
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResponsive pentablock copolymers for siRNA delivery†
Metin
Uz
ab,
Surya K.
Mallapragada
b and
Sacide Alsoy
Altinkaya
*a
aIzmir Institute of Technology, Department of Chemical Engineering, Gulbahce Koyu, Urla, Izmir, 35430-Turkey. E-mail: metinuz@iastate.edu; sacidealsoy@iyte.edu.tr; Fax: +90 232 750 6645; Tel: +90 232 750 6658
bIowa State University, Department of Chemical and Biological Engineering, 2031 Sweeney, Ames, IA 50011-2230, USA. E-mail: suryakm@iastate.edu; Fax: +1-515-294-2689; Tel: +1-515-294-7407, +1 515 441 2046
First published on 28th April 2015
Abstract
In this study, temperature and pH responsive cationic and amphiphilic pentablock copolymers, which consist of the temperature responsive triblock Pluronic F127 sandwiched between pH responsive PDEAEM (poly(2-diethylaminoethyl methacrylate)) end blocks, were used for the first time in the development of polyplex and gold nanoparticle (AuNP) based multicomponent siRNA delivery systems (MCSs). Copolymers in both systems protected siRNA from external effects, provided cell entry and endosomal escape. The thermoreversible micellization of the hydrophobic PPO block facilitated the cellular entry while the PDEAEM blocks enhanced the endosomal escape through protonated tertiary amine groups by pH buffering. The synergistic advantages of the different blocks showed an enhanced effect in the MCSs due to attachment and surface configuration reasons. The siRNA transfection efficiency of MCSs against luciferase expressing SKOV3 cells was 15% higher than both the polyplexes alone and the commercial siRNA transfection agent Lipofectamine RNAiMax at the same applied dose, without any toxicity. The results indicated that the multicomponent systems based on the responsive cationic pentablock copolymers and gold nanoparticles have promising potential as an efficient siRNA delivery vector for future applications.
1 Introduction
The application of small interfering RNA (siRNA) has shown great potential for cancer treatment. Over the last decade, progress in material science and nanotechnology has led to the development of efficient siRNA delivery systems, proposing new approaches to overcome the major challenges and limitations of siRNA delivery.1–3 These challenges include siRNA condensation/attachment to the carrier material, its protection from external effects (such as, degradation by RNase enzymes), stability, cellular entry, endosomal escape and siRNA transfection efficiencies.4–6 In the literature, different siRNA delivery systems have been developed based on different materials such as polymers, lipids, peptides and inorganic nanoparticles.7–12A lot of work has focused on siRNA delivery using polyplex systems that take advantage of the direct electrostatic interactions between siRNA and cationic polymers. However, a general problem in such systems is the poor siRNA release and activity in the cytoplasm due to strong electrostatic interactions resulting from the excess cationic charge of polymers.13–18 Because of this obstacle, high doses of polyplexes are required to achieve efficient siRNA activity, and this brings about the use of excessive polymer amounts which may cause potential toxic effects.15,19–21 As another hurdle, the studies have also indicated that some of the siRNA/polymer polyplex systems are not stable against serum protein-mediated aggregation.13,16,17,21–27 In order to overcome these drawbacks, different types of responsive block copolymers have been developed to form polyplex siRNA delivery systems. The advantage of these polymers stems from the possibility of changing the balance between the blocks with different features, such as electrostatic charge, hydrophilicity, pH and temperature responsiveness, that allows tuning of cytotoxicity, siRNA condensation, cellular uptake, endosomal escape and siRNA transfection efficiency.24,28–36
While synthetic and natural polymers have been investigated as siRNA carriers, recent studies have focused on the application of gold nanoparticles (AuNPs).15,19,37–40 Gold colloids can be used for therapeutic, imaging and diagnostic purposes due to their bio-inertness, biocompatibility, easy synthesis and surface functionalization.41,42 In terms of siRNA delivery applications, AuNPs were mainly used as the core of multicomponent siRNA delivery systems (MCS), on which the subsequent system components (such as, siRNA, polymers, peptides etc.) can be attached easily either with chemical bonds or electrostatic interactions. In literature, siRNA has been mostly attached on the AuNP surfaces through disulfide bonds, which can be cleaved by glutathione in cytoplasm, in order to facilitate its release and enhance its activity.10,19,43 However, since these systems with just siRNA around a AuNP core lack the ability of efficiently protecting the siRNA from external effects and enabling cellular uptake, endosomal escape and siRNA transfection, a functional outer layer (i.e. polymer, liposome, peptide etc.) surrounding the siRNA coated AuNPs through electrostatic interactions or chemical bonds was accompanied.15,19 Therefore, the recent focus has been on the construction of AuNP based multicomponent siRNA delivery systems, along with the use of the cationic functional polymers.15,39,40,44–46
In this work, stimuli responsive pentablock copolymer (PDEAEM–Pluronic F127–PDEAEM) composed of temperature responsive Pluronic F127 (poly(ethyleneoxide)-block-poly(propyleneoxide)-block-poly(ethyleneoxide) (PEO-b-PPO-b-PEO)) and pH responsive cationic PDEAEM poly(2-diethylaminoethyl methacrylate) blocks was used for the first time in the construction of polyplex and multicomponent siRNA delivery systems. In our previous studies, this copolymer has been demonstrated as an efficient DNA or vaccine carrier in the form of polyplex system,47–50 but its potential as a siRNA delivery agent in both polyplex and multicomponent systems has not yet been investigated. Due to apparent differences between siRNA and DNA in terms of size, stability of the formed nucleic acid complexes, and the location and mechanism of action, the same pentablock copolymer efficient in DNA delivery may show different behavior in siRNA delivery.6,51,52 Therefore, it is critical to investigate the eligibility of this copolymer in siRNA delivery systems.
The pentablock copolymers were designed and chosen for the polyplex and multicomponent siRNA delivery systems over the use of cationic polymers, such as polyethylene imine (PEI), because of several advantages. The central triblock Pluronic F127 in the pentablock copolymer contributes to the temperature responsive micellization and has been reported to be able to promote cellular entry while the pH responsive cationic end blocks, PDEAEM, facilitate nucleic acid condensation and endosomal escape.47,48,52–54 Thermoreversible micellization occurs in aqueous solutions because of the lower critical solution temperature (LCST: ∼8 °C) of hydrophobic poly(propyleneoxide) (PPO) block in the middle of Pluronic F127. The presence of hydrophobic PPO chains provide copolymers with the unique ability to be incorporated into cell membranes by enhancing cell interactions and increase translocation of delivery systems into the cells, with minimal damage to the cell membrane integrity.53–55 The end blocks, poly(diethylaminoethyl methacrylate) (PDEAEM), are the essential functional cationic segments (pKa ∼ 7.3) to complex with siRNA and to provide pH buffering capacity at low pH of the endosome with their protonatable tertiary amine groups that aids in the release of entrapped delivery systems from the acidic endosomal vesicles through the proton sponge mechanism.49,55,56 Unlike with the use of cationic polymers such as PEI, the cytotoxicity of these pentablock copolymers can be tuned by changing the balance between the cationic and non-ionic blocks.57 Herein, we present a polyplex system obtained by direct electrostatic interactions between siRNA and pentablock copolymers. We also present a multicomponent system (MCS), developed by the subsequent deposition of siRNA and pentablock copolymers on AuNP surface through cleavable disulfide bonds (to enhance siRNA release) and through the electrostatic interactions (to protect siRNA, enhance cellular uptake, endosomal escape and siRNA activity), respectively (Scheme 1A and B). Both delivery systems were characterized to verify the formation of complexes, the adsorption of each layer on the AuNPs, and the siRNA loading. In addition, their performances in terms of siRNA protection and stability, toxicity, cellular uptake, endosomal escape and transfection efficiencies against luciferase expressing SKOV3 cells were evaluated and compared.
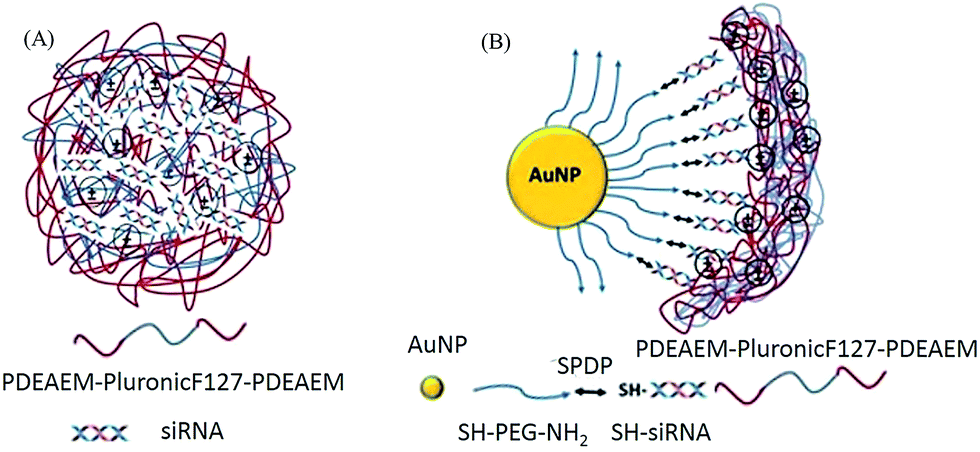 | ||
| Scheme 1 (A) PDEAEM/Pluronic F127/PDEAEM–siRNA polyplexes. (B) AuNP–PEG–siRNA–PDEAEM/Pluronic F127/PDEAEM multicomponent delivery system (MCS). | ||
2 Experimental
2.1 Reagents
Hydrogen tetrachloroaurate(III) trihydrate (99.9%) and sodium citrate used in the preparation of gold nanoparticles were purchased from Sigma-Aldrich. Thiol and amine end group-modified PEG (SH-PEG-NH2: MW 1000) used for gold nanoparticles surface modification was purchased from Creative PEG works. SPDP (N-succinimidyl-3-(2-pyridyldithio)-propionate) cross linker was obtained from Fisher Thermo Scientific. The SKOV3 cell line was purchased from Creative Biogenes. The cell culture media, Dulbecco's Modified Eagle Medium (DMEM) and cell culture media additives, fetal bovine serum (FBS), MEM non-essential amino acids (NEAA) and penicillin streptomycin were obtained from Invitrogen. The fluorescence dyes, Lysotracker Red and Hoechst, used in staining lysosome and nucleus respectively, were obtained from Invitrogen. Cell titer96 and Luciferase assay kits were obtained from Promega. The Quant-iT™ Ribogreen® siRNA detection kit was purchased from Invitrogen. The thiol modified siRNA sequence (siRNA: sense: 5′ HS-GAUUAUGUCCGGUUAUGUA-UU 3′; antisense: 5′ UACAUAACCGGACAUAAUC-UU 3′) was obtained from IDT-DNA. All the buffers were prepared by using RNase free ultrapure water according to standard laboratory procedures.2.2 Preparation of polyplexes
The pentablock copolymers used in this study, PDEAEM–Pluronic F127–PDEAEM, were synthesized by atom transfer radical polymerization (ATRP) as explained elsewhere48 and characterized as described in ESI.† siRNA/Polymer polyplexes at various N/P ratios (N/P: molar ratios of nitrogens (N) in the pentablock copolymer to phosphates (P) in siRNA: 100, 75, 50, 25, 10, 5, 2.5) were prepared by adding appropriate quantities of pentablock copolymer solution in 1× Hepes Buffer, pH 7.0 to siRNA solutions. The mixture was briefly vortexed and allowed to incubate at room temperature for 30 min to ensure complexation.The siRNA condensation and formation of the polyplexes was visualized on a 2% (w/v) agarose gel containing 0.5 µg mL−1 ethidium bromide (EtBr) for 30 min at 100 V in a 1× TAE buffer solution (40 mM TrisHCl, 1% (v/v) acetic acid, and 1 mM EDTA). The samples were loaded in wells and electrophoretic mobility of the polyplexes was visualized and image capture were accomplished using a UV-trans illuminator. In addition, the changes in size and zeta potential of the prepared polyplexes in serum-containing cell culture media were observed with respect to time by dynamic light scattering (DLS).
2.3 Preparation of the multicomponent system
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min and washed with PBS to eliminate weakly bound PEGs.
000 rpm for 15 min and washed with PBS to eliminate weakly bound PEGs.
2.4 Characterization of multicomponent systems
The changes in size, size distribution and zeta potential values of prepared MCSs after each modification were determined by using Dynamic Light Scattering (DLS). In addition, the stability of the MSCs in terms of size and zeta potential in serum containing cell culture media were also evaluated with respect to time by DLS. Bare and modified AuNPs were characterized by observing the shift in the unique surface plasma resonance of AuNPs around 520 nm in the UV-vis spectra upon the modification. The PEG modification of AuNPs was verified by transmission electron microscope (TEM) (Tecnai G2 20 S-TWIN). Samples were prepared by dipping a carbon-coated copper grid in 20 µL colloidal solution of naked or modified AuNPs. For visualizing the PEG layer, 10 µL of AuNPs solution was dropped on TEM grid and waited for 3 min, then excess solution was taken back. 2% (v/v) uranyl acetate solution was used for negative staining. Excess negative solution on the grid was removed using a filter paper and residual solution was air-dried.2.5 Performance evaluation of developed systems
Before the flow cytometry analysis, the cells were seeded (2 × 105 cell per well) and grown to 80% confluency. Then, the siRNA/Polymer polyplexes with different N/P ratios, prepared by using AlexaFluor 488 fluorescence dye attached Polymer A, were added to the cells. After 24 h of incubation at 37 °C under 5% CO2 atmosphere, the cells were harvested and washed with PBS and kept cold until the analysis. Then, the fluorescence intensity of the polyplexes within the cells were measured by flow cytometer (BD Biosciences Facs Canto).
The cellular uptake of the MCS was evaluated by inductively coupled plasma mass spectrometry (ICP-MS) measurements. The SKOV3 cells with a concentration of 1 × 105 cells per well were plated in 6-well plates and incubated 24 h at 37 °C in a 5% CO2 atmosphere. Then, the MCS with an AuNP/Polymer ratio of 1/10, 1/50, 1/100 were administered and further incubated for 24 h under same conditions. After incubation, the cells were washed 3 times with PBS in order to remove the MCS remaining on the outer cell membrane, harvested using Trypsin–EDTA and dispersed in 2 mL of cell culture medium. The cells were collected by centrifugation (1000 rpm, 5 min) and the cell pellet was dispersed in 0.5 mL of concentrated HNO3 at 70 °C for 4 h. At the end of serial dilutions, the amount of gold in the cells was analyzed by ICP-MS. The number of AuNPs in cells was calculated as described in ESI.†
2.6 Statistical analysis
Throughout this study, the significant differences between the groups were evaluated using ANOVA analysis by Tukey's method with 95% confidence interval. The results are presented as average ± standard deviation calculated from at least three independent experiments.3 Results and discussion
The properties of the synthesized pentablock copolymers used to form polyplex and MCSs for siRNA delivery are summarized in Table 1 and illustrated in Fig. S1 through S4 in ESI.† The performances of Polymer A and Polymer C (Table 1) with different molecular weights were evaluated for both of the systems against SKOV3 cells in terms of siRNA protection, cellular entry, endosomal escape and siRNA transfection efficiency.| GPC | NMR | ||||
|---|---|---|---|---|---|
| M w (g mol−1) | M n (g mol−1) | M w/Mn | M n (g mol−1) | DP | |
| Pluronic F127 | 13560 |
12540 |
1.08 | ||
| Pluronic F127 macroinitiator | 14810 |
13150 |
1.12 | ||
| PDEAEM–Pluronic F127–PDEAEM (Polymer A) | 32240 |
26650 |
1.2 | 15650 |
3 |
| PDEAEM–Pluronic F127–PDEAEM (Polymer C) | 72450 |
36050 |
2.01 | 18522 |
1.85 |
3.1 siRNA/polymer polyplex system
The siRNA/polymer polyplex system formed mainly by electrostatic interactions between negatively charged siRNA and positively charged polymers is illustrated in Scheme 1A. The polyplex formation was determined through gel retardation as shown in Fig. 1A. Complete complexation of siRNA with Polymers A and C was observed at N/P ratios of 50, 75 and 100. On the other hand, the decrease in the polymer amount resulted in incomplete condensation of siRNA as demonstrated by similar mobilities of the naked siRNA with that of the siRNA condensed on the polymers when the N/P ratio was 1.25 (Fig. 1A.). Fig. 1 also indicated that there was no significant differences in the amount of condensed siRNA for the different polymer types. | ||
| Fig. 1 (A) Gel electrophoresis of siRNA/Polymer polyplexes with Polymer A and Polymer C at various N/P ratios: 1.25, 2.5, 5, 7.5, 10, 25, 50, 75, 100. Control: naked siRNA. (B) RNase and serum stability of the polyplexes prepared by Polymer A at N/P ratios of 10, 25 and 50. Control: naked siRNA and RNase exposed naked siRNA and serum exposed naked siRNA. | ||
Preliminary screening tests for luciferase expression activity in SKOV3 cells indicated that the polyplexes prepared by Polymers A and C with N/P ratios of 75 and 100 caused a significant decrease in luciferase expression at applied doses. However, the toxicity tests showed that the actual reason for the decrease in the luciferase expression was due to severe toxic effect of the applied dose (Fig. S5 in ESI†). On the basis of these results, we decided to use N/P ratios of 10, 25 and 50, at which the siRNA activity can be observed without significant toxicity. It was noted that the longer PDEAEM chains of Polymer C with higher molecular weight (∼36000) than Polymer A caused significant toxic effects (Fig. S6†), which resulted from its excessive cationic charge, biasing the luciferase suppression results (Fig. S7†) for almost all doses and N/P ratios. This situation makes it difficult to ascertain the pure siRNA delivery efficiency of the system with Polymer C, and makes it less feasible for use in biomedical applications. Therefore, all cytotoxicity and luciferase activity results obtained by Polymer C are included in ESI† and the systems with Polymer A are discussed here.
The stabilities of selected polyplexes against RNase enzymes and serum proteins degradation have been evaluated by gel electrophoresis, size and zeta potential measurements. Polyplexes did not show a significant sign of siRNA degradation, polyplex disassembly or dissociation in the presence of RNase or serum proteins as illustrated in Fig. 1B. In addition, it was noted that the zeta potential and size of polyplexes incubated in serum containing cell culture media for 72 h did not change significantly, except for the particular polyplex with N/P ratio of 25 showing slight decrease in zeta potential upon 72 h of incubation (Fig. S8 and S9,† respectively). The size and zeta potential stabilities of the polyplexes was attributed to the hydrophilic PEO chains of Pluronic F127, which plays a vital role in providing shielding and stealth effects, therefore, preventing the aggregation of particles.55 On the other hand, the stabilities of the polyplexes in RNase and serum containing environment could result from both the strong electrostatic condensation/encapsulation of siRNA in the polyplex matrix and shielding effect of Pluronic F127 block, minimizing its interactions with external effects.13–15,17
Besides providing protection and stability against external effects, a proper siRNA delivery system should also facilitate cellular entry and endosomal escape in order to enable efficient transfection. The cellular accumulation of the polyplexes prepared by attaching a fluorescent dye to Polymer A was investigated through flow cytometry. The results indicated that the increase in N/P ratio induced the cellular accumulation of the polyplexes at the end of 24 h of incubation (Fig. 2A). In addition, it was noted that the internalization of the polyplexes prepared at N/P ratio of 50 increased with time as shown in Fig. 2B. The increased amount of polymer used in the polyplex formulations brings about an augmentation in cationic charge (Fig. S8,† 24 h incubation data), inducing cell membrane interactions, as well as the presence of more hydrophobic PPO groups, capable of promoting cell membrane internalization through thermoreversible micellization and hydrophobic interactions.55
 | ||
| Fig. 2 (A) The cellular accumulation of siRNA/Polymer A polyplexes prepared at different N/P ratios. Incubation time: 24 h. (B) Time dependent cellular uptake of siRNA/Polymer A polyplexes (N/P ratio: 50). * Represents statistical significant difference (p < 0.05). | ||
It is known that after an efficient entry into the cells, the nanocarrier should facilitate escape from the endo-lysosomal pathway in order to prevent degradation of siRNA by many enzymes in the structure of the cell.59 The intracellular distribution of the prepared polyplex systems was investigated by confocal microscopy. Fig. 3 shows that most of the polyplexes were captured by lysosomes preventing their accumulation in the cytoplasm. The quantitative analysis indicated that only 58.2% of polyplexes managed to escape from endosome while the rest were captured. Almost 50% entrapment of the polyplexes could be potentially attributed to the location and number of protonatable free tertiary amine groups in the polyplexes. It is anticipated that the bulk complexation of polymers with siRNA, possibly caused embedding of the responsive blocks in polyplex matrix, resulting in potential obstruction of their responsive properties.
 | ||
| Fig. 3 Confocal images of siRNA/Polymer A polyplexes prepared at N/P ratio of 50. Incubation time: 24 h. Green: polyplexes stained by Alexaflour488, Red: lysosome stained by Lysotracker Red, Blue: nucleus stained by Hoechst. | ||
To evaluate the transfection efficiency and toxicity of the siRNA/Polymer polyplexes prepared with Polymer A, they were administered to the SKOV3 cells at different doses (50, 100, 200 nM). All of the polyplexes with Polymer A were found to be nontoxic at all applied doses except that the polyplex prepared at N/P ratio of 50 reduced cell viability to ∼60% when the applied dose was 200 nM (Fig. 4). This may stem from the use of excess polymer amount and cationic charge for the complexation.20,21 The zeta potential values obtained for 24 h of incubation (Fig. S8†) indicated that the increase in N/P ratio from 10 to 50 resulted in significant increase in cationic charge of the polyplexes. Even though the highest zeta potential value was ∼7 mV, it seemed to be sufficient to show moderate toxic effect at the highest dose of 200 nM. The effect of dose on the toxicity was found to be significant only at the highest N/P ratio which could be due to much higher cellular uptake potential of this polyplex than the others as shown in Fig. 2.
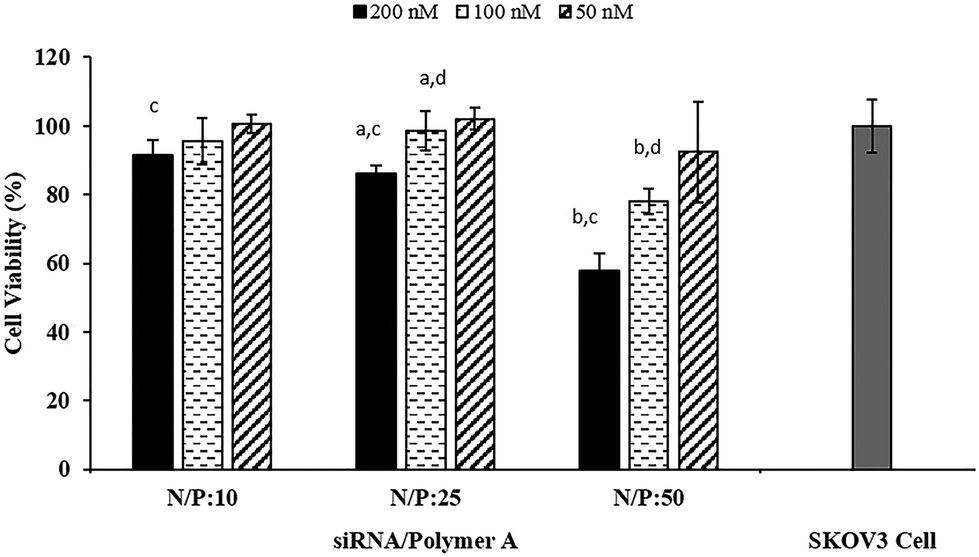 | ||
| Fig. 4 Cell viability of siRNA/Polymer A polyplexes administered to SKOV3 cells. N/P: 10, 25, 50. Applied siRNA dose: 200, 100, 50 nM. Initial SKOV3 cell density: 1.5 × 104 cell per well. Incubation time: 24 h. (a and b) Represent doses of siRNA/Polymer A polyplexes which caused significant difference in cell viability (p < 0.05) for N/P ratios of 25 and 50, respectively. (c and d) Represent the siRNA/Polymer A polyplexes prepared at different N/P ratios which showed significant difference (p < 0.05) in cell viability at doses of 200 and 100 nM, respectively. | ||
Luciferase activity tests indicated that almost all of the polyplexes prepared with Polymer A showed significantly higher luciferase expression suppression without severe toxicity as compared to the naked siRNA (Fig. 5). This result illustrated the necessity of polymer usage to enhance the siRNA transfection efficiency. The polyplexes formed at N/P ratios of 50 were able to suppress more than 60% of the luciferase expression at the maximum applied siRNA dose, 200 nM (Fig. 5). However, the significant toxicity observed at this dose (Fig. 4) led to the conclusion that suppression of the luciferase expression was not completely as a result of siRNA activity but was also due to the toxic effect of the polyplexes. Similarly, gene silencing caused by the polymer alone (without siRNA) at the highest dose of N/P: 50 (control groups in Fig. 5) can be attributed to toxic effect of the polymers on the cells (Fig. 4). The polyplexes induced toxicity since the required amount of polymer to condense 200 nM of siRNA is severely high. At lower doses, the polymers alone (without siRNA) as control exhibited neither significant luciferase expression suppression nor toxicity. Except for this particular case (N/P: 50 and 200 nM dose), for all of the polyplexes selected, ∼50% decrease in luciferase expression was observed without toxicity regardless of the changes in their dose and N/P ratio. The cellular uptake values of the polyplexes varied depending on the N/P ratio (Fig. 2), however, even for the highest uptake (N/P ratio: 50 in Fig. 2A), the most of the complexes were observed to be entrapped in endosome (Fig. 3) which consequently decreased their transfection efficiency. The possible strong electrostatic complexation of siRNA with polymers may provide good siRNA protection, however, it may also prevent efficient siRNA release and transfection efficiency in cytoplasm. Therefore, due to its restricted release, the amount of siRNA required for sufficient gene silencing effect is usually high. This brings about the use of high amount of polymer for a complete condensation of siRNA, which present severe toxicity problems for polyplex systems. Though it is not a direct comparison, the ∼50% decrease in luciferase expression may seem to be relatively low as compared to some findings in the literature obtained with different siRNA sequences and carrier materials against different cell lines.15,19,37,45 Considering this, a direct comparison of the selected polyplexes with a commercial product (Lipofectamine RNAiMax transfection reagent) under same circumstances would give a better idea about the performances of the developed systems. The results in Fig. 5 illustrated that all of the selected polyplexes showed the same luciferase expression suppression (∼50%) as the commercial RNAiMax transfection reagent. Overall, these results suggest that the complexes with Polymer A may be reasonable candidates for siRNA delivery, but without significant advantages over existing systems.
 | ||
| Fig. 5 Luciferase expression suppression of prepared siRNA/Polymer A polyplexes. N/P: 10, 25, 50. Applied siRNA dose: 200, 100, 50 nM. Initial SKOV3 cell density: 1.5 × 104 cell per well. Controls: amount of Polymer A required to form polyplexes possessing N/P ratio 10, 25, 50 (without siRNA), commercial Lipofectamine RNAiMax siRNA transfection reagent, naked siRNA, SKOV3 control cell. Incubation time: 24 h. For N/P ratio of 50, letters a and b on the bars represent doses of siRNA/Polymer A polyplexes or Polymer A alone, respectively, which caused significant difference in luciferase expression suppression (p < 0.05). Letter c represents the significant difference between siRNA/Polymer A polyplex and commercial Lipofectamine RNAiMax transfection reagent in terms of luciferase expression suppression (p < 0.05). *Compared to siRNA/Polymer A polyplexes, Polymer A alone showed significantly lower luciferase expression suppression (p < 0.05) at all N/P ratios and doses applied. ** The luciferase expression suppression values obtained with 200 nM naked siRNA or SKOV3 cells were also found significantly lower (p < 0.05) than the suppression provided by 200 nM siRNA/Polymer A polyplex prepared at all N/P ratios. | ||
3.2 The multicomponent siRNA delivery system
To improve the performance of the pentablock copolymer-based systems, a multicomponent siRNA delivery system based on AuNPs and pentablock copolymers (Scheme 1B) was developed. The first step in the synthesis was modification of the AuNPs with heterobifunctional PEG to increase stability and facilitate the attachment of subsequent layers through cleavable disulfide bonds. In addition to providing conjugation sites for further surface modifications, the PEG layer on AuNPs also acted as a buffer layer avoiding the possible reactions between the bare AuNP surface and disulfide bonds, through which the siRNA attached to the PEG layer, since such interactions have possibility to induce siRNA release.19 The PEG coating resulted in an increase in the cumulative size of the AuNPs from ∼12.5 to ∼18 nm as shown by DLS data represented in Table 2. Despite the increase in size, there was not a significant increase in polydispersity index (PDI) values. The PEG layer coated on the AuNPs was also observed in TEM images (Fig. S10†). The thickness of the PEG layer was evaluated as ∼6 nm from both DLS data (Table 2) and TEM images. Another indication of successful PEG coating was the change in the zeta potential values. Upon PEG attachment, the negatively charged surface of the bare AuNPs (−33.3 mV) became positively charged (+20.2 mV) due to the presence of –NH2 groups at the open end of the PEG (Table 2). The change in the UV-vis spectra of the MCS can also give clues about the success of the surface modifications (Fig. 6). Maximum absorption in the spectra for the 13 nm sized bare AuNPs is observed at 520 nm and a red shift from 520 to 525 nm without severe broadening in the spectra indicated successful PEG coating.| AuNP | AuNP–PEG | AuNP–PEG–SPDP | AuNP–PEG–siRNA | AuNP–PEG–siRNA–Polymer | |
|---|---|---|---|---|---|
| Size (nm) | 12.46 ± 0.66 | 18.09 ± 0.64 | 20.35 ± 0.91 | 31.77 ± 0.11 | 168.93 ± 3.48 |
| PDI | 0.23 | 0.25 | 0.24 | 0.25 | 0.57 |
| Zeta potential (mV) | −33.3 ± 0.98 | 20.23 ± 1.18 | 3.88 ± 1.49 | −2.64 ± 0.67 | 6.98 ± 0.90 |
 | ||
| Fig. 6 UV-vis spectra of AuNP, PEG modified AuNP, SPDP and siRNA modified AuNP–PEG MCS. | ||
In order to introduce disulfide bonds on the particles, the PEG modification was further followed by the SPDP attachment to the AuNP–PEG MCS through the amide bond. The SPDP conjugation was verified by the red shift indicating a movement from 525 (AuNP–PEG) to 527 nm (AuNP–PEG–SPDP) on the absorption maximum in the UV-vis spectra (Fig. 6). Moreover, due to the successful SPDP attachment, there was a decrease in the surface charge from +20.2 to +3.88 mV (Table 2), however, the cumulative size of AuNP–PEG MCS did not increase significantly since the size of the SPDP cross linker is too small.
The deprotected thiol modified siRNA (HS-siRNA) was attached to the AuNP–PEG–SPDP MCS through the thiol-disulfide exchange reaction. The zeta potential of the MCS turned from positive (+3.88 mV) to slightly negative (−2.64 mV) and size increased from ∼20 to ∼32 nm upon attachment of the siRNA (Table 2). In addition, the maximum absorbance wavelength shifted from 527 (AuNP–PEG–SPDP) to 534 nm with significant broadening in the spectra (Fig. 6). The siRNA loaded MCS along with naked siRNA were run in gel electrophoresis. As shown in Fig. 7, the MCS were retarded compared to the naked siRNA and these results provided another evidence of siRNA attachment to the AuNPs.
 | ||
| Fig. 7 Agarose gel electrophoresis retardation of AuNP–siRNA MCS (without polymer) and naked siRNA. RNase and serum protein stabilities of the MCS prepared with Polymer A. AuNP/Polymer ratios: 1/10, 1/50 and 1/100. | ||
The amount of siRNA loaded onto the nanoparticles was determined to be ∼560 nM while the maximum theoretical loading to achieve a complete surface coverage was calculated as 880 nM (see ESI†). The last step in the synthesis of multicomponent system was to coat siRNA loaded AuNPs with pentablock copolymers through electrostatic interactions between negatively charged siRNA and positively charged groups on the copolymer. Table 2 shows that the polymer coating resulted in an increase in zeta potential of the AuNP–PEG–siRNA from −2 mV to +7 mV. The change in the surface charge from negative to positive subsequent to polymer coating is due to the presence of cationic groups of the polymer. In addition, the size of the MCS increased from ∼31 to ∼169 nm (Table 2) and a significant shift in the maximum absorbance wavelength and broadening in the spectra (Fig. 6) were observed upon the assembly of polymer layer on the surface.
The RNase and serum protein stabilities of the MCS were tested through gel retardation. Fig. 7 illustrated that the siRNA loaded MCS with polymer coating on the outer surface showed good siRNA protection against RNAse and serum proteins, since the intensity of these MCS were almost the same with the naked siRNA used as control. This result confirms that the MCS did not show any dissociation in the presence of serum proteins or RNases by protecting loaded siRNA. The stabilities of the multicomponent systems were also observed through the changes in the size and zeta potential values. The results indicated that the zeta potential of MCSs did not change significantly after 72 h of incubation in serum containing cell culture media for the AuNP/Polymer ratios of 1/50 and 1/100 while slight increase in size from ∼160 to ∼170 nm and slight decrease in zeta potential from ∼2 mV to ∼0.5 mV were observed for the MCS prepared with AuNP/Polymer ratios of 1/100 and 1/10, respectively (Fig. S8 and S9†). The amount of polymer used to coat the surface of AuNP–PEG–siRNA was significantly lower than the one required to make polyplexes. It can be claimed that the zeta potential and size stability of MCSs might depend on how the polymers attach to the AuNP–PEG–siRNA surface. Contrary to the case of polyplexes, the polymers are expected to electrostatically attach to the siRNA on the AuNP only from the surface rather than bulk complexation. Therefore, most of the positively charged PDEAEM end blocks would tend to attack siRNA while leaving the middle Pluronic F127 block free close to the outer surface. The hydrophilic PEO blocks of Pluronic F127 on the outer surface enhances the colloidal stability of the system in serum containing cell culture media by providing shielding effect.55 As a result of this shielding effect, it can be said that the MCSs, prepared with Polymer A, possessed relatively better size and zeta potential stability at high AuNP/Polymer ratios (1/50 and 1/100) compared to the polyplexes (Fig. S8 and S9†).
The quantification of MCSs uptaken by the cells was performed by ICP-MS. Fig. 8A shows that the cellular accumulation of MCS significantly increased as the AuNP/Polymer ratio changed from 1/10 to 1/100 at the end of 24 h incubation. With the increased polymer amount, more hydrophobic PPO blocks in Pluronic F127 and more free positively charged PDEAEM blocks on the surface are present and they enhance the cellular entry through hydrophobic and electrostatic interactions, respectively. The time dependent cellular uptake of MCSs prepared with AuNP/Polymer ratio of 1/100 was also investigated (Fig. 8B) indicating that the internalized amount of MCSs increased with time.
 | ||
| Fig. 8 (A) The number of AuNPs in SKOV3 cells measured by ICP-MS. Incubation time: 24 h. (B) Cellular uptake of AuNP–siRNA–Polymer MCS as a function of time. AuNP/Polymer ratio: 1/100. * Represents statistical significant difference (p < 0.05). | ||
Fig. 9 illustrates that the uptaken MCSs were able to escape from endosome and distributed in cytoplasm. Compared to the polyplexes alone shown in Fig. 3, MCSs seemed to provide a better endosomal escape (Fig. 9). The quantitative analysis indicated that 75.6% of the MCSs escaped from endosome while only 58.2% of polyplexes managed to provide endosomal escape. It is anticipated that this is presumably due to the attachment of polymer layer to the AuNP–PEG–siRNA conjugate solely on the surface rather than bulk complexation.60 Alternatively, this situation could have stemmed from the conformation of the polymer on the outer layer of MCSs.61,62 The distribution and therefore, availability of free tertiary amine groups on the surface of MCSs was potentially higher than in the case of the polyplexes alone.
 | ||
| Fig. 9 Confocal images of AuNP–siRNA–Polymer A MCS prepared by AuNP/Polymer ratio of 1/100. Incubation time: 24 h. Green: polyplexes stained by Alexaflour488, Red: lysosome stained by Lysotracker Red, Blue: nucleus stained by Hoechst. | ||
The transfection efficiency and toxicity of AuNP–siRNA–Polymer MCS on the SKOV3 cell line were also evaluated. We have previously shown that the cationic polyplexes are moderately toxic which probably originates from their high doses, excess cationic charge and strong interactions with the cell surface, leading to cell membrane disruption (Fig. 4 and S6†). On the other hand, the AuNP–siRNA–Polymer A MCS, including smaller amounts of cationic polymers in the structure compared to traditional polyplexes, displayed no decrease in cell viability as shown in Fig. 10. Therefore, the developed MCSs provided transfection efficiencies without any toxic damage. The results in Fig. 11 indicated that the MCS without polymer (AuNP–siRNA) did not display any silencing effect while the polymer coated MCSs showed comparatively significant luciferase suppression regardless of their dose or AuNP/Polymer ratio. This result clearly illustrates the benefit of the pentablock copolymer in providing siRNA protection, facilitating cellular entry and endosomal escape and finally enhancing the silencing effect. In general, it was observed that the transfection efficiency of the MCS including polymer A increased with the dose but did not change with the AuNP/Polymer ratio except for the case of AuNP/Polymer ratio of 1/100 at the highest dose (5 nM in terms of AuNP) (Fig. 11). In the literature it was observed that in contrast to the case of polyplex siRNA delivery systems, multicomponent approaches have shown the added advantage of making it possible to achieve higher siRNA activity with lower siRNA loadings and lower polymer concentrations, thereby significantly reducing toxicity.19,37,43,63–67 Herein, we also noticed that compared to the siRNA/Polymer polyplexes, the MCS systems achieved better transfection efficiencies at the same level of siRNA loadings without showing severe toxicity. For instance, the siRNA/Polymer A polyplex formed at N/P ratio of 50 with 100 nM siRNA loading managed to decrease luciferase expression by 50% (Fig. 5) while the multicomponent system prepared with 100 nM siRNA loading (5 nM in terms of AuNP) succeeded in providing ∼65% luciferase expression suppression without causing any toxic effect (Fig. 11). Therefore, to achieve similar gene silencing efficiency with the polyplex systems, larger amounts of siRNA and polymer are needed to form the complexes. However, this may potentially exacerbate the toxicity problem.13 The main reason for better transfection efficiencies of the MCS at the same siRNA doses is the presence of cleavable disulfide bonds in their structure which are broken by the glutathione existing in the cytoplasm facilitating the siRNA release, hence, increasing its luciferase activity within the cell.19,43,63 On the contrary, the strong electrostatic interactions between the polymer and siRNA in the polyplex systems prevent the efficient release of siRNA.17 In addition, the construction of multicomponent systems is also potentially sterically more advantageous by allowing more free blocks of both Pluronic F127 and PDEAEM on the surface as compared to the polyplexes in which part of the blocks are embedded in the structure. The configuration of the polymer on the MCS surface, which stimulates the effect of thermoreversible micellization capability of hydrophobic PPO block, as well as their relatively smaller size and zeta potential values may possibly enhance the cellular entry of the MSC as compared to the polyplexes, leading to higher transfection efficiency. Another reason for higher luciferase suppression of the MCSs was their capability of escaping from endosome and distributing in cytoplasm more efficiently than the polyplexes (Fig. 3 and 9). In order to prove that the developed MCSs are efficient siRNA delivery systems, their performances were compared with the commercial siRNA transfection reagent (Lipofectamine RNAiMax). The results in Fig. 11 showed that the developed MCS (AuNP/Polymer ratio: 1/100 and applied dose 5 nM) provided ∼15% greater decrease in luciferase expression compared to the commercial RNAiMax siRNA transfection reagent. This result also proves the efficiency and superiority of the developed systems.
 | ||
| Fig. 10 Cell viability of AuNP–siRNA–Polymer A MCS treated SKOV3 cells. Applied MCS dose (based on AuNP amount): 5 nM per well. Applied siRNA dose: 100 nM per well. Applied polymer dose: 500 nM per well. Initial SKOV3 cell density: 1.5 × 104 cell per well. Controls: AuNP–PEG, AuNP–PEG–SPDP, uncoated AuNP–siRNA (without Polymer A), Polymer A alone, control SKOV3 cell. Incubation time: 24 h. AuNP–siRNA–Polymer A MCS prepared with different polymer amounts; the layers of this multicomponent system (AuNP–PEG, AuNP–PEG–SPDP and AuNP–siRNA) and Polymer A alone did not cause significantly different cell viabilities compared with the SKOV3 cell alone (p > 0.05). | ||
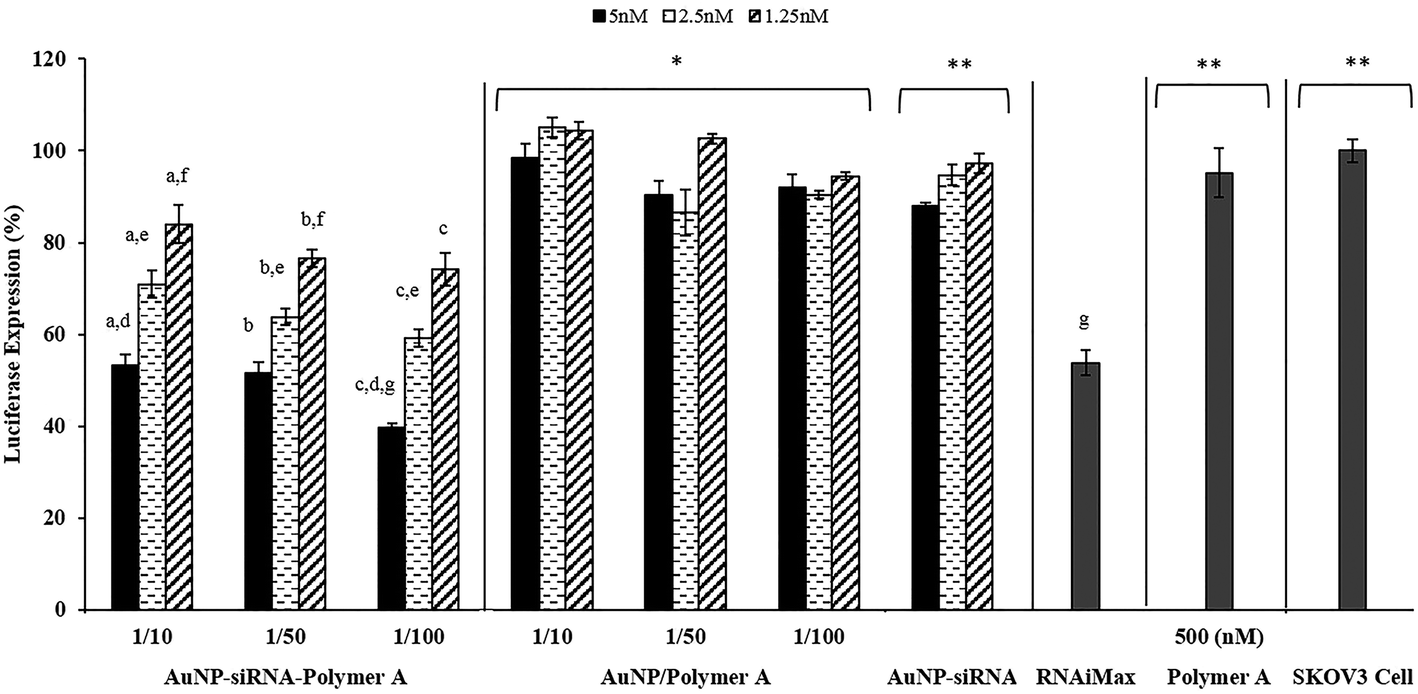 | ||
| Fig. 11 Luciferase suppression of prepared AuNP–siRNA–Polymer A MCS. Applied MCS dose (based on AuNP amount): 5, 2.5, 1.25 nM per well. Applied siRNA dose: 25, 50, 100 nM per well. Initial SKOV3 cell density: 1.5 × 104 cell per well. Controls: AuNP–Polymer A (without siRNA), uncoated AuNP–siRNA (without Polymer A), commercial Lipofectamine RNAiMax siRNA transfection reagent, Polymer A alone, SKOV3 cell. Incubation time: 24 h. (a–c) Represent doses of AuNP–siRNA–Polymer A MCS which caused significant difference in luciferase expression (p < 0.05) for AuNP/Polymer ratios of 1/10, 1/50 and 1/100, respectively. (d–f) Represent the significant difference in luciferase expression suppression of MCSs with different AuNP/Polymer ratio (p < 0.05). (g) Represents the difference between commercial Lipofectamine RNAiMax transfection reagent and prepared MCSs. Only the MCS with AuNP/Polymer ratio of 1/100 and dose of 5 nM significantly decreased the luciferase expression (p < 0.05) compared to RNAiMax while the ones with AuNP/Polymer ratios of 1/10 and 1/50 managed to keep the luciferase expression in the same level as commercial RNAiMax at dose of 5 nM. * Compared to AuNP–siRNA–Polymer A MCS, AuNP/Polymer A MCS showed significantly lower luciferase expression suppression (p < 0.05) at all AuNP/Polymer ratios and doses applied. ** Uncoated AuNP/siRNA MCS, at all doses applied or Polymer A alone or control SKOV3 cell showed significantly lower luciferase expression suppression than AuNP–siRNA–Polymer A MCS. | ||
The MCSs prepared by Polymer C showed severe toxicity as indicated in Fig. S11.† The toxic effect of Polymer C led to conflicting results in luciferase activity of siRNA about the real source of luciferase suppression (due to pure siRNA activity or toxicity of Polymer C). The transfection efficiencies of the MCS fabricated with polymer C were found to be similar to those of the AuNP–siRNA–polymer A MCS (Fig. S12†). On the other hand, when toxicity profile in Fig. S11† is considered, it becomes clear that most of the decrease in the luciferase expression is not due to siRNA activity but because of the cell death resulting from the toxic effect of the MCS. This toxic effect could be due to the massive cationic nature of Polymer C involving longer PDEAEM chains. The result clearly demonstrated that the length of the cationic blocks and the ratio between the cationic and non-cationic blocks in the pentablock copolymers has a significant influence on the toxicity of the MCS as it determines the amount of positively charged groups, hence, the degree of interaction with the cell membrane. This allows us to easily tune the cytotoxicity of the pentablock copolymers by varying the lengths of the cationic blocks and the ratio between the cationic and non-ionic blocks.
4 Conclusion
We have developed and demonstrated the efficacy of PDEAEM–PluronicF127–PDEAEM pentablock copolymer-based multicomponent for siRNA delivery. The potential of the copolymer was evaluated in the development of the polyplex systems formed through the direct electrostatic interactions with siRNA and multicomponent systems prepared by coating the PEG modified AuNPs with siRNA through the cleavable disulfide bonds and subsequently with the pentablock copolymers by electrostatic interactions. The results indicated that both type of delivery systems managed to protect siRNA from external effects and maintain the system stability (in terms of size and zeta potential) in serum supplemented cell culture media. The presence of hydrophobic PPO blocks in both systems enhanced the thermoresponsive micellization and facilitated the cellular entry while the cationic pH responsive PDEAEM block provided the pH buffering in the acidic media of endosome and enhanced the endosomal escape, especially in the multicomponent systems. It was noticed that the nature and form of polymer attachment to siRNA and its conformation upon the attachment is critical for both of the systems in terms of their cell uptake, endosomal escape and transfection efficiency performances. The surface attachment of polymers to siRNA in MCSs reveals the advantages of each block, whereas, the possible embedding of the functional blocks inside the polyplex matrix restricts their properties. Because of these arguments, endosomal escape capabilities of MCSs were seen to be better than that of the polyplex systems alone. These features of MCSs contributed to their higher transfection efficiencies compared to polyplexes. To achieve the same level of luciferase expression suppression, the siRNA loading required in the multicomponent system is lower than that in the polyplex system. Contrary to the case of polyplex systems requiring high polymer amount for complexation with siRNA, this situation brings about lower amounts of polymer use in MCSs to form nontoxic systems. Besides the polymer amounts, it was noted that the manipulation of molecular weight through the ratios or length of each block can be used to tune the toxicity of the polymers. Moreover, the high amount of polymers in polyplex systems forms strong electrostatic complexes avoiding the efficient release and therefore activity of siRNA in cytoplasm. On the other hand, the cleavable disulfide bonds in MCS structure promoting siRNA release in the cytoplasm is one of the key factors enhancing their transfection efficiency. The overall results suggest that the advantage of each block in the pentablock copolymer is highlighted better in the multicomponent conjugate system. When the performance of the developed systems was compared with that of commercial siRNA transfection agent RNAiMax, it was noted that the transfection efficiency with the MCS was ∼15% higher than the commercial product while the efficacy of polyplexes alone were similar to the RNAiMax, indicating the potential of the polymer-based MCS systems. It can be concluded that the responsive pentablock copolymers are promising vectors to deliver siRNA, especially when used in multicomponent systems.Acknowledgements
Metin Uz and Sacide Alsoy Altinkaya would like to thank The Scientific and Technological Research Council of Turkey (TÜBİTAK) Graduate Scholarship Programme and Izmir Institute of Technology (Grant # 2009IYTE01) for their support. Surya K. Mallapragada would like to thank the US Army Medical Research and Materiel Command (Grant number W81XWH-10-1-0806) and the Stanley Endowed Chair in Interdisciplinary Engineering for supporting the work.References
- P. Kesharwani, V. Gajbhiye and N. K. Jain, Biomaterials, 2012, 33, 7138–7150 CrossRef CAS PubMed.
- J. M. Lee, T. J. Yoon and Y. S. Cho, BioMed Res. Int., 2013, 10, DOI:10.1155/2013/782041.
- J. Li, Y. Wang, Y. Zhu and D. Oupicky, J. Controlled Release, 2013, 172, 589–600 CrossRef CAS PubMed.
- R. S. Bora, D. Gupta, T. K. S. Mukkur and K. S. Saini, Mol. Med. Rep., 2012, 6, 9–15 CAS.
- P. Resnier, T. Montier, V. Mathieu, J. P. Benoit and C. Passirani, Biomaterials, 2013, 34, 6429–6443 CrossRef CAS PubMed.
- C. Scholz and E. Wagner, J. Controlled Release, 2012, 161, 554–565 CrossRef CAS PubMed.
- W. J. Dong, Y. J. Zhou and W. Liang, Prog. Biochem. Biophys., 2012, 39, 396–401 CrossRef CAS.
- J. Hoyer and I. Neundorf, Acc. Chem. Res., 2012, 45, 1048–1056 CrossRef CAS PubMed.
- X. Q. Liu, C. Y. Sun, X. Z. Yang and J. Wang, Part. Part. Syst. Charact., 2013, 30, 211–228 CrossRef CAS.
- A. K. R. Lytton-Jean, R. Langer and D. G. Anderson, Small, 2011, 7, 1932–1937 CrossRef CAS PubMed.
- I. Nakase, G. Tanaka and S. Futaki, Mol. BioSyst., 2013, 9, 855–861 RSC.
- Z. W. Wu, C. T. Chien, C. Y. Liu, J. Y. Yan and S. Y. Lin, J. Drug Targeting, 2012, 20, 551–560 CrossRef CAS PubMed.
- B. Ballarin-Gonzalez and K. A. Howard, Adv. Drug Delivery Rev., 2012, 64, 1717–1729 CrossRef CAS PubMed.
- M. Breunig, C. Hozsa, U. Lungwitz, K. Watanabe, I. Umeda, H. Kato and A. Goepferich, J. Controlled Release, 2008, 130, 57–63 CrossRef CAS PubMed.
- A. Elbakry, A. Zaky, R. Liebkl, R. Rachel, A. Goepferich and M. Breunig, Nano Lett., 2009, 9, 2059–2064 CrossRef CAS PubMed.
- C. C. Lee, Y. Liu and T. M. Reineke, Bioconjugate Chem., 2008, 19, 428–440 CrossRef CAS PubMed.
- A. K. Varkouhi, G. Mountrichas, R. M. Schiffelers, T. Lammers, G. Storm, S. Pispas and W. E. Hennink, Eur. J. Pharm. Sci., 2012, 45, 459–466 CrossRef CAS PubMed.
- M. Wagner, A. C. Rinkenauer, A. Schallon and U. S. Schubert, RSC Adv., 2013, 3, 12774–12785 RSC.
- J. S. Lee, J. J. Green, K. T. Love, J. Sunshine, R. Langer and D. G. Anderson, Nano Lett., 2009, 9, 2402–2406 CrossRef CAS PubMed.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- D. Fischer, Y. X. Li, B. Ahlemeyer, J. Krieglstein and T. Kissel, Biomaterials, 2003, 24, 1121–1131 CrossRef CAS PubMed.
- K. Buyens, M. Meyer, E. Wagner, J. Demeester, S. C. De Smedt and N. N. Sanders, J. Controlled Release, 2010, 141, 38–41 CrossRef CAS PubMed.
- J. J. Gu, X. Y. Chen, H. L. Xin, X. L. Fang and X. Y. Sha, Int. J. Pharm., 2014, 461, 559–569 CrossRef CAS PubMed.
- C. E. Nelson, J. R. Kintzing, A. Hanna, J. M. Shannon, M. K. Gupta and C. L. Duvall, ACS Nano, 2013, 7, 8870–8880 CrossRef CAS PubMed.
- S. Prevost, S. Riemer, W. Fischer, R. Haag, C. Bottcher, J. Gummel, I. Grillo, M. S. Appavou and M. Gradzielski, Biomacromolecules, 2011, 12, 4272–4282 CrossRef CAS PubMed.
- T. W. J. Steele, X. B. Zhao, P. Tarcha and T. Kissel, Eur. J. Pharm. Biopharm., 2012, 80, 14–24 CrossRef CAS PubMed.
- C. Troiber, D. Edinger, P. Kos, L. Schreiner, R. Klager, A. Herrmann and E. Wagner, Biomaterials, 2013, 34, 1624–1633 CrossRef CAS PubMed.
- A. Beyerle, O. Merkel, T. Stoeger and T. Kissel, Toxicol. Appl. Pharmacol., 2010, 242, 146–154 CrossRef CAS PubMed.
- A. J. Convertine, D. S. W. Benoit, C. L. Duvall, A. S. Hoffman and P. S. Stayton, J. Controlled Release, 2009, 133, 221–229 CrossRef CAS PubMed.
- S. T. Guo, Y. Y. Huang, T. Wei, W. D. Zhang, W. W. Wang, D. Lin, X. Zhang, A. Kumar, Q. A. Du, J. F. Xing, L. D. Deng, Z. C. Liang, P. C. Wang, A. J. Dong and X. J. Liang, Biomaterials, 2011, 32, 879–889 CrossRef CAS PubMed.
- T. M. Hinton, C. Guerrero-Sanchez, J. E. Graham, T. Le, B. W. Muir, S. N. Shi, M. L. V. Tizard, P. A. Gunatillake, K. M. McLean and S. H. Thang, Biomaterials, 2012, 33, 7631–7642 CrossRef CAS PubMed.
- O. M. Merkel, D. Librizzi, A. Pfestroff, T. Schurrat, K. Buyens, N. N. Sanders, S. C. De Smedt, M. Behe and T. Kissel, J. Controlled Release, 2009, 138, 148–159 CrossRef CAS PubMed.
- M. L. Patil, M. Zhang and T. Minko, ACS Nano, 2011, 5, 1877–1887 CrossRef CAS PubMed.
- T. M. Sun, J. Z. Du, L. F. Yan, H. Q. Mao and J. Wang, Biomaterials, 2008, 29, 4348–4355 CrossRef CAS PubMed.
- M. Y. Zheng, D. Librizzi, A. Kilic, Y. Liu, H. Renz, O. M. Merkel and T. Kissel, Biomaterials, 2012, 33, 6551–6558 CrossRef CAS PubMed.
- L. Zhou, Z. F. Chen, F. F. Wang, X. Q. Yang and B. L. Zhang, Acta Biomater., 2013, 9, 6019–6031 CrossRef CAS PubMed.
- S. H. Lee, K. H. Bae, S. H. Kim, K. R. Lee and T. G. Park, Int. J. Pharm., 2008, 364, 94–101 CrossRef CAS PubMed.
- W. H. Kong, K. H. Bae, S. D. Jo, J. S. Kim and T. G. Park, Pharm. Res., 2012, 29, 362–374 CrossRef CAS PubMed.
- Y. Lee, S. H. Lee, J. S. Kim, A. Maruyama, X. S. Chen and T. G. Park, J. Controlled Release, 2011, 155, 3–10 CrossRef CAS PubMed.
- M. Y. Lee, S. J. Park, K. Park, K. S. Kim, H. Lee and S. K. Hahn, ACS Nano, 2011, 5, 6138–6147 CrossRef CAS PubMed.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- D. Pissuwan, T. Niidome and M. B. Cortie, J. Controlled Release, 2011, 149, 65–71 CrossRef CAS PubMed.
- M. Oishi, J. Nakaogami, T. Ishii and Y. Nagasaki, Chem. Lett., 2006, 35, 1046–1047 CrossRef CAS.
- A. Elbakry, E. C. Wurster, A. Zaky, R. Liebl, E. Schindler, P. Bauer-Kreisel, T. Blunk, R. Rachel, A. Goepferich and M. Breunig, Small, 2012, 8, 3847–3856 CrossRef CAS PubMed.
- S. T. Guo, Y. Y. Huang, Q. A. Jiang, Y. Sun, L. D. Deng, Z. C. Liang, Q. A. Du, J. F. Xing, Y. L. Zhao, P. C. Wang, A. J. Dong and X. J. Liang, ACS Nano, 2010, 4, 5505–5511 CrossRef CAS PubMed.
- L. Han, J. Zhao, X. Zhang, W. P. Cao, X. X. Hu, G. Z. Zou, X. L. Duan and X. J. Liang, ACS Nano, 2012, 6, 7340–7351 CrossRef CAS PubMed.
- A. Agarwal and S. K. Mallapragada, Curr. Top. Med. Chem., 2008, 8, 311–330 CrossRef CAS PubMed.
- M. D. Determan, J. P. Cox, S. Seifert, P. Thiyagarajan and S. K. Mallapragada, Polymer, 2005, 46, 6933–6946 CrossRef CAS.
- B. Q. Zhang, M. Kanapathipillai, P. Bisso and S. Mallapragada, Pharm. Res., 2009, 26, 700–713 CrossRef CAS PubMed.
- J. R. Adams, M. Goswami, N. L. B. Pohl and S. K. Mallapragada, RSC Adv., 2014, 4, 15655–15663 RSC.
- Y. Ping, C. D. Liu, Z. X. Zhang, K. L. Liu, J. H. Chen and J. Li, Biomaterials, 2011, 32, 8328–8341 CrossRef CAS PubMed.
- A. Kwok and S. L. Hart, Nanomedicine, 2011, 7, 210–219 CAS.
- E. V. Batrakova, S. Li, S. V. Vinogradov, V. Y. Alakhov, D. W. Miller and A. V. Kabanov, J. Pharmacol. Exp. Ther., 2001, 299, 483–493 CAS.
- N. S. Melik-Nubarov, O. O. Pomaz, T. Dorodnych, G. A. Badun, A. L. Ksenofontov, O. B. Schemchukova and S. A. Arzhakov, FEBS Lett., 1999, 446, 194–198 CrossRef CAS PubMed.
- A. Agarwal, R. Vilensky, A. Stockdale, Y. Talmon, R. C. Unfer and S. K. Mallapragada, J. Controlled Release, 2007, 121, 28–37 CrossRef CAS PubMed.
- A. Agarwal, R. Unfer and S. K. Mallapragada, J. Controlled Release, 2005, 103, 245–258 CrossRef CAS PubMed.
- A. Agarwal, R. C. Unfer and S. K. Mallapragada, Biomaterials, 2008, 29, 607–617 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 55–75 RSC.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- B. M. Wohl and J. F. J. Engbersen, J. Controlled Release, 2012, 158, 2–14 CrossRef CAS PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- W. Liang and J. K. W. Lam, Endosomal Escape Pathways for Non-Viral Nucleic Acid Delivery Systems, 2012 Search PubMed.
- R. Cheng, F. Feng, F. H. Meng, C. Deng, J. Feijen and Z. Y. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, A. E. Prigodich, P. C. Patel and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 2072–2073 CrossRef CAS PubMed.
- K. Gunasekaran, T. H. Nguyen, H. D. Maynard, T. P. Davis and V. Bulmus, Macromol. Rapid Commun., 2011, 32, 654–659 CrossRef CAS PubMed.
- H. Takemoto, A. Ishii, K. Miyata, M. Nakanishi, M. Oba, T. Ishii, Y. Yamasaki, N. Nishiyama and K. Kataoka, Biomaterials, 2010, 31, 8097–8105 CrossRef CAS PubMed.
- A. K. Varkouhi, R. J. Verheul, R. M. Schiffelers, T. Lammers, G. Storm and W. E. Hennink, Bioconjugate Chem., 2010, 21, 2339–2346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06252g |
| This journal is © The Royal Society of Chemistry 2015 |