
Thermoresponsive polymer coated gold nanoparticles: from MADIX/RAFT copolymerization of N-vinylpyrrolidone and N-vinylcaprolactam to salt and temperature induced nanoparticle aggregation†
Samarendra Majia,
Zhiyue Zhangb,
Lenny Voorhaara,
Sophie Pietersc,
Birgit Stubbec,
Sandra Van Vlierberghec,
Peter Dubruelc,
Bruno G. De Geest*b and
Richard Hoogenboom*a
aSupramolecular Chemistry Group, Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4, 9000 Ghent, Belgium. E-mail: richard.hoogenboom@ugent.be
bFaculty of Pharmaceutical Sciences, Department of Pharmaceutics, Ghent University, Ottergemsesteenweg 460, 9000 Ghent, Belgium. E-mail: br.degeest@ugent.be
cPolymer Chemistry and Biomaterials Group, Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4, 9000 Ghent, Belgium
First published on 1st May 2015
Abstract
In the present contribution, we report the MADIX/RAFT polymerization for the synthesis of thermoresponsive homo and statistical copolymers of N-vinylcaprolactam (NVCL) and N-vinylpyrrolidone (NVP). The conditions for the polymerization of NVP were optimized using an automated parallel synthesizer and these optimal conditions were applied for preparing copolymers with systematical variation in composition. The cloud point temperatures (TCP's) of aqueous solutions of PNVCL and P(NVCL-stat-NVP)’s (CP1–CP5) were found to be tuneable between 40 °C and >95 °C at 5 mg mL−1. Next, stable colloidal solutions of AuNPs coated with PNVCL and CP1–CP5 were obtained via an exchange reaction of pre-synthesized citrate stabilized AuNPs with PNVCL and CP1–CP5 by a direct ‘grafting to’ approach. The maximum absorbance wavelength (λmax) of the surface plasmon resonance (SPR) band and size of all the thermoresponsive polymer coated AuNPs were found to be almost unchanged up to 65 °C (above the TCP of PNVCL and CP5) in MilliQ water which is presumably due to electrostatic stabilization of the AuNPs by residual citrate groups on the surface. However, in 0.1 M NaCl aqueous solution the λmax of the thermoresponsive AuNPs were red shifted when heated up to 65 °C which is attributed to the screening of the citrate negative charges on the surface of AuNPs that suppress electrostatic stabilization enabling T-induced aggregation leading to a shift in the SPR band. These thermoresponsive AuNPs may find applications as colorimetric temperature and/or salt sensors.
Introduction
Thermoresponsive polymers presenting a lower critical solution temperature (LCST) in aqueous solution have received great interest in recent decades, due to their wide range of potentially biological relevant applications, like controlled drug delivery, bio-separation, filtration, smart surfaces, and regulating enzyme activity.1 Poly(N-isopropylacrylamide) (PNIPAM) is one of the best known thermoresponsive polymers whose LCST is around 32 °C, which is very close to the human body temperature.2,3 In addition to PNIPAM, poly(oligoethylene glycol(meth) acrylate)s,4 poly(propylene oxide),5 polyvinylether6,7 and poly(2-oxazoline)8,9 also demonstrate LCST behavior in water. Poly(N-vinylcaprolactam) (PNVCL) also attracted significant attention due to its solubility in water, biocompatibility, and hydrolytic stability while it can be synthesized from the easily available inexpensive monomer NVCL by controlled free radical polymerization methods.10–14 In addition to that, PNVCL demonstrates an LCST which is close to body temperature (∼36 °C) and has proven to be a valuable alternative to PNIPAM (LCST ∼ 32 °C) due to its higher biocompatibility than PNIPAM as confirmed by cytotoxicity assays.12Controlled radical polymerization methods (CRP), such as atom transfer radical polymerization (ATRP), nitroxide-mediated polymerization (NMP), reversible addition fragmentation chain transfer (RAFT) and macromolecular design via the interchange of xanthates (MADIX) polymerizations have become key tools for the polymerization of a wide range of conjugated monomers to get well defined polymers with predetermined molecular weight, narrow molecular weight distribution, and high degree of chain end functionality.15–22 However, CRP for unconjugated monomers like N-vinylpyrrolidone (NVP) and NVCL are challenging due to the less activated vinyl groups. In recent years the synthesis of well-defined polymers of these unconjugated monomers via CRP is also getting more successful. CRP of NVP has mostly been reported using MADIX/RAFT.23–25 CRP of NVCL has been reported using cobalt mediated CRP,26–31 ATRP24,32–35 and MADIX/RAFT.36–45 Recently, Liu et al. reported the synthesis of well-defined thermoresponsive block copolymer of PEG-b-PNVCL synthesized by RAFT using a PEG Macro-CTA. Later, this block copolymer was used for coating gold nanorods (GNRs), via a ‘grafting-to’ approach, that were used as smart drug delivery systems (DDS).43 Ponce-Vargas et al. investigated RAFT polymerization of NVCL using two trithiocarbonate CTAs and two initiators 4,4′-azobis(4-cyanovaleric acid) and 4,4′-azobis(4-cyano pentanol) as well as a statistical copolymer of NVCL and NVP.44 However, a systematic kinetic study for the copolymerization of NVCL and NVP has not been reported to the best of our knowledge.
High-throughput parallel synthesis represents a very promising approach in polymer chemistry for speeding-up preparation, increasing reproducibility and enabling investigation of structure–property relationships of the new polymeric materials. Further, parallel synthesis has the advantage of continuous sampling over longer time periods making it ideally suited for studying kinetics of CRP. Although several attempts have been reported for CRP via parallel synthesis, namely ATRP,46,47 NMP48,49 and RAFT,50–53 we are only aware of one paper in which the homo- and copolymerization of butyl acrylate in an automated parallel synthesizer by MADIX polymerization was reported.54
Thermoresponsive polymer coated AuNPs have attracted much interest due to their response to external stimuli, such as temperature, pH and salt.43,55–63 This kind of “smart” nanomaterials open up a new area of intense research due to its potential application as an active delivery vehicle in biomedical science, sensors, catalysis and nanoelectronics.56 Different methods have been used for the synthesis of thermoresponsive polymer coated AuNPs.39,57–59 Yusa et al. showed that the SPR of 18 nm diameter AuNPs (prepared by citrate reduction) coated with thiol functionalized PNIPAM showed a red-shift with increasing NaCl concentration, However, without NaCl, PNIPAM@AuNPs does not show thermoresponsive aggregation behavior upon heating up to 40 °C.60 We also observed that in pure water PNIPAM@AuNPs prepared via ‘grafting to’ approach were found insensitive to temperature up to 40 °C, whereas in 0.1 M aqueous NaCl, it showed temperature-responsive behavior.58 Beija et al. demonstrated thermoresponsive PNVCL coated AuNPs (prepared by NaBH4 reduction) that presented a sharp reversible response to temperature.39
In the present contribution, we report the kinetic investigations for the polymerization of NVP with seven different monomer/initiator ([M]/[I]) ratios to optimize the polymerization to obtain well defined PNVP by MADIX/RAFT polymerization, using an automated parallel synthesizer (Fig. 1). The optimal reaction conditions resulting from the kinetic investigations were then used for the synthesis of thermoresponsive PNVCL and several statistical copolymers of NVCL and NVP, P(NVCL-stat-NVP) (CP1–CP5) with variable TCP's by varying the [NVCL]/[NVP] ratio (Scheme 1). These thermoresponsive PNVCL and (CP1–CP5) were used for the preparation of thermoresponsive polymer coated AuNPs without the need to reduce the xanthate end groups. In addition, we will discuss the thermoresponsive aggregation behavior of PNVCL and P(NVCL-stat-NVP) coated AuNPs induced by aqueous NaCl solution.
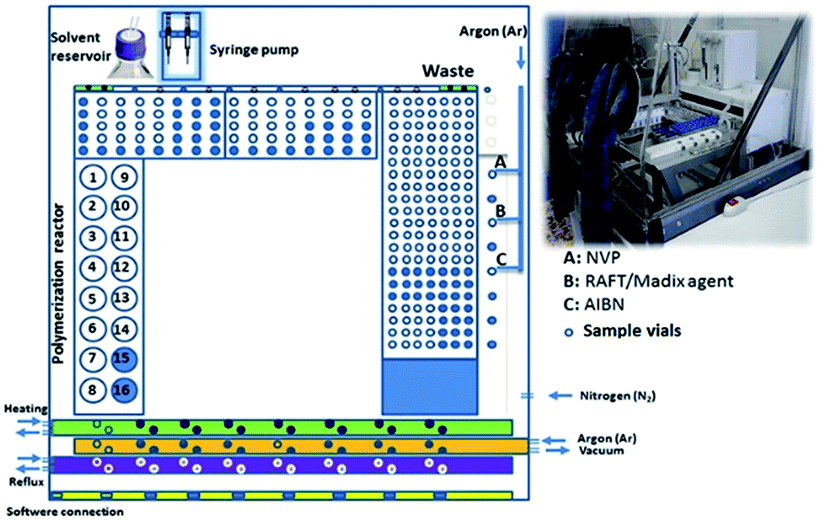 | ||
| Fig. 1 Layout of the ASW2000 synthesizer of 14 parallel NVP polymerizations in a 16-vessel reactor block as used for the kinetic screening. | ||
 | ||
| Scheme 1 Reaction scheme for the MADIX/RAFT homo-polymerization of NVP and homo- and copolymerization of NVCL. | ||
Experimental section
Materials
N-Vinylpyrrolidone (NVP, 99%, Acros) was purified by vacuum distillation. N-Vinylcaprolactam (NVCL) was purified by recrystallization from hexane and stored at 4 °C. 2,2′-Azobis(isobutyronitrile) (AIBN), gold(III)chloride trihydrate and trisodium citrate dihydrate were obtained from Sigma-Aldrich. AIBN was recrystallized twice from methanol before use. HPLC grade solvents N,N-dimethylacetamide (DMA), diethyl ether and dichloromethane were obtained from Sigma Aldrich, N,N-dimethylformamide (DMF) from Biosolve and n-hexane from Fischer Scientific. All other solvents and chemicals used in this work were analytical grade and used without further purification. MilliQ water was used for all the experiments. The xanthate CTA [(S)-2-(ethyl propionate)-(O-ethyl xanthate)] was synthesized by a general procedure described previously.64Instrumentation
The main paragraph text follows directly on here. Gas chromatography was performed on a 7890A from Agilent Technologies with an Agilent J&W Advanced Capillary GC column (30 m, 0.320 mm, and 0.25 mm). Injections were performed with an Agilent Technologies 7693 auto sampler. Detection was done with a FID detector. Injector and detector temperatures were kept constant at 250 and 280 °C, respectively. The column was initially set at 50 °C, followed by two heating stages: from 50 °C to 120 °C with a rate of 20 °C min−1 and from 100 °C to 300 °C with a rate of 50 °C min−1, and then held at this temperature for 0.5 minutes. Conversions of the monomers were determined based on the integration of monomer peaks using the polymerization solvent, anisole, as an internal standard.Size-exclusion chromatography (SEC) was performed on a Agilent 1260-series HPLC system equipped with a 1260 online degasser, a 1260 ISO-pump, a 1260 automatic liquid sampler, a thermostatted column compartment, a 1260 diode array detector (DAD) and a 1260 refractive index detector (RID). Analyses were performed on two Mixed-D (Agilent) 30 cm columns and a Mixed-D precolumn (Agilent) in series at 50 °C. DMA containing 50 mM of LiCl was used as eluent at a flow rate of 0.593 mL min−1. The spectra were analysed using the Agilent Chemstation software with the GPC add on. Molar mass and dispersity (Đ) values were calculated against PMMA standards. The size and zeta potential of the AuNPs (concentration of 0.09 mg mL−1) were measured using a Malvern Zetasizer Nano Series operating a 4 mW He–Ne laser at 633 nm. All samples were filtered through a 0.2 μm filter prior to analysis. Analysis was performed at an angle of 173° at temperatures from 20 to 65 °C at a heating rate of 1.0 °C min−1. Measurements at a 90° scattering angle were performed in a Zetasizer Nano S90 (backscattering mode). Both methods measure the rate of the intensity fluctuation and the size of the particles is determined through the Stokes–Einstein equation (d(H) = kT/3πηD) where d(H) is the mean hydrodynamic diameter, k is the Boltzmann constant, T is the absolute temperature, η is the viscosity of the dispersing medium, and D is the apparent diffusion coefficient.
Cloud points were measured on a Crystal16™ parallel crystallizer turbidimeter developed by Avantium Technologies connected to a recirculation chiller and dry compressed air. Aqueous polymer solutions (5 mg mL−1) were heated from 20 to 95 °C with a heating rate of 1.0 °C min−1 followed by cooling to 2 °C at a cooling rate of 1.0 °C min−1. This cycle was repeated two times. The cloud point temperatures (TCP's) are reported as the 50% transmittance temperature in the 2nd heating run.
UV-vis spectra were measured with a Cary 300 Bio UV-visible spectrophotometer with a Peltier temperature controlled cuvette holder. Citrate and polymer coated AuNPs were placed in plastic cuvettes, and spectral analysis was performed in the 300–800 nm range at 25 °C. In the case of temperature-dependent measurements (AuNPs concentration of 0.09 mg mL−1), the temperatures were varied from 20 to 65 °C.
TEM specimens were prepared by immersion of a nickel grid with carbon-support film into the polymer solution followed by careful wiping off the excess solution. Polymer coated AuNPs that had adhered to the carbon film were investigated by means of various TEM modes: conventional and high-resolution (HR) TEM. A microscope JEOL JEM2200FS-Cs-corrected, operated at 200 kV and equipped with Schottky-type FEG, JEOL JED-2300D and JEOL in-column omega filter was used.
General procedure for the parallel MADIX/RAFT polymerizations
The main paragraph text follows directly on here. Reactions were performed using a Chemspeed ASW2000 automated synthesizer equipped with 16 parallel reactors with a volume of 13 mL, a Huber Petite Fleur thermostat for heating/cooling, a Huber Ministat 125 for reflux and a Vacuubrand PC 3000 vacuum pump (Fig. 1).65 Stock solutions of all components were prepared and bubbled with argon for at least 30 minutes before being introduced into the robot system and then kept under an argon atmosphere. The hood of the automated synthesizer was continuously flushed with nitrogen and the reactors were flushed with argon to ensure an inert atmosphere. Before starting the polymerization, the reactors were degassed through ten vacuum–argon cycles. Stock solutions were transferred to the reactors using the syringe of the automated synthesizer leading to a polymerization mixture with the desired ratio of reagents and a total volume of 6 mL. During the reactions, 150 μL samples were taken into 1.5 mL sample vials, at preset time intervals.Kinetic investigation for the MADIX/RAFT polymerization of NVP using the automated parallel synthesizer
Stock solutions of the monomer (NVP), CTA [(S)-2-(ethyl propionate)-(O-ethyl xanthate)] and the initiator AIBN in anisole were transferred into the 13 mL reaction vessels resulting in different ratios of [CTA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[AIBN] of 1:0.1, 1:0.2, 1:0.3, 1:0.4, 1:0.5, 1:0.7, and 1:1, a [NVP]:[CTA] ratio of 150 and 3 M monomer concentration (Scheme 1). All reactions were performed in duplicate for checking the reproducibility (Fig. 1). The mixtures were heated to 60 °C and vortexed at 600 rpm for 24 h with the reflux condensers set to 5 °C. During the 24 h, samples were automatically taken from the reaction mixtures at suitable time intervals. The samples were analyzed by GC to determine the conversion and theoretical molar masses, and SEC (DMA) to determine the molar mass with respect to PMMA calibration and dispersity (Đ).
:[AIBN] of 1:0.1, 1:0.2, 1:0.3, 1:0.4, 1:0.5, 1:0.7, and 1:1, a [NVP]:[CTA] ratio of 150 and 3 M monomer concentration (Scheme 1). All reactions were performed in duplicate for checking the reproducibility (Fig. 1). The mixtures were heated to 60 °C and vortexed at 600 rpm for 24 h with the reflux condensers set to 5 °C. During the 24 h, samples were automatically taken from the reaction mixtures at suitable time intervals. The samples were analyzed by GC to determine the conversion and theoretical molar masses, and SEC (DMA) to determine the molar mass with respect to PMMA calibration and dispersity (Đ).
MADIX/RAFT statistical copolymerization of NVCL and NVP [poly(N-vinylcaprolactam-stat-N-vinylpyrrolidone)] using the automated parallel synthesizer
Stock solutions of NVCL and NVP, CTA [(S)-2-(ethyl propionate)-(O-ethyl xanthate)] and the initiator AIBN in anisole were transferred into the 13 mL reaction vessels at different ratios of monomers [NVCL]/[NVP]: 30:120, 60:90, 75:75, 90:60, 120:30 and 150:0 with respect to CTA and a [CTA]:[AIBN] ratio of 1:0.3 resulting in 3 M monomer concentration (Scheme 1). All reactions were performed in duplicate to check the reproducibility. The mixtures were heated to 60 °C and vortexed at 600 rpm for 24 h with the reflux condensers set to 5 °C (Fig. 1). During the 24 h, samples were automatically taken from the reaction mixtures at suitable time intervals. All the samples were analyzed by GC to determine the conversion and theoretical molecular weights, and SEC (DMA) to determine the molecular weight with respect to PMMA calibration and dispersity (Đ). Finally, the reactions were quenched by exposing the reactors to air and the polymers were precipitated in hexane. Further, the polymers were dissolved in dichloromethane (DCM) and re-precipitated in hexane three times and were recovered as a white powder. The purified polymers were analyzed by SEC and 1H NMR spectroscopy (ESI Fig. S1†).
Synthesis of citrate stabilized AuNPs
Citrate stabilized AuNPs (citrate@AuNPs) were synthesized by the Turkevich method following reported literature procedures.58,66 All glassware was first washed with aqua regia and then several times rinsed with MilliQ water prior to synthesis. Briefly, 20 mL of 1 mM HAuCl4 was refluxed for 30 min. Then 2 mL of 1 wt % sodium citrate was quickly added and the color of solution changed from yellow to wine red within 5 min. After cooling, the reaction solution was stored at 4 °C for next experiments.Preparation of PNVCL and P(NVCL-stat-NVP) coated AuNPs
Thermoresponsive polymer coated gold nanoparticles (AuNPs) were prepared in the presence of sodium citrate according to the literature procedure.58 A total of 9 mL of a citrate stabilized gold nanoparticles solution was mixed with 200 μL of an aqueous solution containing 8 mg of PNVCL/P(NVCL-stat-NVP) (CP1–CP5) and stirred overnight at room temperature. The resulting polymer coated AuNPs were three times purified by centrifugation at 4 °C at 10000 rpm for 20 min followed by redispersion in pure water.
Results and discussion
Optimization of MADIX/RAFT polymerization of NVP via automated parallel synthesizer
We report on the systematic kinetic investigation of the MADIX/RAFT polymerization of NVP ([NVP] = 3 M in anisole) using [(S)-2-(ethyl propionate)-(O-ethyl xanthate)] as chain transfer agent (CTA) with a [NVP]:[CTA] ratio of 150 and different ratios of [CTA]:[AIBN] of 1:0.1, 1:0.2, 1:0.3, 1:0.4, 1:0.5, 1:0.7, and 1:1 in anisole at 60 °C (Scheme 1). The amount of initiator was varied to optimize the polymerizations with regard to control over the polymerization as well as reaction time. Polymerizations were performed in an automated parallel synthesizer to minimize batch-to-batch variation. In all the 7 combinations of [CTA]/[AIBN], NVP polymerizations were well controlled, as demonstrated by the linear pseudo-first-order kinetics, at least at the early stages of polymerization, and fairly linear evolution of Mn with conversion as well as relatively low Đ’s as shown in Fig. 2A and B. All reactions were performed in duplicate showing good reproducibility. One representative duplicate kinetic studies is plotted in Fig. 2A. From the kinetic plot (Fig. 2A) it is evident that the polymerization rate increases with decreasing [NVP]/[AIBN] ratio, which can be explained by the increasing initiator concentration and thus higher free radical concentration. In the early stages of the reaction the rate of the polymerization increases with increasing the amount of AIBN. However, at higher conversion, the fastest reactions showed more termination, as is evident from a decrease in the slope of the first order kinetic plot, yielding terminated polymer chains and higher dispersities (Đ). As such it can be concluded that a too high AIBN concentration, i.e. more than 0.4 eg relative to CTA, leads to less controlled polymerizations. SEC traces of the PNVP obtained at different times revealed a continuous growth of the polymer chains in time (representative SEC traces for the polymerization of NVP with [CTA]/[AIBN] = [1]/[0.3] are shown in Fig. 2C). Fig. 2D shows the SEC traces of the final polymers (P1–P7) after the end of the reaction. It is evident that a too low amount of AIBN results in very slow polymerization while the SEC traces corresponding to high AIBN concentrations show a high molecular weight shoulder resulting from termination by recombination. Table 1 summarizes the properties of all the polymers demonstrating that the best reaction condition for the polymerization of NVP is [CTA]/[AIBN] = [1]/[0.3] as it showed comparatively good conversion after 24 hours (68%) with the best dispersity (Đ = 1.20) in the series of reactions.
 | ||
| Fig. 2 (A) Pseudo first order kinetic plot for MADIX/RAFT polymerization of NVP in anisole ([NVP] = 3 M) at 60 °C using [(S)-2-(ethyl propionate)-(O-ethyl xanthate)] as chain transfer agent (CTA) with a [NVP] : [CTA] ratio of 150 and different ratios of [CTA] : [AIBN] of 1 : 0.1, 1 : 0.2, 1 : 0.3, 1 : 0.4, 1 : 0.5, 1:0.7, and 1 : 1; (B) corresponding number average molar mass and dispersity (Đ) vs. conversion plot; (C) representative SEC traces (DMA) obtained during the kinetic study of (P3), ([NVP] : [CTA] : [AIBN] = [150] : [1] : [0.3]) and (D) SEC (DMA) traces of the PNVP (P1–P7) obtained after 24 h. | ||
:[AIBN] ratios
| Polymer | [NVP] : [CTA] : [AIBN]a | Convb. [%] | DPc | Mn(GC)d (g mol−1) | Mn(SEC)e (g mol−1) | Đe |
|---|---|---|---|---|---|---|
| a Polymerization in anisole at 60 °C for 24 h.b Determined by GC with anisole as internal standard.c Degree of polymerization (DP) calculated from conversion and the used ratio of [NVP]/[CTA].d Mn,theo. = ([NVP]/[CTA] × conversion × MNVP) + MCTA.e Determined by SEC in DMA containing 50 mM of LiCl at a flow rate of 0.593 mL min−1 using PMMA calibration. | ||||||
| P1 | 150 : 1 : 0.1 | 22 | 33 | 3800 | 3200 | 1.36 |
| P2 | 150 : 1 : 0.2 | 48 | 72 | 8200 | 6300 | 1.28 |
| P3 | 150 : 1 : 0.3 | 68 | 102 | 11500 |
7550 | 1.20 |
| P4 | 150 : 1 : 0.4 | 83 | 125 | 13950 |
9850 | 1.25 |
| P5 | 150 : 1 : 0.5 | 78 | 117 | 13 500 | 8900 | 1.30 |
| P6 | 150 : 1 : 0.7 | 92 | 138 | 15 600 | 10 950 | 1.26 |
| P7 | 150 : 1 : 1.0 | 97 | 146 | 16 400 | 11 000 | 1.26 |
Synthesis of NVCL and NVP copolymers [poly(N-vinylcaprolactam-stat-N-vinylpyrrolidone)] via MADIX/RAFT copolymerization
The optimized experimental procedure for the polymerization of NVP was further used in this work for the synthesis of PNVCL, which is a thermoresponsive polymer, and statistical copolymers of NVCL and NVP (CP1–CP5), as both the monomers belong to the same family of non-conjugated vinyl monomer that can be polymerized via MADIX/RAFT (Scheme 1). The incorporation of the more hydrophilic NVP into PNVCL will allow increasing the TCP. PNVCL and several P(NVCL-stat-NVP) copolymers were prepared using [(S)-2-(ethyl propionate)-(O-ethyl xanthate)] as CTA with different ratios of the monomers [NVCL]/[NVP]: 30:120, 60:90, 75:75, 90:60, 120:30 and 150:0 in respect to CTA and a [CTA]:[AIBN] ratio of 1:0.3 in anisole at 60 °C (Scheme 1). The kinetic plot of the homopolymerization of NVCL is shown in Fig. S2 (see ESI†). Fig. 3A demonstrates the kinetic plots for the copolymerization of NVCL with NVP. The kinetics of the homo and copolymerization revealed linear first order kinetic plots for both monomers indicating a constant free radical concentration indicative for the absence of significant-termination reactions. A linear increase of experimental Mn versus Mn,theo as well as the relatively narrow molar mass distributions (Đ < 1.45) further demonstrate good control over the copolymerization of the two monomers (Fig. 3B). All reactions performed in duplicate showed good reproducibility. One representative duplicate of the copolymer is plotted in Fig. 3A for clarity. The first order kinetic plots for the copolymerisation of NVCL with NVP revealed that NVP is incorporated faster than the NVCL. The kinetic study with equimolar amounts of the monomers revealed a ratio of the NVP and NVCL apparent polymerization rate constants of as high as 1.53 resulting from the ratio of the individual monomer conversion slopes. Polymerization was stopped after 24 h yielding polymers with different molar masses by changing the [NVCL]/[NVP] ratios due to the decrease in polymerization rates with increasing NVCL content. All the obtained polymers exhibit relatively narrow dispersities (Đ = 1.23 − 1.44). The deviation of Mn (measured by SEC) from the theoretical values is attributed to the relative determination of Mn versus PMMA standards in SEC and is also evident in the Mn versus Mn,theo plot of the kinetic study (Fig. 3B). Representative SEC traces of the copolymerization of NVCL and NVP (CP1) are shown in Fig. 3C revealing a continuous increase in molar mass of CP1 during the reaction. Fig. 3D shows the SEC traces of the final polymers PNVCL and (CP1–CP5) after purification. The TCP values of these polymers were measured in MilliQ water by turbidimetry (Table 2) and the corresponding transmittance versus temperature curves are shown in Fig. 4A. Fig. 4 clearly reveals that TCP decreases with higher amounts of hydrophobic NVCL. Furthermore, the good overlap between heating and cooling curves demonstrates that there is nearly no hysteresis in contrast to PNIPAM.67 Ponce-Vargas et al., also observed that copolymers of NVCL and NVP exhibited temperature-responsive behavior with the phase transition temperature (TCP) depending on the NVCL/NVP ratio (Note that TCP values were measured at pH 7.4).44 Among the copolymers CP1 does not show any TCP up to 95 °C due to its too high hydrophilicity. The effect of aqueous NaCl (salt) solution (0.1 and 1.0 M respectively) on the TCP's of the polymers was also investigated. The TCP values were found to decrease only to a minor extent with 0.1 M NaCl and 10–15 °C with 1.0 M NaCl (Fig. 4B). The addition of NaCl leads to a decrease in TCP value due to dehydration of the hydrophobic polymer regions, resulting in more polymer–polymer interactions than polymer water interactions, which is also called as the salting out effect (see ESI Fig. S3†).68
 | ||
| Fig. 3 (A) Kinetic plot of ln([M]0/[M]t) vs. time in the MADIX/RAFT copolymerizations of N-vinylcaprolactam (NVCL) and N-vinylpyrrolidone (NVP) (open symbols: kinetic plots of NVCL and closed symbols: kinetic plots of NVP); (B) experimental number-average molecular weight (Mn) and dispersity (Đ) versus theoretical number-average molecular weight Mn,theo (Mn,theo was calculated from the monomer conversion measured by GC); (C) Representative SEC traces (DMA) obtained at different times during the kinetic study of the statistical copolymer (CP1), ([NVCL] + [NVP]) = 3 M, ([NVCL]:[NVP]:[CTA]:[AIBN] = [30]:[120]:[1]:[0.3]) and (D) SEC (DMA) traces of the statistical copolymers (CP1–CP5) and PNVCL after 24 h. | ||
| Polymer | [NVCL] : [NVP] : [CTA] : [AIBN]a | Conv.b [%] | Molar composition (%) [NVCL]/[NVP](Theo.) | Molar compositionb (%) [NVCL]/[NVP](Expt.) | DPc | Mn(GC)d (g mol−1) | Mn(SEC)e (g mol−1) | Đe | TCPf (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Polymerization in anisole at 60 °C for 24 h.b Determined by GC with acetone as internal standard.c Degree of polymerization (DP) calculated from conversion.d Mn,theo = [([NVCL] × Conv.NVCL × MNVCL) + ([NVP] × Conv.NVP × MNVP)]/[CTA] + MCTA.e Determined by SEC in DMA containing 50 mM of LiCl at a flow rate of 0.593 mL min−1 using PMMA calibration; (Here the reported Mn and dispersity values for CP5 and PNVCL are less accurate as the SEC system peak partially overlaps with the polymer peaks) and.f Cloud point temperature (TCP) determined by turbidimetry with 5 mg mL−1 polymer concentration in water (2nd heating cycle). | |||||||||
| CP1 | 30 : 120 : 1 : 0.3 | 72/85 | 20/80 | 17/83 | 22/102 | 14 550 | 9400 | 1.23 | — |
| CP2 | 60 : 90 : 1 : 0.3 | 63/77 | 40/60 | 35/65 | 38/70 | 13 200 | 8100 | 1.26 | 89 |
| CP3 | 75 : 75 : 1 : 0.3 | 51/66 | 50/50 | 44/56 | 38/49 | 11 000 | 5600 | 1.43 | 75 |
| CP4 | 90 : 60 : 1 : 0.3 | 44/58 | 60/40 | 53/47 | 39/35 | 9500 | 4700 | 1.39 | 72 |
| CP5 | 120 : 30 : 1 : 0.3 | 36/48 | 80/20 | 75/25 | 43/15 | 7800 | 3000 | 1.44 | 49 |
| PNVCL | 150 : 00 : 1 : 0.3 | 30/00 | 100/0 | 100/0 | 45/00 | 6050 | 2200 | 1.40 | 40 |
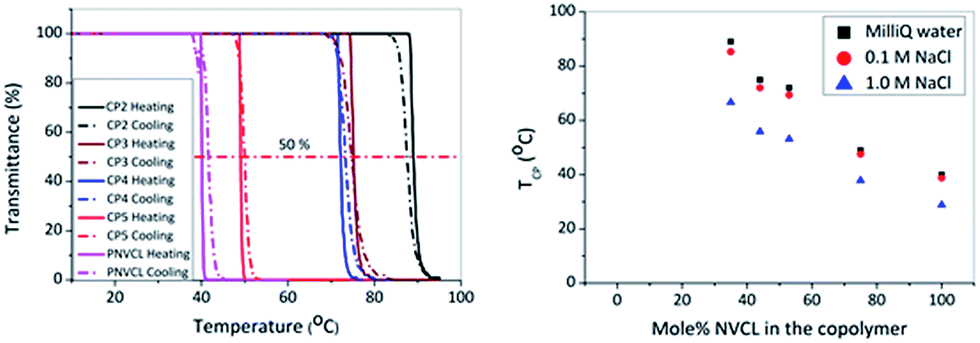 | ||
| Fig. 4 (A) Transmittance vs. temperature plot [overlapping heating (full line) and cooling (dashed line) cycles] of the PNVCL and poly(NVCL-stat-NVP) copolymers and (B) Detected cloud point temperatures (TCP's) in MilliQ water, 0.1 M NaCl and 1.0 M NaCl of the PNVCL and P(NVCL-stat-NVP) copolymers (5 mg mL−1, heating rate 1 °C min−1). | ||
Preparation of characterization of statistical copolymer (CP1–CP5) and PNVCL coated AuNPs
Citrate stabilized AuNPs were synthesized by adopting the Turkevitch and Frens method66 where gold(III)chloride trihydrate was reduced with trisodium citrate dihydrate in aqueous medium under reflux. After refluxing for 30 minutes, a deep red colored solution of citrate@AuNPs was obtained. It is worth to mention that no additional stabilizing agent is required for stabilizing the AuNPs as citrate acts as both a reducing agent as well as a stabilizing agent. The synthesized citrate@AuNPs were first investigated by UV-vis-spectroscopy (Fig. 5A) revealing a comparatively narrow absorption spectrum in MilliQ water corresponding to the surface plasmon resonance (SPR) band with a maximum absorbance wavelength (λmax) at 522 nm when measured at 25 °C (Table 3). It is well-known that the SPR is highly dependent on the shape and the dielectric constant of the microenvironment surrounding the nanoparticle.69 Agglomeration of the AuNPs induces a red-shift of the SPR band yielding a purple/blue solution. Importantly, the synthesized citrate stabilized AuNPs do not show any significant change of the plasmonic properties (SPR) upon heating from 20 °C to 65 °C (Fig. 5B). The size of the citrate@AuNPs was measured by TEM and DLS. TEM analysis reveals a diameter of 15 ± 2 nm (Fig. 6), whereas a mean (intensity) diameter of ca. 29 nm was measured by DLS (scattering angle 90 deg.) at 25 °C (Fig. 5C) (Table 3). This observed difference in the size of the citrate@AuNPs is attributed to the different methods of analysis. Size based on the DLS measurements always shows a higher diameter due to the measurements of hydrodynamic diameter in aqueous medium while TEM was measured in dry state. In addition to the main distribution of the synthesized colloidal AuNPs, an additional smaller size distribution was observed by DLS (below 10 nm), which was demonstrated to be an artefact arising from rotational diffusion of the particles70 and will be discussed in detail later. The citrate@AuNPs showed a zeta potential of ca. −41 mV, indicating the effective stabilization via repulsive electrostatic interactions (Table 3). In order to investigate the effect of the thermoresponsive PNVCL and its statistical copolymers (CP1–CP5) on the AuNPs, MADIX/RAFT functionalized polymer coated AuNPs were prepared by replacing the weakly bound citrate on the AuNPs via a ‘grafting to’ approach. It has been reported that the MADIX/RAFT end group of polymers can efficiently be used to attach them to the gold surface without the need for reduction of the MADIX/RAFT end groups into thiols.57 Excess non-grafted polymers were removed from the polymer coated AuNPs by three centrifugation–redispersion cycles using MilliQ water. Finally the purified polymer coated AuNPs were characterized by UV-vis, TEM, and DLS. | ||
| Fig. 5 (A) Normalized UV-vis absorbance spectra of all the AuNPs measured at 25 °C [inset: Zoomed image of the UV-vis absorbance spectra]; (B) λmax and absorbance as function of temperature of citrate@AuNPs in MilliQ water; (C) Intensity size distribution from of citrate@AuNPs measured at 25 °C [conditions: scattering angle: 90 deg., (backscattering mode)] and D) Photographs of plastic cuvettes at 25 °C containing citrate@AuNPs, (CP1-CP5)@AuNPs and PNVCL@AuNPs in MilliQ water [concentration of the AuNPs: 0.09 mg mL−1]. | ||
| Polymer | Plasmon peak (nm)@ 25 °C (MilliQ water) | Dominant peak size (d nm) (intensity) (DLS)a | PDI (DLS) | Zeta-potential (mV) @25 °C | |
|---|---|---|---|---|---|
| MilliQ water | 0.1 (M) NaCl | ||||
| a Hydrodynamic diameter of polymer@AuNPs at 25 °C were measured by dynamic light scattering (scattering angle: 90 deg.). The concentrations of AuNPs were 0.09 mg mL−1.b Could not measure due to the lack of sample. | |||||
| Citrate@AuNPs | 522 | 29.0 | 0.28 | −41 ± (3) | —b |
| CP1@AuNPs | 526 | 42.3 | 0.38 | −27 ± (2) | —b |
| CP2@AuNPs | 527 | 39.6 | 0.37 | −25 ± (1) | —b |
| CP3@AuNPs | 527 | 36.1 | 0.56 | −29 ± (1) | −12 ± (3) |
| CP4@AuNPs | 526 | 40.9 | 0.50 | −21 ± (1) | −14 ± (1) |
| CP5@AuNPs | 526 | 41.6 | 0.62 | −27 ± (2) | −16 ± (2) |
| PNVCL@AuNPs | 525 | 40.8 | 0.51 | −27 ± (1) | −14 ± (2) |
 | ||
| Fig. 6 Bright field scanning transmission electron microscopy (STEM) picture of the citrate@AuNPs (scale bar = 50 nm) and transmission electron microscopy (TEM) pictures of PNVP-stat-PNVCL (CP1-CP5) and PNVCL coated AuNPs [colored pictures highlight the polymer corona around the AuNPs (scale bar = 10)]. | ||
UV-vis measurements of the polymer coated AuNPs revealed that the polymer modification induced a red shift of the SPR band up to 5 nm and showed comparatively broader spectra than citrate stabilized AuNPs (Fig. 5A) (Table 3). This change in the SPR band is attributed to the grafting of the polymer around the AuNPs that leads to a minor reduction in polarity around the AuNPs compared to citrate.71,72 All the polymer coated AuNPs showed a red color in solution (Fig. 5D), which is in agreement with the UV-vis results.
TEM data of the polymer coated AuNPs undoubtedly confirm the presence of a polymer corona surrounding the AuNPs (Fig. 6). All the polymer coated AuNPs showed an increase in hydrodynamic diameter around 40 nm than the citrate@AuNPs (ca. 29 nm) measured by DLS (Table 3). Fig. 7A shows the main size distribution of the DLS measurements at 90 deg. scattering angle of all the polymer modified AuNPs used in the present investigation. Unexpectedly, all the polymer@AuNPs also showed a smaller particle distribution below 10 nm by DLS in addition to the main distribution of the particles as we also observed for citrate@AuNPs. It has been reported that colloidal gold can reveal a second peak at a smaller size (below 10 nm) in DLS, which can be attributed to the rotational diffusion of the particles if the AuNPs are anisotropic in shape.70 As the relaxation time of translational diffusion is angular dependent whereas rotational diffusion is not, DLS measurements were performed at two different scattering angles (90 deg. and 173 deg.) (Fig. 7B).
 | ||
| Fig. 7 (A) Size distribution of citrate and polymer coated AuNPs obtained via DLS measured at a 90° scattering angle (concentration of the AuNPs = 0.09 mg mL−1 in MilliQ water; temperature = 25 °C) and (B) Normalized intensity vs. relaxation time plot for citrate and the polymer coated AuNPs, measured at two different scattering angles (90 and 173 deg.). | ||
It is clear that the relaxation time of the smaller peak does not show angular dependence confirming that it is a measuring artefact resulting from rotational diffusion.73 The stabilization of the polymer@AuNPs was assessed by zeta potential measurements. As seen in Table 3, the polymer@AuNPs have a zeta potential in the range of −21 to −29 mV, indicating the presence of remaining citrate groups on the AuNP surfaces that may effectively stabilize the AuNPs via repulsive electrostatic interactions besides the steric stabilization from the grafted polymer chains.
Thermoresponsive behavior of the polymer coated AuNPs
Next, we focused on the thermoresponsive behavior of the polymer coated AuNPs. To check the phase transition behavior of the polymer coated AuNPs in aqueous solution, (gold concentration 0.09 mg mL−1) the polymer@AuNPs were heated up to 65 °C which is above the TCP of the statistical copolymer CP5 and PNVCL (Note that DLS measurements were done in plastic cuvettes) limiting the maximum temperature. No phase transition was observed for all the polymer coated AuNPs when measured by turbidimetry. For further investigation we have evaluated the SPR absorption of all the polymer coated AuNPs as a function of temperature. The SPR band of the polymer@AuNPs alters insignificantly when heated from 20 to 65 °C. These results support that no self-association occurs between the particles. As representative example, the effect of temperature on the SPR band maximum wavelength for the CP2@AuNPs is shown in Fig. 8A and B. Fig. 8B reveals, a slight decrease in the absorbance of the SPR band which can be attributed to the solvent thermal expansion that results in a slightly lower concentration of nanoparticles.73 Furthermore, the statistical copolymer (CP1–CP5) coated AuNPs did not show any significant difference in hydrodynamic size (both intensity and volume) measured by DLS (173 deg. scattering angle) upon heating from 20 to 65 °C. As representative example, the effect of temperature on the hydrodynamic size of the statistical copolymer CP2 coated AuNPs at two different temperatures (20 and 65 °C) is shown in Fig. 8C. The hydrodynamic size remains invariable in both intensity and volume distribution when DLS was measured at 20 and 65 °C (Fig. 8C and D). However, a significantly smaller particle size was observed by DLS for PNVCL@AuNPs at 65 °C (ca. 47 nm) in comparison to 20 °C (ca. 66 nm). At 20 °C, which is below TCP, the PNVCL chains are effectively hydrated whereas at 65 °C (above the TCP of PNVCL) the PNVCL chains are dehydrated leading to their collapse onto the AuNPs.60 Despite the collapse of the PNVCL chains, no interparticle aggregation was observed even at 65 °C. This can be ascribed to the presence of excess citrate on the AuNPs that gives electrostatic stabilization as confirmed by the zeta potential of −27 mV (Table 3). We recently reported on a similar observation for PNIPAM@AuNPs.58 Finally, we have investigated the effect of temperature on the thermoresponsive aggregation behavior of the polymer coated AuNPs in 0.1 M NaCl aqueous solution. Addition of 0.1 M NaCl does not have a significant effect on TCP (Fig. 4B) but should be sufficient for screening of excess charges on the AuNPs and to disrupt their electrostatic stabilization. Fig. 8E and F show the representative variable temperature UV-vis absorption spectra of the CP2@AuNPs in aqueous 0.1 M NaCl. A continuous decrease of absorption intensity was observed in addition to the red shift of the SPR band due to interparticle association, driven by dehydration and aggregation of the polymer chains. This is confirmed by the zeta potential value that drops to −12 to −16 mV for the polymer coated AuNPs in aqueous 0.1 M NaCl (Table 3). From Fig. 8G and H it's evident that hydrodynamic size of the particles in aqueous 0.1 M NaCl changes with temperature due to aggregation of the AuNPs. | ||
| Fig. 8 (A) and (B) UV-vis absorbance spectra of CP2@AuNPs in MilliQ water and the corresponding variation of maximum absorbance and maximum absorbance wavelength (λmax) of the SPR band versus temperature; (C) and (D) DLS data of CP2@AuNPs measured at two different temperatures (20 and 65 °C) in milliQ water (scattering angle: 173 deg.); (E) and (F) UV-vis absorbance spectra of CP2@AuNPs in 0.1 M NaCl and the corresponding variation of maximum absorbance and maximum absorbance wavelength (λmax) of the SPR band versus temperature plot; (G) and (H) DLS data of CP2@AuNPs measured at two different temperatures (20 and 65 °C) in 0.1 M NaCl (scattering angle: 173 deg.). | ||
To look in detail, UV-vis spectroscopy measurements were performed to understand the effect of salt on the aggregation behavior of PNVCL@AuNPs and (CP1–CP5)@AuNPs at 25 °C. Fig. 9A shows the comparison of the SPR of CP1@AuNPs and PNVCL@AuNPs in MilliQ water and in 0.1 M aqueous NaCl solution at 25 °C. The drop in absorbance of the PNVCL@AuNPs in 0.1 M NaCl is more significant in comparison to CP1@AuNPs. This could be due to the lower TCP of PNVCL leading to particle agglomeration and macroscopic precipitation of PNVCL@AuNPs. A series of snapshots were also taken from all of the polymer coated AuNPs in 0.1 M NaCl aqueous solution at 25 °C (Fig. 9B). A clear red shift is visible for the CP5@AuNPs and PNVCL@AuNPs. All the polymer coated AuNPs exhibited temperature induced aggregation, in presence of 0.1 M NaCl as shown by the change of the λmax of the SPR band upon heating in a temperature range from 25 to 60 °C, due to the interparticle association (Fig. 9C). In contrast to the precursor polymers, all polymer@AuNPs revealed temperature induced agglomeration below 60 °C. This discrepancy can be ascribed to the high local polymer concentration at the surface of the AuNPs which significantly lowers TCP. Fig. 9D compares the colors of PNVCL coated AuNPs in aqueous 0.1 M NaCl at 20 and 65 °C. The red color of the PNVCL@AuNPs turned to a dark insoluble precipitate when the solution was heated to 65 °C.
 | ||
| Fig. 9 (A) UV-vis absorbance spectra of CP1@AuNPs and PNVCL@AuNPs at 25 °C in MilliQ water and in 0.1 M NaCl; and (B) photographs of plastic cuvettes at 25 °C containing (CP1-CP5)@AuNPs and PNVCL@AuNPs in 0.1 M NaCl; (C) the variation of maximum absorbance wavelength (λmax) of the SPR band of the polymer@AuNPs versus temperature in 0.1 M NaCl and D) Effect of temperature and salt on the PNVCL. | ||
Conclusions
In this work, we have reported the successful synthesis of thermoresponsive homo- and several statistical copolymers of PNVCL. The utilized MADIX/RAFT polymerization protocol was optimized for the synthesis of PNVP using an automated parallel synthesizer revealing an optimal [CTA]:[AIBN] ratio of 1:0.3 with regard to control of reaction speed. The synthesized polymers displayed a broad range of TCP values (from 40 to >95 °C) in water when measured at 5 mg mL−1 polymer concentration. All the TCP values of the polymer solution were found to decrease when measured in 1.0 M NaCl concentration, due to salting out while 0.1 M NaCl did not significantly influence TCP. We have also demonstrated the formation of polymer coated AuNPs using the synthesized thermoresponsive PNVCL and P(NVCL-stat-NVP) via a ‘grafting to’ approach. All the polymer coated AuNPs showed a larger hydrodynamic size in comparison to the citrate@AuNPs as measured by DLS and the polymer coating was also visualized by TEM. The SPR band revealed a slight red shift after polymer coating of the AuNPs due to a decrease in local polarity. Upon heating from 20 to 65 °C, the PNVCL@AuNPs and (CP1–CP5)@AuNPs do not show any color change in water (red) due to the presence of excess citrate on the AuNPs that imparts repulsive electrostatic stabilization. In contrast, in presence of 0.1 M NaCl the red color of the polymer@AuNP solutions turned blue-purple and finally the color fades as a dark precipitate was formed due to macroscopic phase separation. This color change was found to depend on the copolymer composition and ultimately on the TCP of the polymers resulting in a color change of the polymer coated nanoparticles in between 25 °C and 60 °C. This concept of tuneable temperature and salt induced thermoresponsive behavior of AuNPs may be useful for development of colorimetric salt, pH and/or temperature sensors which is the focus of our ongoing work.
Acknowledgements
S.M. gratefully acknowledges FWO for the Pegasus Marie Curie Fellowship. L.V. thanks Ghent University (BOF) and SIM. Z.Z. gratefully acknowledge the Chinese Scholarship Council for a PhD scholarship and Ghent University for BOF co-funding. B.G.D.G. and R.H. acknowledge Ghent University (BOF-GOA) and the FWO Flanders for funding. P.D. thanks Ghent University for funding (i.e. Multidisciplinary Research Partnership Nano-and biophotonics and BOF10/GOA/005). S.V.V. acknowledges FWO Flanders for funding. We are grateful to Prof. Niek Sanders for providing the facility of DLS measurement (90° scattering angle) and Dr Vitaliy Bliznuk for the TEM measurement. R.H. acknowledges the Belgian Program on Interuniversity Attraction Poles initiated by the Belgian State, the Prime Minister's office (P7/541 05) and the European Science Foundation – 542 Precision Polymer Materials (P2M) program for financial support.References
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- C. de las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- B. Lee, A. Jiao, S. Yu, J. B. You, D. H. Kim and S. G. Im, Acta Biomater., 2013, 9, 7691–7698 CrossRef CAS PubMed.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- Y. C. Wang, H. Xia, X. Z. Yang and J. Wang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6168–6179 CrossRef CAS.
- O. Confortini and F. Du Prez, Macromol. Chem. Phys., 2007, 208, 1871–1882 CrossRef CAS.
- Y. Maeda, Langmuir, 2001, 17, 1737–1742 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- L. T. T. Trinh, H. M. L. Lambermont-Thijs, U. S. Schubert, R. Hoogenboom and A. L. Kjøeniksen, Macromolecules, 2012, 45, 4337–4345 CrossRef CAS.
- L. M. Mikheeva, N. V. Grinberg, A. Y. Mashkevich, V. Y. Grinberg, L. T. M. Thanh, E. E. Makhaeva and A. R. Khokhlov, Macromolecules, 1997, 30, 2693–2699 CrossRef CAS.
- F. Meeussen, E. Nies, H. Berghmans, S. Verbrugghe, E. Goethals and F. Du Prez, Polymer, 2000, 41, 8597–8602 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS PubMed.
- H. Vihola, A. Laukkanen, H. Tenhu and J. Hirvonen, J. Pharm. Sci., 2008, 97, 4783–4793 CrossRef CAS PubMed.
- S. T. Sun and P. Y. Wu, J. Phys. Chem. B, 2011, 115, 11609–11618 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Chem.–Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- S. Maji, G. Vancoillie, L. Voorhaar, Q. Zhang and R. Hoogenboom, Macromol. Rapid Commun., 2014, 35, 214–220 CrossRef CAS PubMed.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- A. Can, E. Altuntas, R. Hoogenboom and U. S. Schubert, Eur. Polym. J., 2010, 46, 1932–1939 CrossRef CAS.
- H. Zhang, Eur. Polym. J., 2013, 49, 579–600 CrossRef CAS.
- T. Bilgic and H.-A. Klok, Eur. Polym. J., 2015, 62, 281–293 CrossRef CAS.
- R. Devasia, R. L. Bindu, R. Borsali, N. Mougin and Y. Gnanou, Macromol. Symp., 2005, 229, 8–17 CrossRef CAS.
- K. Nakabayashi and H. Mori, Eur. Polym. J., 2013, 49, 2808–2838 CrossRef CAS.
- D. C. Wan, K. Satoh, M. Kamigaito and Y. Okamoto, Macromolecules, 2005, 38, 10397–10405 CrossRef CAS.
- A. Debuigne, A. N. Morin, A. Kermagoret, Y. Piette, C. Detrembleur, C. Jérôme and R. Poli, Chem.–Eur. J., 2012, 18, 12834–12844 CrossRef CAS PubMed.
- A. Kermagoret, C. A. Fustin, M. Bourguignon, C. Detrembleur, C. Jérôme and A. Debuigne, Polym. Chem., 2013, 4, 2575–2583 RSC.
- M. Hurtgen, J. Liu, A. Debuigne, C. Jérôme and C. Detrembleur, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 400–408 CrossRef CAS.
- J. Liu, C. Detrembleur, M. Hurtgen, A. Debuigne, M. C. D. Pauw-Gillet, S. Mornet, E. Duguet and C. Jérôme, Polym. Chem., 2014, 5, 77–88 RSC.
- J. Liu, C. Detrembleur, A. Debuigne, M. C. D. Pauw-Gillet, S. Mornet, L. Vander Elst, S. Laurent, E. Duguet and C. Jérôme, J. Mater. Chem. B, 2014, 2, 1009–1023 RSC.
- A. Kermagoret, K. Mathieu, J. M. Thomassin, C. A. Fustin, R. Duchêne, C. Jérôme, C. Detrembleur and A. Debuigne, Polym. Chem., 2014, 5, 6534–6544 RSC.
- P. Singh, A. Srivastava and R. Kumar, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1503–1514 CrossRef CAS.
- X. Jiang, Y. Li, G. Lu and X. Huang, Polym. Chem., 2013, 4, 1402–1411 RSC.
- X. Jiang, G. Lu, C. Feng, Y. Li and X. Huang, Polym. Chem., 2013, 4, 3876–3884 RSC.
- I. Negru, M. Teodorescu, P. O. Stanescu, C. Draghici, A. Lungu and A. Sarbu, Mater. Plast., 2010, 47, 35–41 CAS.
- X. Liang, V. Kozlovskaya, C. P. Cox, Y. Wang, M. Saeed and E. Kharlampieva, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2725–2737 CrossRef CAS.
- R. Devasia, R. Borsali, S. Lecommandoux, R. L. Bindu, N. Mougin and Y. Gnanou, Polym. Prepr., 2005, 46, 448–449 CAS.
- D. Wan, Q. Zhou, H. Pu and G. Yang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3756–3765 CrossRef CAS.
- M. Beija, J. D. Marty and M. Destarac, Chem. Commun., 2011, 47, 2826–2828 RSC.
- L. Shao, M. Hu, L. Chen, L. Xu and Y. Bi, React. Funct. Polym., 2012, 72, 407–413 CrossRef CAS.
- Y. C. Yu, G. Li, J. Kim and J. H. Youk, Polymer, 2013, 54, 6119–6124 CrossRef CAS.
- Y. C. Yu, H. U. Kang and J. H. Youk, Colloid Polym. Sci., 2012, 290, 1107–1113 CAS.
- J. Liu, C. Detrembleur, M. C. De Pauw-Gillet, S. Mornet, E. Duguet and C. Jérôme, Polym. Chem., 2014, 5, 799–813 RSC.
- S. M. Ponce-Vargas, N. A. Cortez-Lemus and A. Licea-Claverie, Macromol. Symp., 2013, 325–326, 56–70 CrossRef CAS.
- W. Tian, X. Lv, L. Huang, N. Ali and J. Kong, Macromol. Chem. Phys., 2012, 213, 2450–2463 CrossRef CAS.
- H. Zhang, M. W. M. Fijten, R. Hoogenboom, R. Reinierkens and U. S. Schubert, Macromol. Rapid Commun., 2003, 24, 81–86 CrossRef CAS.
- H. Zhang, H. Abeln Caroline, M. W. M. Fijten and U. S. Schubert, e-Polym., 2006, 6, 90–98 Search PubMed.
- A. W. Bosman, A. Heumann, G. Klaerner, D. Benoit, J. M. J. Frechet and C. J. Hawker, J. Am. Chem. Soc., 2001, 123, 6461–6462 CrossRef CAS PubMed.
- T. M. Eggenhuisen, C. R. Becer, M. W. M. Fijten, R. Eckardt, R. Hoogenboom and U. S. Schubert, Macromolecules, 2008, 41, 5132–5140 CrossRef CAS.
- M. W. M. Fijten, M. A. R. Meier, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5775–5783 CrossRef CAS.
- C. R. Becer, A. M. Groth, R. Hoogenboom, R. M. Paulus and U. S. Schubert, QSAR Comb. Sci., 2008, 27, 977–983 CAS.
- C. G. Sanchez, L. O'Brien, C. Brackley, D. J. Keddie, S. Saubern and J. Chiefari, Polym. Chem., 2013, 4, 1857–1862 RSC.
- J. J. Haven, C. G. Sanchez, D. J. Keddie and G. Moad, Macromol. Rapid Commun., 2014, 35, 492–497 CrossRef CAS PubMed.
- P. Chapon, C. Mignaud, G. Lizarraga and M. Destarac, Macromol. Rapid Commun., 2003, 24, 87–91 CrossRef CAS.
- J. Raula, J. Shan, M. Nuopponen, A. Niskanen, H. Jiang, E. I. Kauppinen and H. Tenhu, Langmuir, 2003, 19, 3499–3504 CrossRef CAS.
- D. X. Li, Q. A. He and J. B. Li, Adv. Colloid Interface Sci., 2009, 149, 28–38 CrossRef CAS PubMed.
- C. Boyer, M. R. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917–6926 CrossRef CAS.
- Z. Zhang, S. Maji, A. B. d. F. Antunes, R. D. Rycke, Q. Zhang, R. Hoogenboom and B. G. D. Geest, Chem. Mater., 2013, 25, 4297–4303 CrossRef CAS.
- M. Q. Zhu, L. Q. Wang, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2004, 126, 2656–2657 CrossRef CAS PubMed.
- S. Yusa, K. Fukuda, T. Yamamoto, Y. Iwasaki, A. Watanabe, K. Akiyoshi and Y. Morishima, Langmuir, 2007, 23, 12842–12848 CrossRef CAS PubMed.
- C. Boyer, M. R. Whittaker, K. Chuah, J. Q. Liu and T. P. Davis, Langmuir, 2010, 26, 2721–2730 CrossRef CAS PubMed.
- S. Sistach, M. Beija, V. Rahal, A. Brûlet, J. D. Marty, M. Destarac and C. Mingotaud, Chem. Mater., 2010, 22, 3712–3724 CrossRef CAS.
- A. Aqil, H. Qiu, J. F. Greisch, R. Jérôme, E. D. Pauw and C. Jérôme, Polymer, 2008, 49, 1145–1153 CrossRef CAS.
- G. Pound, Z. Eksteen, R. Pfukwa, J. M. Mckenzie, R. F. M. Lange and B. Klumperman, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6575–6593 CrossRef CAS.
- L. Voorhaar, S. Wallyn, F. Du Prez and R. Hoogenboom, Polym. Chem., 2014, 5, 4268–4276 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- J. F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- M. M. Bloksma, D. J. Bakker, C. Weber, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 724–728 CrossRef CAS PubMed.
- T. Pradeep, et al., A Textbook of Nanoscience and Nanotechnology, Tata McGraw-Hill Education, 2012, p. 622 Search PubMed.
- B. N. Khlebtsov and N. G. Khlebtsov, Colloid J., 2011, 73, 118–127 CrossRef CAS.
- D. X. Li, Q. He, Y. Yang, H. Mohwald and J. B. Li, Macromolecules, 2008, 41, 7254–7256 CrossRef CAS.
- M. I. Gibson, D. Paripovic and H. A. Klok, Adv. Mater., 2010, 22, 4721–4725 CrossRef CAS PubMed.
- V. R. De la Rose, Z. Zhang, B. G. De Geest and R. Hoogenboom, Adv. Funct. Mater., 2015, 25, 2511–2519 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data including 1H NMR spectra of PNVCL and P(NVCL-stat-NVP), kinetic plot for the homopolymerization of NVCL and transmittance versus temperature of solutions of CP3 in MilliQ water, 0.1 M NaCl and 1 M NaCl. See DOI: 10.1039/c5ra06559c |
| This journal is © The Royal Society of Chemistry 2015 |