). The sol has peaks that are a combination of the constituents of the solution. There are almost no free hydroxyl groups as evidenced by the down shifted, almost indistinct –OH peak at 3150 cm−1. Both AA and CA present –OH groups and the position of this peak suggests that there are several varieties of hydrogen bonding occurring in the solution.38 There are also two peaks assigned to the CO stretching mode: one is located at 1754 cm−1 associated with the carbonyl of the CA and the other more intense peak at 1711 cm−1 stems from the carbonyl of the AcAcACAC and AA.39 The peaks at 1576, 1530, and 1385 cm−1 relate to the CO symmetric and asymmetric stretch of the Zr(IV)AcAc.40 In addition, the AcAc has two bands that arise due to mixed vibrations of the –CH3 and –CH2 out of plane bending interacting with –OH at 1421 and 1293 cm−1. Finally, we see a peak at 774 cm−1 due to the –CH3 out of plane rocking.39 The bands assigned to the Zr–O are at 664 and 544 cm−1 and the peaks characteristic for CA are located at 1363 cm−1 for the –CH in plane bending, 1225 cm−1 for the C–O stretch of the carboxylate, and 1160 cm−1 for the asymmetrical C–O–C (bridge) stretch. Finally, the pryanose ring C–O–C band is at 1051 cm−1 and the out of plane –OH is located at 600 cm−1.38 The FTIR spectrum of ZrO fibers is very similar to the sol except the –OH stretch has shifted to higher wavenumbers from 3150 to 3495 cm−1 indicating there are more free –OH and less hydrogen bonding. The carbonyl peaks for the CA and AcAc have become indistinguishable and shifted to higher wave numbers (i.e., 1737 cm−1) indicating less hydrogen bonding to the AcAc. This is most likely due to the release of the AcAc from the Zr ion. A new peak at 1560 cm−1 is assigned to the Zr–O–C stretch from the CA forming a complex with the Zr ion. In addition, a new band at 840 cm−1 is due to the formation of Zr–O–H, an intermediate step in the condensation reaction. There is also significant broadening of the bands below 750 cm−1 assigned to the formation of the O–Zr–O network.41
The remaining organic groups in the fiber, after the condensation reaction is completed, are converted to carbon during carbothermal reduction at 1500 °C for 2 h. The resulting carbon is slightly more organized than the carbon in the TiC/C nanocomposite, as seen from TEM (Fig. 3F). This observation is supported by Raman spectroscopy (Fig. 4B, Table 2) where a ID/IG-ratio of 1.7 was indicative of a higher degree of carbon ordering. In the spectra, the D-band is located at 1337 ± 1.3 cm−1 (FWHM: 66 ± 3.6 cm−1) and the G-band at 1594 ± 0.5 cm−1 (FWHM: 63 ± 2.2 cm−1). Besides the partially graphitic carbon, there is only one fully crystalline phase, namely ZrC, as identified with XRD (Fig. 4C). Rietveld analysis confirmed the space group Fmm typical for zirconium carbide and yielded a lattice parameter of 0.469 nm (cf. PDF-35-0784: 0.469 nm).34 The calculated average coherence length was 65 nm which is in agreement with TEM micrographs (Fig. 3E and F), but larger than what we observed for TiC/C nanocomposite fibers.
3.3. Niobium carbide/carbon nanocomposite fibers
Electrospinning NbBO with PVP was particularly challenging because of NbBO's strong tendency towards hydrolysis and condensation in the presence of ambient water. The solution droplet at the spinneret tip gels as soon as it is exposed to ambient moisture making it difficult to electrospin because the viscosity increases dramatically with time. As a result, the yield of fibers is very low and impossible to remove from the collector. To retard gelation, acetic anhydride was added to the spin dope because the Ac2O forms a bidentate ligand with the hydrolyzed niobium ion.42 This ligand inhibits the condensation reaction and, in effect, slows down the gelation rate so that the viscosity remains in a suitable range during electrospinning. This way, freestanding nonwoven textile mats can be effectively obtained.Adopting the outlined synthesis procedure, we obtained smooth and cylindrical as-spun fibers with an average diameter of 124 ± 74 nm (Fig. 5A and 7A). After pyrolysis at 400 °C, the fibers maintain their smooth morphology and their diameter does not change significantly (Fig. 5B and 7A). Electron microscopy shows a uniform amorphous structure after heat treatment at 400 °C building a free-standing network of partially sintered fibers (Fig. 5B).
 | ||
| Fig. 5 Scanning electron and transmission electron micrographs of (A) NbO as spun, and (B) after 400 °C in argon. (C) FTIR spectra of the NbO sol and the as spun fibers. | ||
Analysis of the FTIR spectrum (Fig. 5C) of the NbBO sol is complicated because of overlapping bands arising from DMF, PVP, and the carboxylates from the Ac2O. The bands in the 3500–3200 cm−1 can be assigned to –NH stretch of the PVP. In addition, absorbed water is present in the samples as evidenced by the free water rotations between 3900–3500 cm−1. The –CH2 and –CH3 stretch modes are assigned to the bands between 3000–2800 cm−1 and the prominent band at 1667 cm−1 is a convolution of the CO stretch peaks from the PVP, DMF, and Ac2O. The bands assigned to the DMF below 1550 cm−1 are the –CH3 bending at 1506 cm−1, the symmetric bending of the methyl groups at 1394 cm−1, and the –CN at 1255 cm−1. These bands are not present in the spectra of the fibers because the DMF evaporates during electrospinning. PVP is present in both the sol and the fibers and the bands assigned to the PVP are the ring CH2 scissoring at 1489, 1456, and 1423 cm−1. The NbBO in the sol has assigned bands at 1472 cm−1 for the –CH bending and at 1086 cm−1 and 1055 cm−1 assigned to the Nb–O in the butoxide. In addition, we see peaks assigned to PVP and Nb–O from the butoxide between 1200–1000 cm−1 and also very broad peaks below 900 cm−1 assigned to the formation of poorly organized NbO.43
Scanning electron microscopy presents cylindrical fibers with rough surface structure for all temperatures (Fig. 6). After carbothermal reduction at 1100 °C, the fiber diameter decreases to 80 ± 44 nm and further annealing at 1700 °C reaches 65 ± 36 nm fiber diameter (Fig. 7A). To the best of our knowledge, these are the thinnest such fibers reported in the literature so far.
 | ||
| Fig. 6 (A, C, E and G) Scanning and (B, D, F and H) transmission electron micrographs of NbC/C synthesized at 1100, 1300, 1500, and 1700 °C. | ||
 | ||
| Fig. 7 (A) Fiber diameter distributions of NbO as spun and after different annealing temperatures. (B) Raman spectra, (C) XRD diffractograms, and (D) calculated NbC nanodomains sizes (i.e., average coherence length) for NbC/C nanocomposite nanofibers obtained after carbothermal reduction at different temperatures. | ||
To evaluate the effect of the carbothermal reduction temperature on the resulting carbide/carbon composite, fiber mats from the NbBO precursor were subjected to carbothermal reduction in argon at 1100, 1300, 1500, or 1700 °C. The free carbon content increases from 15 mass% for 1100 °C synthesis temperature to finally 29 mass% at 1700 °C (Table 2). The mats remained structurally intact after all temperatures; however, the material became more brittle as a result of increased carbon graphitization. The latter can be seen in the Raman spectra of the samples taken after each heat treatment (Fig. 7B).
Aligning with the results for TiC/C and ZrC/C, NbC/C MCC also exhibits a uniform distribution of metal carbide nanocrystals engulfed by a matrix of disordered carbon (Fig. 6B, D, F and H). Thermal treatment at 1100 °C yields highly disordered NbC particles (Fig. 6B), but the crystallinity increases significantly at 1300 °C (Fig. 6D) and larger crystals can be seen for temperatures of 1500 °C and 1700 °C (Fig. 6F and H). NbC nanocrystals were engulfed or covered by partially graphitic and disordered carbon coming from the carrier polymer. The nanocomposite character of NbC/C nanofibers is corroborated by Raman spectroscopy (Fig. 7B, Table 2) and XRD measurements (Fig. 7C). These data identify partially graphitic carbon and nanocrystalline niobium carbide as the two components of the composite nanofibers. In particular, the Raman spectra exhibit the characteristic D- and G-modes indicating the presence of free-carbon and the disordered nature (Fig. 7B). Using different annealing temperatures, the ID/IG-ratio decreases dramatically from 2.6 to 1.2 from 1100 °C to 1700 °C, indicating the increasing carbon ordering (Table 2).
XRD data was subject to Rietveld analysis yielding an average coherence length of 38 nm and a lattice parameter of 0.447 nm for Fmm niobium carbide (cf. PDF-38-1364: 0.447 nm) (Fig. 7C and D).34 No changes in lattice spacing or crystal structure for the samples derived by carbothermal reduction at 1300, 1500, or 1700 °C were observed. In contrast, the XRD diffractogram after treatment at 1100 °C shows only broad peaks associated with partially graphitic carbon with a very small (111) peak emerging at around 34.8° 2θ. Partially graphitic carbon is also encountered for the other samples; yet, the high signal intensity of metal carbide peaks down-scale the carbon-related signal so strongly that it appears from first inspection that only metal carbides would be present. Thus, it is important to complement XRD with Raman data, as done in this study. We also see a continuous increase in the NbC domain size (i.e., average coherence length) with temperature, increasing from 27 nm at 1300 °C and 38 nm at 1500 °C to finally 65 nm at 1700 °C what is in agreement with transmission electron microscopy (Fig. 7D).
Gas sorption analysis presents specific surface areas of 151 m2 g−1 for 1100 °C which decreases to 39 m2 g−1 for 1700 °C. Due to the more disordered carbon nature at 1100 °C, a higher pore volume is expected and shown by the gas sorption isotherms (ESI, Fig. S1B†). NbC/C synthesized at 1100 °C shows a characteristic type I isotherm shape indicating a microporous material. With increasing synthesis temperature the surface area decreases and the isotherm changes to type II with a small hysteresis characteristic for non-porous materials. These findings are in agreement with Raman spectroscopy presenting the transition of a highly disordered and porous material to a higher degree of carbon ordering with less pores at 1700 °C.
4. Conclusions
In our study, we have presented the first comprehensive comparison of three different metal oxide/carbon nanocomposite fibers from a sol–gel-approach that can easily be converted to metal carbide/carbon nanocomposite nonwoven textiles via carbothermal reduction. In all cases, we obtained submicrometer-sized fibers; more precisely, we were able to obtain the following average fiber diameters: 294 ± 108 nm for ZrC/C, 122 ± 28 nm for TiC/C, and 65 ± 36 nm for NbC/C. All MCC nanocomposite fibers were homogenously composed of nanodomains of metal carbide in the range between 20 nm (TiC/C), 38 nm (NbC/C), and 65 nm (ZrC/C) engulfed in a matrix of highly disordered carbon. This combination yields a relatively large specific surface area of 450 m2 g−1 for TiC/C (1400 °C), 224 m2 g−1 for ZrC/C (1500 °C), and 121 m2 g−1 for NbC/C (1500 °C). These high values are related to the presence of disordered and highly porous free carbon, varying between 15 and 32 mass%. This array of MCC nanocomposite fibers was obtained in the form of freestanding nonwoven textiles which may serve as an ideal precursor for the synthesis of highly porous carbide-derived carbon electrodes for electrochemical applications.Acknowledgements
The INM (www.inm-gmbh.de) is part of the Leibniz Research Alliance Energy Transition (LVE). We acknowledge funding from the German Federal Ministry for Research and Education (BMBF) in support of the nanoEES3D project (award number 03EK3013) as part of the strategic funding initiative energy storage framework. LMF acknowledges financial support by the Manfred Lautenschläger Foundation via the MINT Excellence program. The authors thank Prof. Eduard Arzt for his continuing support and thank Dr Elmar Kroner for his support (both INM). Dr Mesut Aslan (INM) is thanked for his help with experimental work, Rudolf Karos (INM) for XRD measurements, and the group of Prof Frank Mücklich (Chair of Functional Materials, Saarland University) for supplying focused ion beam operations.References
- Z.-M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- L. F. Zhang, A. Aboagye, A. Kelkar, C. L. Lai and H. Fong, J. Mater. Sci., 2014, 49, 463–480 CrossRef CAS.
- G. Wang, Q. Dong, Z. Ling, C. Pan, C. Yu and J. Qiu, J. Mater. Chem., 2012, 22, 21819–21823 RSC.
- D. Li and Y. Xia, Nano Lett., 2003, 3, 555–560 CrossRef CAS.
- J. Watthanaarun, V. Pavarajarn and P. Supaphol, Sci. Technol. Adv. Mater., 2005, 6, 240 CrossRef CAS.
- L. Zhang, J. Hu, A. A. Voevodin and H. Fong, Nanoscale, 2010, 2, 1670–1673 RSC.
- V. Presser, L. F. Zhang, J. J. Niu, J. McDonough, C. Perez, H. Fong and Y. Gogotsi, Adv. Energy Mater., 2011, 1, 423–430 CrossRef CAS.
- V. Presser, M. Heon and Y. Gogotsi, Adv. Funct. Mater., 2011, 21, 810–833 CrossRef CAS.
- Y. Gao, V. Presser, L. Zhang, J. J. Niu, J. K. McDonough, C. R. Pérez, H. Lin, H. Fong and Y. Gogotsi, J. Power Sources, 2012, 201, 368–375 CrossRef CAS.
- H. A. Liu and K. J. Balkus Jr, Mater. Lett., 2009, 63, 2361–2364 CrossRef CAS.
- M. Rose, E. Kockrick, I. Senkovska and S. Kaskel, Carbon, 2010, 48, 403–407 CrossRef CAS.
- A. Guo, M. Roso, M. Modesti, J. Liu and P. Colombo, J. Eur. Ceram. Soc., 2014, 34, 549–554 CrossRef CAS.
- A. Guo, M. Roso, M. Modesti, J. Liu and P. Colombo, J. Appl. Polym. Sci., 2014, 131, 39836 Search PubMed.
- C. Wessel, R. Ostermann, R. Dersch and B. M. Smarsly, J. Phys. Chem. C, 2010, 115, 362–372 Search PubMed.
- X. Zhang, V. Aravindan, P. S. Kumar, H. Liu, J. Sundaramurthy, S. Ramakrishna and S. Madhavi, Nanoscale, 2013, 5, 5973–5980 RSC.
- C. Tekmen, A. Suslu and U. Cocen, Mater. Lett., 2008, 62, 4470–4472 CrossRef CAS.
- S. Cavaliere, S. Subianto, L. Chevallier, D. J. Jones and J. Roziere, Chem. Commun., 2011, 47, 6834–6836 RSC.
- J. R. Martin, L. Borchardt, M. Oschatz, G. Mondin and S. Kaskel, Chem. Ing. Tech., 2013, 85, 1742–1748 CrossRef CAS.
- E. Davies, A. Lowe, M. Sterns, K. Fujihara and S. Ramakrishna, J. Am. Ceram. Soc., 2008, 91, 1115–1120 CrossRef CAS.
- C. Shao, H. Guan, Y. Liu, J. Gong, N. Yu and X. Yang, J. Cryst. Growth, 2004, 267, 380–384 CrossRef CAS.
- N. Dharmaraj, C. H. Kim and H. Y. Kim, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2006, 36, 29–32 CrossRef CAS.
- E. Formo, M. S. Yavuz, E. P. Lee, L. Lane and Y. Xia, J. Mater. Chem., 2009, 19, 3878–3882 RSC.
- Y. S. Nam, X. M. Cui, L. Jeong, J. Y. Lee and W. H. Park, Thin Solid Films, 2009, 517, 6531–6538 CrossRef CAS.
- X. M. Cui, Y. S. Nam, J. Y. Lee and W. H. Park, Mater. Lett., 2008, 62, 1961–1964 CrossRef CAS.
- F. Li, Z. Kang, X. Huang and G.-J. Zhang, Ceram. Int., 2014, 40, 10137–10141 CrossRef CAS.
- K. Nakane, M. Morinaga and N. Ogata, J. Mater. Sci., 2013, 48, 7774–7779 CrossRef CAS.
- M. Macias, A. Chacko, J. P. Ferraris and K. J. Balkus Jr, Microporous Mesoporous Mater., 2005, 86, 1–13 CrossRef CAS.
- P. Viswanathamurthi, N. Bhattarai, H. Y. Kim, D. R. Lee, S. R. Kim and M. A. Morris, Chem. Phys. Lett., 2003, 374, 79–84 CrossRef CAS.
- L. M. Berger, W. Gruner, E. Langholf and S. Stolle, Int. J. Refract. Met. Hard Mater., 1999, 17, 235–243 CrossRef CAS.
- W. S. Rasband, ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, 1997–2014 Search PubMed.
- P. Lespade, R. Al-Jishi and M. S. Dresselhaus, Carbon, 1982, 20, 427–431 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2477–2512 CrossRef CAS PubMed.
- C. Portet, G. Yushin and Y. Gogotsi, J. Electrochem. Soc., 2008, 155, A531–A536 CrossRef CAS.
- H. Nowotny and G. Z. Glenk, Metallkd, 1947, 38 Search PubMed.
- F. X. Perrin, V. Nguyen and J. L. Vernet, J. Sol-Gel Sci. Technol., 2003, 28, 205–215 CrossRef CAS.
- D. C. Bradley, R. C. Mehrotra and D. P. Gaur, Metal Alkoxides, Harcourt Brace Jovanovich, London, 1978 Search PubMed.
- L. S. Taylor, F. W. Langkilde and G. Zografi, J. Pharm. Sci., 2001, 90, 888–901 CrossRef CAS PubMed.
- L. M. Ilharco and R. Brito de Barros, Langmuir, 2000, 16, 9331–9337 CrossRef CAS.
- S. F. Tayyari and F. Milani-nejad, Spectrochim. Acta, Part A, 2000, 56, 2679–2691 CrossRef CAS.
- R. C. Fay and T. J. Pinnavaia, Inorg. Chem., 1968, 7, 508–514 CrossRef CAS.
- B. S. Buyuktas and O. Aktas, Transition Met. Chem., 2006, 31, 56–61 CrossRef CAS.
- M. Nabavi, S. Doeuff, C. Sanchez and J. Livage, J. Non-Cryst. Solids, 1990, 121, 31–34 CrossRef CAS.
- Y.-n. Cai, S.-h. Yang, S.-m. Jin, H.-p. Yang, G.-f. Hou and J.-y. Xia, J. Cent. South Univ. Technol., 2011, 18, 73–77 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra05409e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |