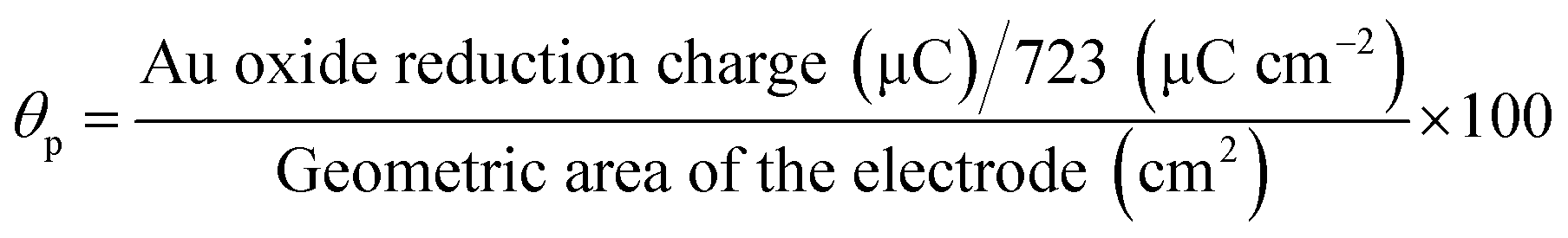DOI:
10.1039/C5RA06537B
(Paper)
RSC Adv., 2015,
5, 42369-42375
Modification of a glassy carbon electrode with gold–platinum core–shell nanoparticles by electroless deposition and their electrocatalytic activity†
Received
12th April 2015
, Accepted 5th May 2015
First published on 5th May 2015
Abstract
Gold–platinum (Au@Pt) nanoparticles with a core–shell structure were fabricated on a glassy carbon electrode (GCE) by an electroless deposition method. Initially, gold nanoparticles (AuNPs) were deposited on GCE by reducing HAuCl4 with NH2OH. SEM studies showed that the size of the deposited AuNPs was found to be 30 nm. The deposited AuNPs on GCE act as the nucleation centre for the deposition of platinum nanoparticles (PtNPs) in the presence of H2PtCl6 and NH2OH. The size of the PtNP increases on increasing the electroless deposition time. The SEM studies demonstrated that the electroless deposition of Pt on Au was isotropic and uniform. Further, Au@Pt modified substrates were characterized by X-ray photoelectron spectroscopy (XPS), an X-ray diffraction method (XRD), energy dispersive X-ray analysis (EDAX) and cyclic voltammetry (CV). XPS showed characteristic binding energies at 71.2 and 74.4 eV for PtNPs and 83.6 and 87.3 eV for AuNPs indicating the zero valent nature of both Au and Pt. Further, the electrocatalytic activity of the Au@Pt modified electrode was examined by studying the reduction of dioxygen and oxidation of hydrazine. The modified electrode showed higher electrocatalytic activity towards the oxidation of hydrazine not only by shifting its oxidation potential towards a less positive potential but also enhancing the oxidation peak current. It also exhibited higher electrocatalytic activity towards the reduction of dioxygen by shifting its reduction overpotential by 650 mV towards a less positive potential when compared to bare GCE.
Introduction
The field of heterogeneous catalysis using bimetallic nanoparticles as catalysts has received great interest in recent years because they offer increased catalytic activity with enhanced stability as compared to their monometallic counterparts.1–3 It has been well established that bimetallic NPs often show novel properties that are not exhibited by either of the monometallic NP.4,5 However, it is difficult to tune the electronic and chemical properties of particular bimetallic NPs relative to the respective monometallic NP. For this reason, the study of bimetallic NPs in the field of catalysis has gained considerable interest. However, it is a important task for researchers to prepare bimetallic NPs with excellent electronic and chemical properties that differ from their individual counterparts.6–8
Among the different bimetallic NPs, Au–Pt bimetallic NPs received huge attention because they can be used for both fuel cell and biosensors applications.9–12 Several methods have been reported in the literature for the synthesis of Au–Pt bimetallic NPs which include radiolytic reduction, photolytic reduction and sol–gel synthesis.13–15 However, these bimetallic NPs are often synthesized in the form of colloidal dispersion or matrix materials by these methods. It is well known that for the fabrication of solid devices for different applications, these Au–Pt NPs must be attached.16–20 Therefore, it is a great challenge for the researchers to develop a simple and efficient method to modify solid substrates with Au–Pt NPs.
“Electroless deposition” is a term coined by Brenner and Riddell to describe the spontaneous reduction of metal ions to metallic particles in the absence of an external source of electric current.21 It includes autocatalytic, substrate catalyzed and galvanic displacement processes.22–27 In the substrate catalyzed deposition, the substrate surface catalyzes the reduction of the metal salt in the presence of a reducing agent. Once the film is completely covered the substrate, reduction ceases because the substrate is no longer exposed.25,26 Electroless deposition has been demonstrated as a feasible method for the preparation of bimetallic catalysts with more targeted placement of even small amounts of the secondary metal.27 Bimetallic catalysts prepared by electroless deposition have been shown to have more intimate contact between the metals than catalysts prepared using traditional wet methods.28 Most importantly, the secondary metal species in these new bimetallic compounds is preferentially deposited onto certain sites of the primary metal, leaving other active sites available for catalysis.28
Synthesis of bimetallic Au–Pt NPs has been already reported in the literature by several methods. Lou et al. prepared the carbon supported Au–Pt bimetallic NPs by two phase synthesis with organic monolayer encapsulation on the nanocrystal core, followed by assembling on carbon black support material by thermal calcinations. They showed that the lattice parameter of bimetallic NPs scales varies linearly with respect to Au–Pt content in the composition.29 De and Rao reported the sol–gel oriented synthesis of Au–Pt alloys embedded in thin SiO2 films and showed the formation of core–shell NPs of Au and Pt at elevated temperatures.30 Li et al. prepared the Au–Pt core shell NPs by immobilizing Au colloids onto ethylenediamine electrografted glassy carbon electrode (GCE) followed by underpotential deposition of copper by galvanic exchange method.31 Schmid et al. reported the preparation of Au–Pt bimetallic NPs with an Au core surrounded by Pt shell via reducing PtCl62− by NH2OH in the presence of gold sol.32 The Au–Pt core–shell NPs were also synthesized through chemical deposition of PtNPs on self-assembled AuNPs.33 Although Au–Pt bimetallic NPs were successfully prepared by different methods, no report is available in the literature for the formation of both Au and PtNPs by electroless deposition method. Thus, the objective of the present study is to prepare Au–PtNPs by electroless deposition and examine their electrocatalytic activity towards oxygen reduction reaction (ORR) and hydrazine oxidation. The Au–Pt NPs prepared by electroless deposition were characterized by XPS, XRD, SEM, EDAX and CV. The SEM images showed that the deposited PtNPs on AuNPs show isotropic structure. The Au–Pt NPs modified electrode showed greater electrocatalytic activity towards ORR and hydrazine oxidation when compared to Au or Pt deposited GC electrodes.
Experimental
Chemicals
Platinic acid (H2PtCl6·6H2O) and hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O) were purchased from Sigma-Aldrich and were used as received. Hydroxylamine hydrochloride (NH2OH·2HCl), hydrazine dihydrochloride (NH2NH2·2HCl) and sulfuric acid (H2SO4) were purchased from Merck, India and were used as received. 0.2 M phosphate buffer (PB) solution was prepared using Na2HPO4 and NaH2PO4. All other chemicals were of analytical grade and were used as received. Double distilled water was used for preparing all solutions. Indium tin oxide (ITO) plates were purchased from Asahi Beer Optical Ltd., Japan.
Instrumentation
XPS measurements were carried out by using Shimadzu Axis 165 high performance multi-technique analysis using an Al Kα source with pass energy of 80 eV, where the pressure in the analysis chamber was lower than 1 × 10−8 Torr and the dwell time was 458 ms. The binding energies for identical samples were reproducible within ±0.10 eV. X-Ray diffraction analysis was carried out with a Rigaku X-ray diffraction unit using Ni-filtered Cu Kα (λ = 1.5406 Å) radiation. Scanning electron microscope (SEM) measurements were carried at VEGA3 TESCAN, USA. Energy dispersive X-ray analyses were carried out using Bruker Nano, Germany. The electrochemical measurements were carried out with CHI electrochemical workstation (Model 643B, Austin, TX). Electrochemical measurements were performed in a conventional two-compartment three-electrode cell with GCE as a working electrode, platinum wire as a counter electrode and NaCl-saturated Ag/AgCl as a reference electrode. All the electrochemical experiments were carried out under nitrogen atmosphere at room temperature.
Electroless deposition of Au–PtNPs on GC electrode
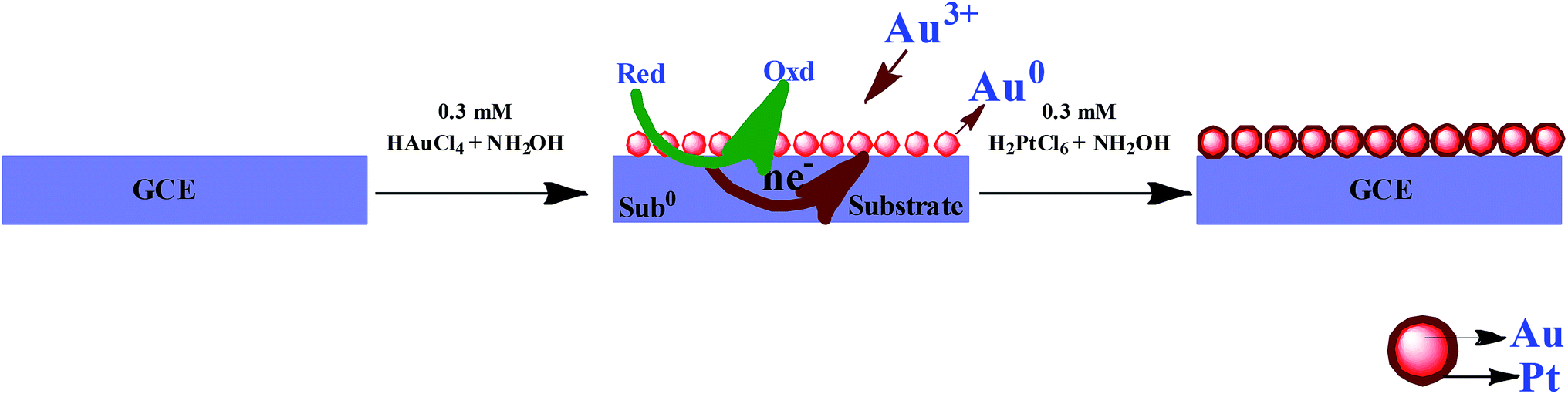
Prior to experiments, the GC electrode was polished with 0.5 μm alumina and then ultrasonically cleaned in water for 5 min. The electroless deposition of AuNPs on GC electrode was carried out by immersing the well cleaned GCE into an aqueous solution of each 0.3 mM HAuCl4 and NH2OH for 30 min. Then, the electrode was removed from the solution and rinsed with double distilled water. The AuNPs were deposited on the GC electrode due to the reduction of Au3+ ions by NH2OH. This electrode is termed as GC/AuNPs electrode. The GC/AuNPs electrode was then immersed into a solution containing 0.01% each H2PtCl6 and NH2OH for 90 min to grow PtNPs. This electrode is termed as GC/Au–PtNPs electrode. Scheme 1 illustrates the schematic representation for the modification of GCE with Au–Pt NPs. For XPS, XRD, SEM and EDAX characterizations, ITO substrates with similar modification were used.
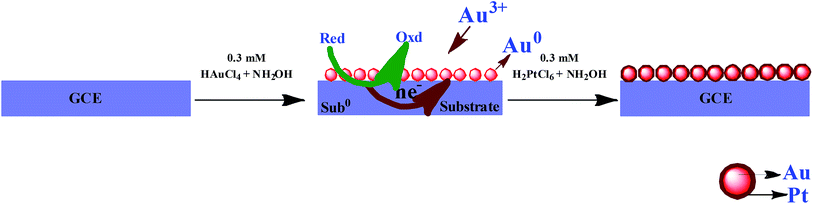 |
| | Scheme 1 Schematic representation for the mechanism of electroless deposition of Au@Pt NPs on GCE. | |
Results and discussion
Mechanism for electroless deposition of Au–PtNPs on GCE
The GC/AuNPs electrode was fabricated by electroless deposition method.26,27 The deposition bath contains HAuCl4 and NH2OH. Since NH2OH is thermodynamically capable of reducing Au3+ ions, the deposition of AuNPs takes place on GC electrode. The initially deposited AuNPs catalyzes further growth of AuNPs and hence the size of AuNPs was increased. The PtNPs were grown on GC/AuNPs electrode by immersing it into the solution containing H2PtCl6 and NH2OH. The AuNPs on GCE act as the nucleation centre for the deposition of PtNPs.33
Characterization of Au–Pt NPs by UV-vis spectroscopy
Fig. S1† shows UV-visible absorption spectra obtained for AuNPs and Au–PtNPs modified ITO substrates. The AuNPs modified substrate shows the characteristic SPR band at 581 nm (Fig. S1,† curve a). When the PtNPs were grown on AuNPs by immersing into a solution containing H2PtCl6 and NH2OH for 30 min, the absorption intensity was decreased. While increasing the immersion time to 45, 90 and 120 min, the SPR band was blue shifted and suppression of the Au plasmon peak was noticed (Fig. S1,† curves b–d). This is in accordance with the report by Liz-Marzan and Philipse.34 They suggested that the Au plasmon absorption peak became suppressed with the increase of surface coverage of Pt and finally vanished when a complete Pt monolayer was formed.34 The obtained spectral results confirmed that the PtNPs were deposited on the AuNPs modified substrate.
Characterization of Au–Pt NPs by XPS
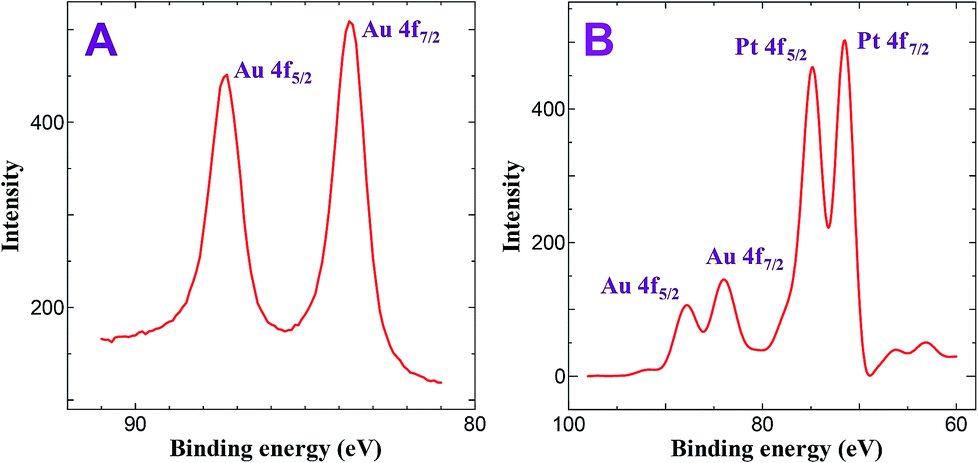
Fig. 1 shows XPS spectrum of AuNPs and bimetallic Au–Pt NPs. XPS of AuNPs deposited on ITO substrate for 30 min shows peaks at 83.6 and 87.3 eV with a difference of 3.7 eV corresponds to the 4f7/2 and 4f5/2 of Au, respectively which is consistent with the zero valent nature of Au (Fig. 1A).35,36 On the other hand, deposition of PtNPs on ITO/AuNPs for 45 min shows four peaks at 71.2, 74.4, 83.6 and 87.3 eV. The obtained 71.2 and 74.4 eV are the characteristic peaks of 4f7/2 and 4f5/2 of zero valent nature of Pt, respectively (Fig. 1B).37 In contrast to Fig. 1A, the intensity of the Au peaks were decreased after the deposition of PtNPs. It has been already reported that when the Pt layer is not thick enough the XPS instrument can detect the signal of Au whereas it cannot detect XPS signal when thick layer of Pt covered the AuNPs.33 Based on this result, it is proposed that the Au–Pt bimetallic nanoparticles should have a core–shell structure.33 Similarly, in the present study, the intensity of the Au signal (Fig. 1A) was dramatically decreased after the deposition of PtNPs on ITO/Au (Fig. 1B). From the decrease in intensity of Au signal, it is assumed that the present method of electroless deposition of PtNPs on AuNPs leads to the formation of core–shell structure, in which Au core being coated by a shell of Pt metal.
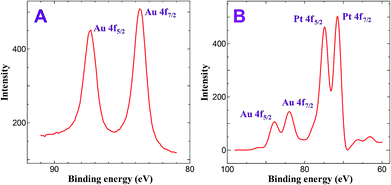 |
| | Fig. 1 XPS obtained for (A) ITO/AuNPs and (B) ITO/Au@Pt NPs. | |
Characterization of Au–Pt NPs by XRD and EDAX
Fig. S2† shows the XRD pattern of the Au–Pt NPs. It illustrates the diffraction features appearing at 34.97°, 38.16°, 44.35°, 50.34°, 55.73°, 60.01°, 74.08° and 77.28°. The peaks at 34.97°, 44.35°, 55.73° and 74.08° corresponding to Au (111), Au (200), Au (220) and Au (311) planes, respectively (JCPDS card no. 04-0802). The peaks at 38.16°, 50.34°, 60.01° and 77.28° corresponding to Pt (111), Pt (200), Pt (220) and Pt (311) planes, respectively (JCPDS card no. 03-065-2870). The peak corresponding to the Pt (111) plane is more intense than the other planes, suggesting that it is a predominant orientation. Besides, compared with Au (111), the peak intensity of Pt (111) is much higher, implying that Au (core)–Pt (shell) orientation. Further, the Au–Pt NPs modified substrate was characterized by EDAX analysis. Fig. S3† shows EDAX spectrum obtained for Au–Pt NPs modified ITO substrate. The peaks at 2.12 and 9.71 keV are the characteristic peaks for Au and 2.05 and 9.44 keV are the characteristic peaks for Pt, confirming the presence of both Au and PtNPs on the ITO substrate and the weight percentage of Pt and Au loading level is 19.5 and 17.28%, respectively.
Characterization by SEM
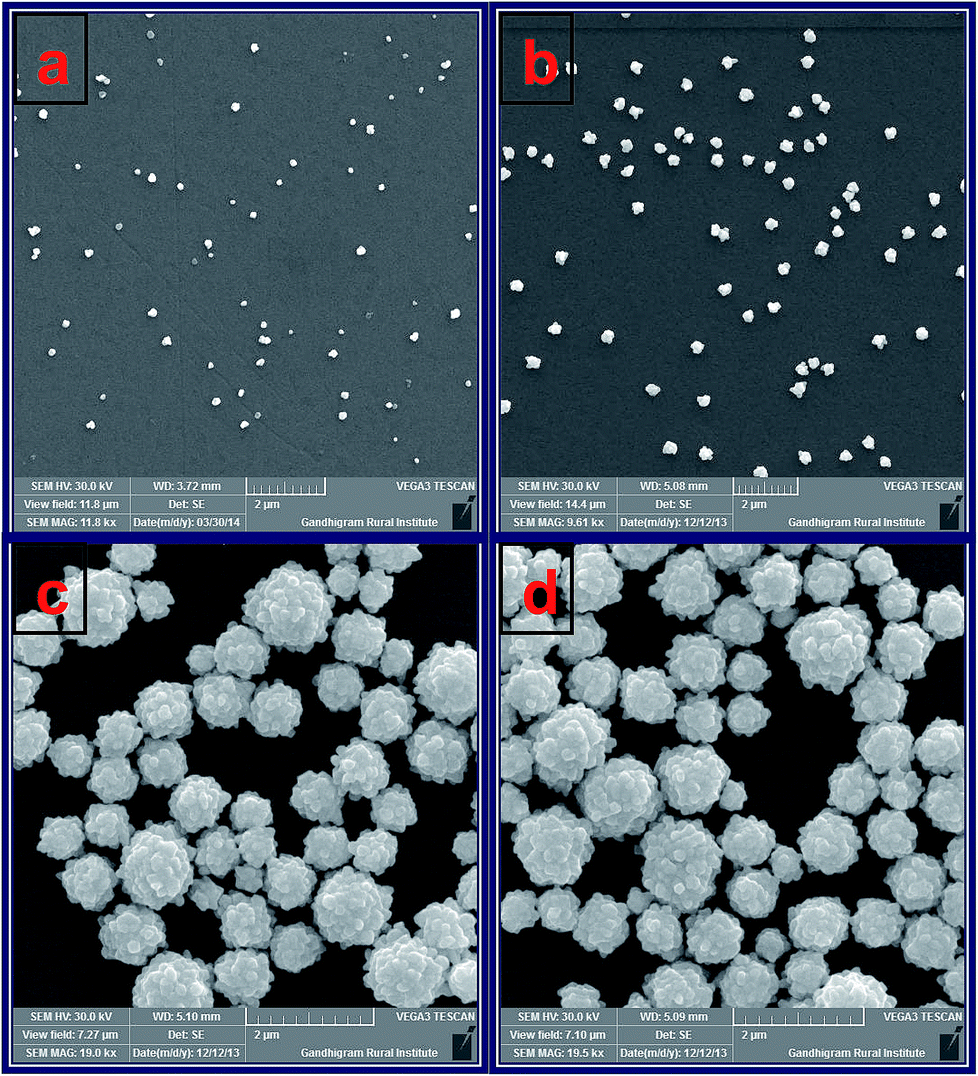
Further, the morphology and size of Au–Pt NPs were characterized by SEM. Fig. 2 shows the SEM images obtained for ITO/AuNPs and ITO/AuNPs immersed into a solution containing H2PtCl6 and NH2OH for 45, 90 and 120 min. The SEM image of ITO/AuNPs shows that the deposited AuNPs were spherical in shape and the size of the AuNP was found to be 30 nm (Fig. 2a). When the AuNPs modified substrate was immersed into the H2PtCl6 and NH2OH solution, the PtNPs were grown on the surface of the AuNPs and this was monitored by SEM. For 45 min immersion, the size of the NPs was found to be increased to 50 nm compared to AuNPs alone. This is due to the reduction of PtCl62− by NH2OH, catalyzed by the AuNPs seeds which are deposited on the ITO substrate. The AuNPs seeds act as the nucleation centre. Hence, the growth of PtNPs was facilitated by the Au seeds deposited on the ITO surface (Fig. 2b). When the immersion time was increased to 90 min, the size of the PtNPs was further increased and the growth of PtNPs was promoted by the initially deposited PtNPs on the AuNPs modified substrate. Here, the initially deposited PtNPs act as the nucleation centre and the growth of PtNPs was found to be high (Fig. 2c). For 120 min, the growth process was attained the saturation due to large size of initially deposited PtNPs (Fig. 2d). These results suggested that the growth of PtNPs on the surface of the AuNPs takes place at the optimum time of 90 min and the formed nanostructures are in the form of Au(core)–Pt(shell). Similar deposition of Au–Pt NPs was also carried out on GC substrate. Fig. S4† shows the SEM images obtained for (a) GCE/AuNPs and (b) GCE/Au@PtNPs for the optimum deposition time of 30 min for Au and 90 min for Pt. The AuNPs modified GCE shows that the NPs were spherical with the average size of 30 nm (Fig. S4a†). When the GCE/AuNPs was modified with PtNPs the size of the NPs was found to be increased to 90 nm (Fig. S4b†).
 |
| | Fig. 2 SEM images obtained for (a) ITO/AuNPs and ITO/AuNPs immersed into the solution containing H2PtCl6 and NH2OH for (b) 45, (c) 90 and (d) 120 min. | |
Electrochemical characterization of GC/Au–PtNPs electrode
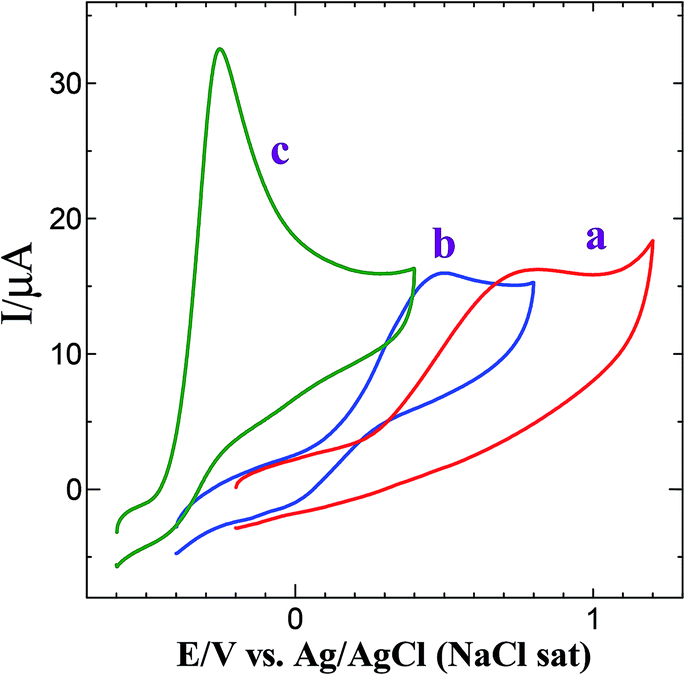
Fig. S5† shows the CVs obtained for AuNPs deposited GC electrode with different electroless deposition time. The oxidation and reduction peaks were observed at +0.90 V and +0.40 V, corresponding to the oxidation and subsequent reduction of gold oxide, respectively. The surface coverage was calculated using the following equation.28,36| |
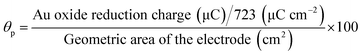 | (1) |
where, 723 μC cm−2 corresponds to the charge of an electrochemically grown monolayer of gold oxide.36,37 The particle coverage was found to be 2.7, 7,1 and 9.2% corresponding to the immersion time of 10, 20 and 30 min, respectively.
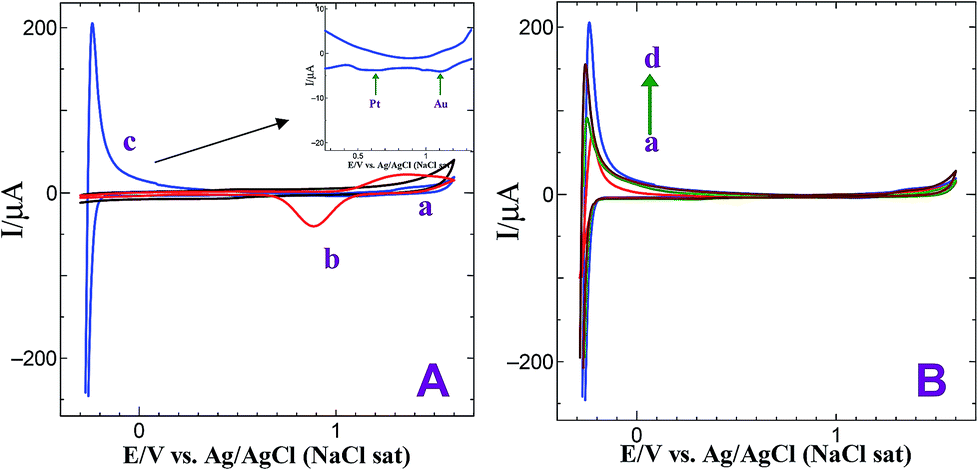
Fig. 3 shows CVs obtained for bare GCE, GCE/AuNPs and GC/Au@Pt NPs electrode prepared with electroless deposition time of 120 min in 0.5 M H2SO4 at a scan rate of 10 mV s−1. Bare GCE does not show any obvious oxidation and reduction peak response in 0.5 M H2SO4 (Fig. 3A, curve a). When the electrode was modified with AuNPs, it shows Au oxidation peak at +1.25 V and Au oxide reduction peak at +0.90 V (Fig. 3A, curve b). On the other hand, when the PtNPs were deposited on AuNPs, exhibit the characteristic hydrogen adsorption–desorption peaks at −0.25 V and the Pt oxide reduction peak at 0.60 V (Fig. 3A, curve c). For different times of immersion, the hydrogen adsorption and desorption peaks were increased and after 90 min time of deposition, were decreased. The deposition of PtNPs was facile on AuNPs deposited at 30 min compared to high or less deposition time of AuNPs. For less than 30 min deposition time, the rate of growth of PtNPs on AuNPs modified substrate is very less whereas for more than 30 min the size of AuNP was found to be high and thus it does not catalyze the reduction effectively. Hence, the optimization time of 30 min was fixed for the deposition of AuNPs. For the GC/Au–Pt electrode, the Au oxide reduction peak at +0.9 V still can be observed but it becomes suppressed with an increasing amount of Pt (Fig. 3A and B). When the Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio is raised by increasing the immersion time, the Au oxide reduction peak was completely disappeared. This reveals that Pt is deposited on the Au surface instead of forming its own nucleus. Moreover, the presence of the Pt oxide reduction peak at +0.60 V along with the less intense Au oxide reduction peak at +0.90 V suggests that few exposed Au sites still remain on the surface. Based on the Au oxide reduction peak areas before and after the deposition of Pt shell, it is estimated that 10% of the originally existed Au is electrochemically active after the deposition of the Pt shell.30
:Au ratio is raised by increasing the immersion time, the Au oxide reduction peak was completely disappeared. This reveals that Pt is deposited on the Au surface instead of forming its own nucleus. Moreover, the presence of the Pt oxide reduction peak at +0.60 V along with the less intense Au oxide reduction peak at +0.90 V suggests that few exposed Au sites still remain on the surface. Based on the Au oxide reduction peak areas before and after the deposition of Pt shell, it is estimated that 10% of the originally existed Au is electrochemically active after the deposition of the Pt shell.30
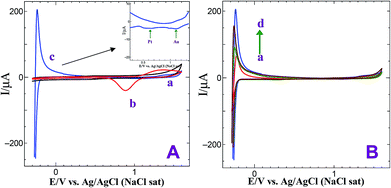 |
| | Fig. 3 (A) CVs obtained for (a) bare GCE, (b) GCE/AuNPs and (c) GC/Au@Pt NPs electrode prepared with electroless deposition time of 120 min in 0.5 M H2SO4 at a scan rate of 10 mV s−1. Inset: gold oxide and platinum oxide reduction peaks. (B) CVs obtained for GC/Au@Pt NPs electrode prepared with electroless deposition time of 30, 45, 90 and 120 min in 0.5 M H2SO4 at a scan rate of 10 mV s−1. | |
Electrocatalytic reduction of dioxygen
The oxygen reduction reaction (ORR) is one of the most important reactions in life processes including biological respiration and in energy converting systems such as fuel cells.| | |
O2 + 4H+ + 4e− → 2H2O
| (2) |
| | |
O2 + 2H+ + 2e− → H2O2
| (3) |
| | |
H2O2 + 2H+ + 2e− → 2H2O
| (4) |
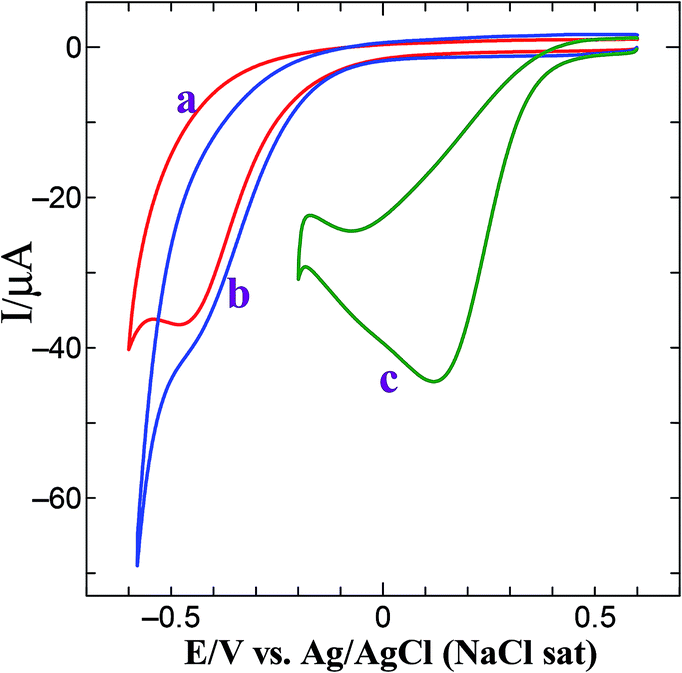
The generalized mechanism for the ORR in acid medium is shown in eqn (2)–(4).38,39 Bulk oxygen approaches the surface by diffusive mass transport where two parallel reaction pathways occur and the dominant reaction pathway is determined by the type of molecular chemisorptions. Depending on the interaction of O2 with the catalyst surface, the oxygen reduction will either proceed via the direct four-electron reduction of oxygen to water (eqn (2)) or two-electron reduction of oxygen as peroxide route (eqn (3) and (4)).38,39 The electrocatalytic activity of the Au@PtNPs towards ORR was examined by cyclic voltammetry. Fig. 4 shows the CVs obtained for bare GCE, GCE/AuNPs and GCE/Au@PtNPs in an oxygen saturated 0.5 M H2SO4. At bare GCE, dioxygen reduction occurs at −0.45 V (Fig. 4, curve a). When AuNPs were deposited on the GCE, the dioxygen reduction overpotential was reduced by 50 mV (−0.40 V) (Fig. 4, curve b). The onset potential for ORR at the GC/Au–PtNPs electrode (0.20 V) (Fig. 4, curve c) is significantly more positive than the bare GCE (−0.45 V) and GC/AuNPs (−0.40 V) electrodes. The GC/Au@PtNPs electrode not only shifted the dioxygen reduction potential to more positive potential but also markedly increased its reduction current when compared to bare and AuNPs modified GCE. The obtained large shift in the onset as well as the reduction peak potential at the GC/Au@PtNPs electrode indicate that the Au@PtNPs efficiently catalyzed the reduction of dioxygen. The electrocatalytic activity of the GC/Au@PtNPs electrode towards dioxygen reduction was further examined by varying the deposition time (30, 45, 90 and 120 min) of PtNPs. The electroless deposition of PtNPs on GC/AuNPs electrode at the deposition time of 90 min shows higher catalytic activity towards dioxygen reduction (Fig. S6,† curve c) compared to other deposition times. This is due to the complete coverage and size effect of the PtNPs. The catalytic activity towards dioxygen reduction decreases when the deposition time was increased to more than 90 min (curve d). This is due to the overgrowth of PtNPs on GC/AuNPs electrode.
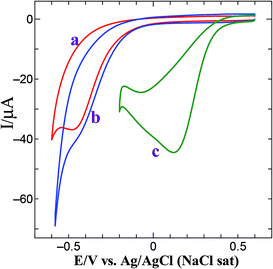 |
| | Fig. 4 CVs obtained for dioxygen reduction at (a) bare GCE, (b) GCE/AuNPs and (c) GCE/Au@Pt NPs electrode in O2 saturated 0.5 M H2SO4 at a scan rate of 50 mV s−1. | |
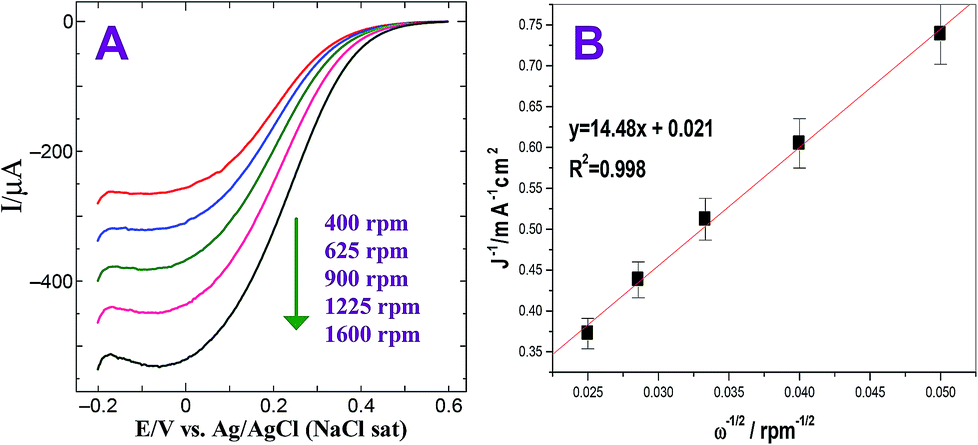
The ORR was also carried out at Au@Pt modified electrode by rotating disc electrode (RDE). Fig. 5A shows the hydrodynamic voltammograms for the ORR at GC/Au@PtNPs electrode in O2 saturated 0.5 M H2SO4 at electrode rotation rates in the range of 400 to 1600 rpm and at a potential scan rate of 50 mV s−1. A gradual increase in the limiting current was observed while increasing the rotation owing to the increase in the mass transport to the electrode surface (Fig. 5A). The observed current density is normalized to the geometric area of the electrode in the respective curves. According to the Koutecky–Levich equation (eqn (5)), the inverse of current density could be given as the sum of the inverses of the above current components. It could also be given in terms of jk, B and ω expressed in revolution per minute (rpm).
where,
j is the current density,
jk is the kinetic current density.
B is evaluated by
eqn (5a).
| | |
B = 0.2nFCO2(DO2)2/3ν−1/6
| (5a) |
where, 0.2 is a constant.
n is the number of electrons transferred per mole of oxygen,
F is Faraday constant (96
485 C mol
−1),
CO2 is the bulk concentration of the oxygen (1.1 × 10
−6 mol cm
−3),
DO2 is the diffusion coefficient of oxygen in sulfuric acid (1.4 × 10
−5 cm
2 s
−1) and
ν is kinematic viscosity of sulfuric acid (1.0 × 10
−2 cm
−2 s
−1).
40,41 The Au@Pt NPs modified electrode (
Fig. 5B) shows the linear relationship of
j−1 vs. ω−1/2. The slope (
B) of the straight line of K–L plot could be used to calculate the number of electrons involved in ORR. The Au@Pt NPs modified electrode shows
B value of 14.48 × 10
−2 mA cm
−2 rpm
−1/2 and its corresponding
n value is 2.46. The theoretical
B value calculated for 2-electron transfer process is 17.2 × 10
−2 mA cm
−2 rpm
−1/2.
40 The experimental
B and
n values are close to the theoretical values. This observation confirms that the Au@Pt NPs modified GCE follows two electron pathway for reduction of oxygen as shown in
eqn (3).
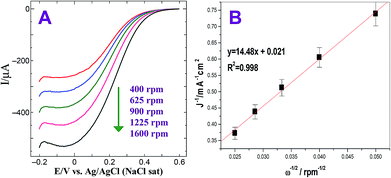 |
| | Fig. 5 (A) Steady-state voltammograms for the ORR obtained at GC/Au@PtNPs electrode in O2 saturated 0.5 M H2SO4. Rotation rate: 400, 625, 900, 1225 and 1600 rpm. Scan rate: 50 mV s−1. (B) Corresponding K–L plot. | |
Electrocatalytic oxidation of hydrazine
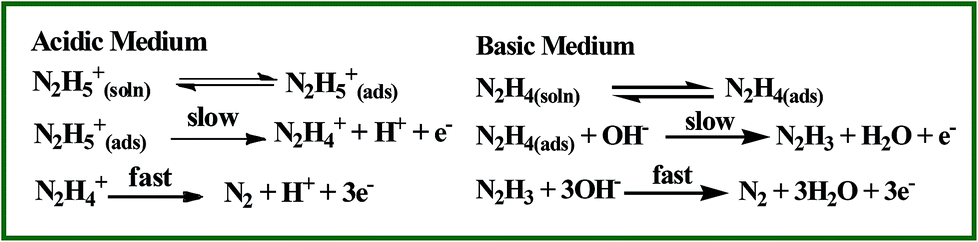
Hydrazine is a highly reactive base and is used as an ideal fuel for direct hydrazine fuel cell. It is a promising one due to its more hydrogen content, higher electromotive force and absence of exhaust of greenhouse gases during oxidation when compared to other fuel cells.42 Thus, the electrochemical oxidation of hydrazine received great interest in the field of fuel cells. The proposed mechanism for electrochemical oxidation of hydrazine in acidic and basic solution at platinum electrode is given in Scheme 2.43 Fig. 6 shows the CVs obtained for bare GCE, AuNPs and Au@PtNPs modified GCEs in the presence of 0.5 mM hydrazine in 0.2 M PBS (pH 7.2) at a scan rate of 50 mV s−1. The bare GCE and AuNPs modified GCE show broad oxidation peaks for hydrazine at +0.73 V and +0.48 V, respectively (Fig. 6; curves a and b). Interestingly, the oxidation peak for hydrazine was shifted towards less negative potential (−0.26 V) with enhanced current at GC/Au@PtNPs modified electrode (Fig. 6; curve c). This infers that the Au@PtNPs modified electrode effectively enhanced the catalytic activity towards hydrazine oxidation when compared to bare and AuNPs modified GCEs. It has been already reported that the hydrazine oxidation depends on the pH of the electrolyte solution.44 Therefore, oxidation of hydrazine at different pH was studied using CV. Fig. S7† shows the oxidation of 0.5 mM hydrazine at Au–PtNPs modified GCE in different pH at a scan rate of 50 mV s−1. The oxidation potential was shifted towards less positive potential upon increasing the pH from 3 to 8 (curves a–f). The plot of potential vs. pH gives a slope value of 90 mV per pH. This value is close to the Nernstian slope of 59 mV per pH at 25 °C, indicating the number of proton and electron in the electrode process is equal. Fig. S8† shows the CVs obtained for 0.5 mM hydrazine at Au@PtNPs modified GCE in PBS (pH 7.2) at different scan rates. The oxidation current of hydrazine was increased while increasing the scan rate. A good linearity between the anodic current of hydrazine and square root of scan rate was obtained with a correlation coefficient of 0.993 (Fig. S8,† inset) at scan rates from 25 to 250 mV s−1. This indicates that the oxidation of hydrazine is a diffusion controlled process at GCE/Au@PtNPs.
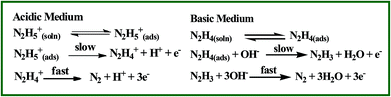 |
| | Scheme 2 Mechanism for electrochemical oxidation of hydrazine in acidic and basic solutions at platinum electrode. | |
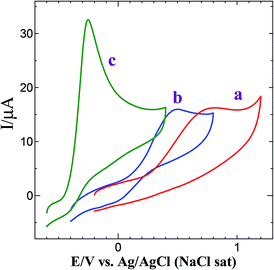 |
| | Fig. 6 CVs obtained for 0.5 mM hydrazine at (a) bare GCE, (b) GCE/AuNPs and (c) GCE/Au@Pt NPs in 0.2 M PBS (pH 7.2) at a scan rate of 50 mV s−1. | |
The Au@Pt modified electrode prepared by the present method showed excellent performance towards hydrazine oxidation compared to the existing Pt and other nanoparticle modified electrodes (Table 1).45–51 The electrode which shows the oxidation of hydrazine at less positive potential is preferred for the analysis of hydrazine. The oxidation potential of hydrazine obtained at the present modified electrode is remarkably less positive potential with respect to the reported modified electrodes.45–51 Although Pd and Au nanoparticle modified electrode could detect hydrazine in the potential range of −0.175 to 0.35 V, the electrode fabrication is time consuming (13–15 h).
Table 1 Comparison of oxidation potential of hydrazine oxidation over various modified electrodesa
| S. no. |
Electrodes |
pH |
Oxidation potential (vs. Ag/AgCl), V |
Reference |
| nRh/CNF – rhodium nanoparticles loaded carbon nanofibers, Au/PdNPs/graphene – gold palladium bimetallic nanoparticles loaded graphene, Ac/Au – gold nanospheres – activated carbon nanocomposites, PtNPs/CNF – platinum nanoparticles coated CNF, PAni/PtNPs – polyaniline – PtNPs nanocomposites, PtNPs/MWCNT – PtNPs loaded multiwalled carbon nanotubes, Au/TiO2NTs – gold–titanium dioxide nanotubes. |
| 1 |
nRh/CNF |
Neutral |
0.38 |
45 |
| 2 |
Au/PdNPs/graphene |
Neutral |
0.2 |
46 |
| 3 |
AC/Au |
Neutral |
0.13 |
47 |
| 4 |
PtNPs/CNF |
Neutral |
1 |
48 |
| 5 |
PAni/PtNPs |
Neutral |
0.439 |
49 |
| 6 |
PtNPs/MWCNT |
Neutral |
−0.35 |
50 |
| 7 |
Au/TiO2NTs |
Alkaline |
0.2 |
51 |
| 8 |
Au@PtNps |
Neutral |
−0.25 |
This work |
Conclusions
The assemblies of Au@Pt core–shell NPs were prepared by electroless deposition method on GC electrode using the concept of substrate catalyzed electroless deposition. XPS results confirmed the zero valent nature of both Au and Pt. SEM images revealed that the deposited AuNPs on GCE and PtNPs on AuNPs/GCE were uniform and isotropic. The GC/Au@PtNPs modified electrode was effectively catalyzed the dioxygen reduction by not only shifting its reduction potential towards less positive potential but also enhanced its reduction current when compared to bare GC electrode. The oxidation of hydrazine was also studied using the present modified electrode. It was found that Au@PtNPs modified electrode effectively catalyzed the oxidation of hydrazine by shifting the oxidation potential to less negative potential with enhanced current.
Acknowledgements
N. S. K. Gowthaman thanks the University Grants Commission (UGC), New Delhi, India for the award of a Meritorious Student Fellowship (F. 7-225/2009(BSR)). Financial support from Department of Biotechnology (BT/PR10372/PFN/20/904/2013), New Delhi, India is gratefully acknowledged.
References
- J. H. Sinfelt, Acc. Chem. Res., 1977, 10, 15 CrossRef CAS.
- J. H. Sinfelt, Bimetallic Catalysts: Discoveries, Concepts and Applications, John Wiley and Sons, New York, 1983 Search PubMed.
- J. H. Sinfelt and G. W. Dembinski, U.S. Pat., US3442973, 1969.
- C. T. Campbell, Annu. Rev. Phys. Chem., 1990, 41, 775 CrossRef CAS.
- J. A. Rodriguez, Surf. Sci. Rep., 1996, 24, 225 CrossRef.
- J. G. Chen, C. A. Menning and M. B. Zellner, Surf. Sci. Rep., 2008, 63, 201 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93 Search PubMed.
- B. N. Wanjala, J. Luo, B. Loukrakpam, B. Fang, D. Mott, P. Njoki, M. Engelhard, J. K. Wu, O. Malis and C. J. Zhong, Chem. Mater., 2010, 22, 4282 CrossRef CAS.
- J. Luo, M. M. Maye, N. N. Kariuki, L. Y. Wang, P. Njoki, Y. Lin, M. Schadt, H. R. Naslund and C. J. Zhong, Catal. Today, 2005, 99, 91 CrossRef PubMed.
- X. H. Kang, Z. B. Mai, X. Y. Zou, P. X. Cai and J. Y. Mo, Anal. Biochem., 2007, 369, 71 CrossRef CAS PubMed.
- N. Kristian, Y. Yan and X. Wang, Chem. Commun., 2008, 353 RSC.
- G. C. Bond, Platinum Met. Rev., 2007, 51, 63 CrossRef CAS.
- M. Harada, K. Asakura and N. Toshima, J. Phys. Chem., 1993, 97, 5103 CrossRef CAS.
- H. Y. Heng, P. C. Gibbons, K. F. Kelton and W. E. Buhro, J. Am. Chem. Soc., 1997, 119, 10382 CrossRef.
- F. Baletto, C. Mottet and R. Ferrando, Phys. Rev. Lett., 2003, 90, 135504 CrossRef CAS.
- R. Crooks and M. Zhao, Adv. Mater., 1999, 11, 217 CrossRef.
- R. Dey and C. R. Raj, Chem.–Asian J., 2012, 7, 417 CrossRef CAS PubMed.
- B. K. Jena and C. R. Raj, Langmuir, 2007, 23, 4064 CrossRef CAS PubMed.
- B. K. Jena and C. R. Raj, Chem. Mater., 2008, 20, 3546 CrossRef CAS.
- H. Ye and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 4930 CrossRef CAS PubMed.
- A. Brenner and E. Riddell, J. Res. Natl. Bur. Stand., 1946, 37, 3 Search PubMed.
- Y. Okinaka and T. Osaka, Adv. Electrochem. Sci. Eng., 1994, 3, 55 CAS.
- Y. Okinaka and M. Hoshino, Gold Bull., 1998, 31, 3 CrossRef CAS.
- G. O. Mallory and J. B. Hajdu, Electroless Plating: Fundamentals and Applications, AESF, Orlando, FL, 1990 Search PubMed.
- M. Kato, J. Sato, H. Otani, T. Homma, Y. Okinaka and O. Yoshioka, J. Electrochem. Soc., 2002, 149, 164 CrossRef PubMed.
- J. Sato, M. Kato, H. Otani, T. Homma, Y. Okinaka and O. Yoshioka, J. Electrochem. Soc., 2002, 149, 168 CrossRef PubMed.
- G. Oskam, J. G. Long, A. Natarajan and P. C. Searson, J. Phys. D: Appl. Phys., 1998, 31, 1927 CrossRef CAS.
- S. Kumar and S. Zou, J. Phys. Chem. B, 2005, 109, 15707 CrossRef CAS PubMed.
- Y. Lou, M. M. Maye, L. Han, J. Luo and C.-J. Zhong, Chem. Commun., 2001, 473 RSC.
- G. De and C. N. R. Rao, J. Mater. Chem., 2005, 15, 891 RSC.
- Y. Li, Q. Lu, S. Wu, L. Wang and X. Shi, Biosens. Bioelectron., 2013, 41, 576 CrossRef CAS PubMed.
- G. Schmid, A. Lehnert, J. Malm and J. Bovin, Angew. Chem., Int. Ed. Engl., 1991, 7, 30 Search PubMed.
- L. Cao, L. Tong, P. Diao, T. Zhu and Z. Liu, Chem. Mater., 2004, 16, 3239 CrossRef CAS.
- L. M. Liz-Marzan and A. P. Philipse, J. Phys. Chem., 1995, 99, 15120 CrossRef CAS.
- P. Kannan and S. A. John, Electrochim. Acta, 2010, 55, 3497 CrossRef CAS PubMed.
- N. Alexeyeva and K. Tammeveski, Anal. Chim. Acta, 2008, 618, 140 CrossRef CAS PubMed.
- T. Matsumoto, T. Komatsu, K. Arai, T. Yamazaki, M. Kijima, H. Shimizu, Y. Takasawab and J. Nakamura, Chem. Commun., 2004, 840 RSC.
- R. A. Sidik and A. B. Anderson, J. Electroanal. Chem., 2002, 528, 69 CrossRef CAS.
- R. S. Arnald, J. Shi, E. Murad, A. M. Whalen, C. Q. Sun, R. Polavarapu, S. Parthasarathy and J. D. Lambeth, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5550 CrossRef PubMed.
- J. J. Salvador-Pascual, S. Citalan-Cigarroa and O. Solorza- Feria, J. Power Sources, 2007, 172, 229 CrossRef CAS PubMed.
- J. Zeng, S. Liao, J. Y. Lee and Z. Liang, Int. J. Hydrogen Energy, 2010, 35, 942 CrossRef CAS PubMed.
- A. Serov and C. Kwak, Appl. Catal., B, 2010, 98, 1 CrossRef CAS PubMed.
- S. Hwang, J. Lee and J. Kwak, J. Electroanal. Chem., 2005, 579, 143 CrossRef CAS PubMed.
- S. V. Guerra, C. R. Xavier, S. Nakagaki and L. T. Kubota, Electroanalysis, 1998, 10, 462 CrossRef CAS.
- G. Hu, Z. Zhou, Y. Guo, H. Hou and S. Sho, Electrochem. Commun., 2010, 12, 422 CrossRef CAS PubMed.
- Y. Liu, B. Li, W. Wei, Q. Wan and N. Yang, Adv. Mater. Res., 2013, 704, 246 CrossRef CAS.
- R. Madhu, V. Veeramani and S.-M. Chen, Sens. Actuators, B, 2014, 204, 382 CrossRef CAS PubMed.
- W. Zhou, L. Xu, M. Wu, L. Xu and E. Wang, Anal. Chim. Acta, 1994, 299, 189 CrossRef CAS.
- A. P. O'Mullane, S. E. Dale, J. V. Macpherson and P. R. Unwin, Chem. Commun., 2004, 1606 RSC.
- S. Chakraborty and C. R. Raj, Sens. Actuators, B, 2010, 147, 222 CrossRef CAS PubMed.
- L. T. Tamsiunaite, J. Rakauskar, A. Balciunaite, A. Zabielaite, J. Vaiciuniene, A. Selskis, R. Juskenas, V. Pahstar and E. Norkus, J. Power Sources, 2014, 272, 362 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis absorption spectra, X-ray diffraction pattern and EDAX spectrum obtained for ITO/Au–PtNPs, SEM images and CVs obtained for GC/AuNPs at different deposition times, dioxygen reduction at GC/Au@PtNPs at different deposition times and effect of pH and scan rates on hydrazine oxidation for Au@PtNPs modified electrode is available in the online version of this article. See DOI: 10.1039/c5ra06537b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.