
Advanced analysis of nanoparticle composites – a means toward increasing the efficiency of functional materials†
C. R. Crick*a,
S. Noimarkb,
William J. Pevelerb,
J. C. Bearb,
A. P. Ivanova,
J. B. Edela and
I. P. Parkinb
aDepartment of Chemistry, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: c.crick@imperial.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
First published on 9th June 2015
Abstract
The applications of functional materials containing nanoparticles are rapidly increasing. This area is especially relevant to the healthcare industry and the design of new light activated antimicrobials. Wider application of these materials will require quantification of localised nanoparticle concentration, which is currently only available through indirect estimates (including functional testing and bulk spectroscopy). The work presented uses direct visualisation of embedded cadmium selenide quantum dots (Ø – 13.1 nm) using fluorescence lifetime imaging. The nanoparticles used in this study are embedded into a polydimethylsiloxane host matrix via swell encapsulation. The swell encapsulation of the particles is shown to achieve the highest concentration of material at the polymers surface, while a lower concentration is found in the bulk. Fluorescence imaging provides direct comparison of nanoparticle concentration between samples. A comparative swell encapsulation of titanium dioxide nanoparticles (Ø – 12.6 nm) provides further analysis, including photocatalytic dye degradation, water contact angle measurement and energy-dispersive X-ray analysis. The techniques demonstrated allow quantification of nanoparticle concentration within a host matrix, both the functional nanoparticles at the materials' surface and the redundant particles within the bulk.
Introduction
The investigation of sub-micron scale materials and their unique properties is a burgeoning field in scientific research.1–3 A drive towards incorporating nanoparticles into devices and materials for real-world applications has resulted in the generation of many nanoparticle containing commercial products.4–6 Examples include: drug delivery agents, medical imaging mediators, computers, solar cells and strengthening additive in building materials.7–12 These applications illustrate some of the many applied uses of nanocomposite materials, demonstrating material stabilisation, filling a space within a material or imparting properties not intrinsic to a host material. Indeed, a range of medical devices which are tactile and designed to minimise surface acquired infections have been trialled, including catheters, computer equipment, furniture and clothing.13–16 There are many literature reports regarding the advanced application of nanoparticulate materials and their composites, these include a range of materials for sensing applications.17–24The main method of nanoparticle incorporation is via direct addition into the fabrication process, usually through mixing with the matrix material.24–26 As a result, an even concentration of nanoparticles is obtained throughout the matrix. This can be inefficient for many antimicrobial materials, as the active nanoparticles are only required at the surface, rendering deep nanoparticle incorporation superfluous. This is particularly an issue if the nanoparticles are fabricated from precious metals, rare elements or require expensive manufacturing techniques.27,28
A range of post-treatments for nanoparticle incorporation are reported in the literature including: swell-encapsulation, thermal deposition and electrochemical deposition.29–33 These methods focus the nanoparticle placement at the surface of the material. Swell encapsulation can be applied to polymeric materials and expands the polymer using a solvent. This creates space for small molecules and nanoparticles to permeate the matrix, becoming embedded in the polymer. The removal of the swelling solvent via evaporation causes the polymer to shrink, trapping the incorporated materials (Fig. 1). The swelling volume of the polymer, and the diffusion of the nanoparticle through the swollen matrix control the penetration depth of nanomaterials or small molecules into the host matrix. Successful antimicrobial surfaces developed using this strategy have been reported in the literature.29–31 Medical grade polymers such as polyurethane, silicone and polyvinyl chloride have been treated using a swell-encapsulation shrink strategy to incorporate a range of photosensitiser dyes, in addition to nanoparticles which include, zinc oxide, gold and titania.24,25,29–31 These surfaces demonstrate efficacious antimicrobial activity when tested against a range of bacteria, under laser, white light and UVA illumination. These materials are based on and have been designed for medical device and hospital tactile surface applications. Although the incorporation of nanoparticles into polymeric matrices have enhanced the antimicrobial properties, full surface characterisation of these materials remains elusive, with no reported method for accurate and direct quantification of the nanoparticle surface concentration. Previous analysis of these materials have been founded upon secondary observations, such as monitoring the improvement in antimicrobial activity, photosensitiser triplet state production and photocatalytic activity.24
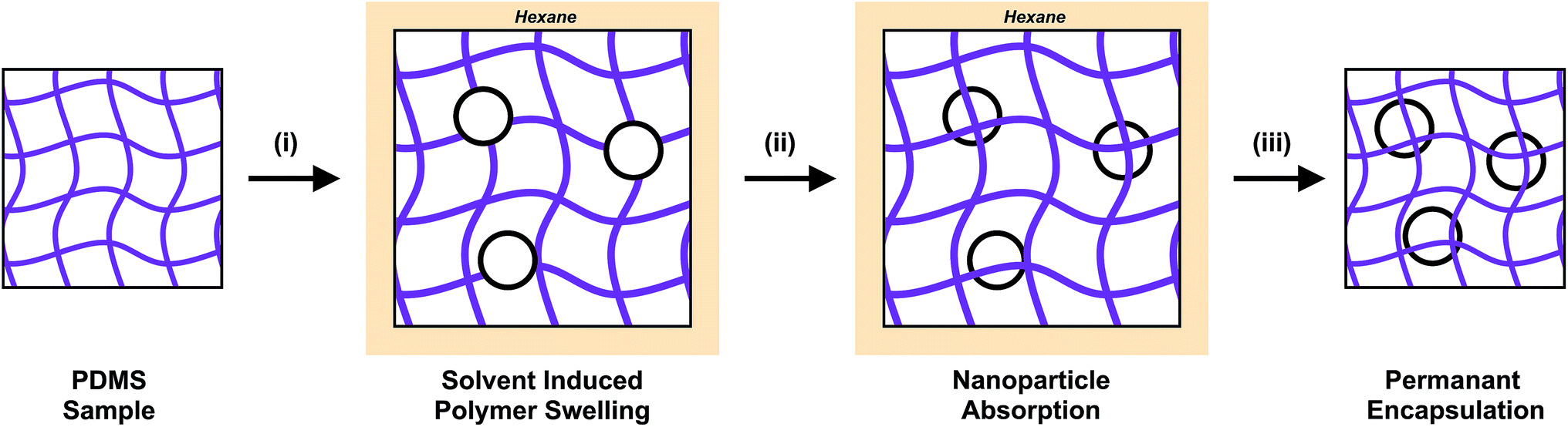 | ||
| Fig. 1 Scheme showing the swell encapsulation process. (i) The PDMS samples were placed in a nanoparticle dispersion. The solvent acts to swell the polymer, increasing the separation of the chains of the polymer matrix. (ii) The samples are left for the define amount of time, throughout which the nanoparticles are absorbed. (iii) The samples are remove from the swell encapsulation solution, rinsed and left to dry. The nanoparticles that entered the material are now fixed in place. | ||
Herein we present a method for visualising the swell encapsulation of nanoparticles, and for the first time the surface coverage is then related to the functional activity. Cadmium selenide quantum dots (QDs) were swell encapsulated into a silicone polymer matrix, and the uptake of these nanoparticles was directly monitored using cross-sectional fluorescence imaging. The swelling time and concentration of nanoparticles in the swelling solution were varied to find the optimal conditions for increasing the surface concentration of particles. These optimum swell encapsulation conditions were also carried forward in experiments using photoactive (titanium dioxide) nanoparticles. The surface concentration was explored through experiments and examination of wetting behaviour of the composite materials. The techniques reported in this manuscript demonstrate accurate quantification of nanoparticle concentration within a host matrix. The authors believe this is the first time such quantification has been carried out on materials fabricated through swell encapsulation. As this technology is currently being explored commercially, the reported analysis and further advances in this area are key to understanding and obtaining optimal functional properties in nanoparticle encapsulated materials.
Experimental
Materials
Polydimethylsiloxane sheets were purchased from NuSil, Polymer Systems Technology Ltd. Anatase nanoparticles were provided by Prof. Jawwad Darr and Dr Peter Marchand (UCL). Oleylamine (technical grade, ≤70%), trioctylphosphine (technical grade, 90%), trioctylphosphine oxide (technical grade, 90%), 1-octadecene (technical grade, 90%), zinc diethyldithiocarbamate (97%), oleic acid (technical grade, 90%) and triethylamine (99.5%) were purchased from Sigma-Aldrich and used as received. Cadmium oxide (98.9%), hexadecylamine (technical grade, 90%) and 1-dodecylphosphonic acid (95%) were purchased from Alfa Aesar and used as received. Selenium powder (99.5+%, 200 mesh) was purchased from Acros and used as received. Laboratory solvents of the highest possible grade were purchased from Fisher Scientific Limited.Nanoparticle synthesis
CdSe QDs were synthesised according to an adapted procedure derived from Bear et al.34,35 Cadmium oxide (51 mg, 0.4 mmol), trioctylphosphine oxide (3.7 g, 9.6 mmol), hexadecylamine (1.93 g, 8 mmol) and 1-dodecylphosphonic acid (0.22 g, 0.88 mmol) were placed into a nitrogen-purged 250 mL, 3-neck flask fitted with a condenser. The flask was evacuated and back-filled with nitrogen five times, before heating to 320 °C and stirring for 1 hour. A 0.5 M solution of selenium powder in trioctylphosphine solution (8 mL) was injected rapidly, instantaneously lowering the temperature to 270 °C. The reaction was stirred for 9 minutes, to generate a red dispersion of CdSe QD cores. The flask was then cooled rapidly to 100 °C in boiling water before addition of chloroform (10 mL). The QD cores were precipitated with ethanol (ca. 100 mL), and centrifuged at 3600 × g. The supernatant was discarded and the precipitated CdSe QD cores re-suspended in n-hexane (10 mL).In order to create a passivating ZnS shell on the QDs, the CdSe cores in n-hexane were mixed with zinc diethyldithiocarbamate (0.5 g, 1.4 mmol), oleylamine (3 mL, 9.12 mmol), 1-octadecene (10 mL) and trioctylphosphine (3 mL, 6.73 mmol). The mixture was heated at 3.3 °C min−1 under partial vacuum then under a flow of nitrogen to 120 °C. After 2 hours, the reaction was cooled in air, and the QDs were precipitated with ethanol (ca. 100 mL), and centrifuged at 3600 × g. The QD slurry was dried in air, and suspended in n-hexane (10 mL), before centrifuging again to remove any insoluble impurities. The QDs were then stored at 4 °C for further use.
Anatase TiO2 nanoparticles were synthesized hydrothermally under the following reaction conditions: pressure = 24.1 MPa, supercritical water temperature = 400 °C, flow rate = 400 mL min−1. This was done using [TiOSO4] (aq., 0.925 M) and [KOH] (aq., 2 M) precursors. The particles were then heated to 80 °C in excess oleic acid (120 mmol, 38.1 mL), with a catalytic amount of triethylamine (8 mmol, 1.12 mL) added to encourage ester formation between the titanol groups on the particle surfaces and the oleic acid. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy observed free and bound oleic acid signals, indicating ionic attraction of carboxylate groups to titanol (Ti-(OH2)+) groups on the particle surface.24
Swell encapsulation
QD polymer samples were prepared by swelling 1 × 1 cm squares of PDMS polymer in n-hexane solutions of varying concentration, for varying amounts of time. n-Hexane (9 mL) was mixed with the QD dispersion (1 mL) to give a stock and then diluted with n-hexane to give a 66% (v/v) solution, 50% solution and 33% solution, to generate the 4 swelling dispersions. The polymer squares were swelled in each dispersion for 24 hours, before removal and drying. In addition squares were swelled in the most concentrated dispersion for 1, 3, 6 and 24 hours. After drying the samples were rinsed with deionised water to remove any surface bound materials.TiO2 functionalised with oleic acid was suspended in toluene (20 mL) and this mixture was used to swell 1 × 1 cm PDMS squares for 1, 3, 6 and 24 h.36 The squares were allowed to dry before rinsing with deionised water to remove surface bound material.
Characterisation techniques
X-ray diffraction (XRD) studies were carried out using a Bruker-Axs D8 (GADDS) diffractometer. The instrument operates with a Cu X-ray source, monochromated (Kα1 and Kα2) and a 2D area X-ray detector with a resolution of 0.01° (glancing incident angle, θ = 5°). The diffraction patterns obtained were compared with database standards. UV/vis absorption spectra were obtained using a Perkin-Elmer Lambda 25 UV/vis spectrometer single beam instrument over a range of 250–1000 nm. TEM samples were prepared by dropping a small amount of particles in solution onto holey carbon-coated copper grids (Agar Scientific) and drying in air. TEM micrographs were collected using a Jeol 2100 microscope, fitted with a Gatan Orius digital camera at a beam acceleration of 200 kV. Particle counting/sizing was performed using ImageJ software. Scanning electron microscopy (SEM) was carried out on the polymer samples using a LEO Gemini 1525 FEGSEM using an acceleration voltage of 5 kV. Energy-dispersive X-ray (EDS) analysis was carried out using the same instrument at an acceleration voltage of 20 kV. Fluorescence imaging was utilised to visualise cadmium selenide QD encapsulation into the polymer matrix. PDMS samples (10 × 10 × 1 mm) were cut into half (providing two 5 × 10 × 1 mm portions), the freshly cut cross-sections were used for fluorescent imaging.Fluorescent lifetime imaging
A spectrally filtered supercontinuum laser (SC450, Fianium) producing 5 ps pulses at a rate of 20 MHz was used as the excitation source for all lifetime fluorescence measurement. An Acousto-Optic Tuneable Filter (AOTF) system, directly coupled to the laser output was used to select a 488 nm laser line. The laser beam was directed via a long pass filter (LP02-488RU-25, RazorEdge, Semrock) towards a custom-built laser-scanning unit (based on a FV300 inverted scanning microscope, Olympus).37 A dichroic mirror (AH/FV1000/DM/11, Olympus) was used to reflect the laser beam into the back aperture of a 10× objective and finally onto the sample. The same objective and same dichroic mirror we used to collect fluorescence emission, which was then focused by a lens onto a 100 μm confocal pinhole (P100S, Thorlabs). A second dichroic mirror (630DCXR, Chroma) was used to direct fluorescence towards an avalanche photodiode (SPCM-AGR-13, Perkin-Elmer Optoelectronics) operating in single photon counting mode. A time-correlated single photon counting board (TimeHarp 200, Picoquant GmbH) was used for lifetime measurements. Lifetime data was analyzed with custom Matlab scripts, allowing to extract fluorescence lifetimes, and to construct two-dimensional fluorescence intensity, lifetime and intensity weighted lifetime maps. Each map was reconstituted from 5 minutes continuous scanning consisting of 264 images with size 512 × 512 pixels and calculated using a maximum likelihood estimator (MLE) with threshold of 150 photons.38,39 The MLE algorithm determines the occurrence probability of a specific lifetime and is given by:where, ni is the number of photon counts in channel i, k is the number of channels (or bins) for each fluorescence decay, pi(j) is the probability that a group of photons will fall in channel i if the particles have a lifetime j, and N is the total number of counts for a given decay. Matlab scripts were also used to map out the sample area and calculate the intensity weighted lifetime per sample area.
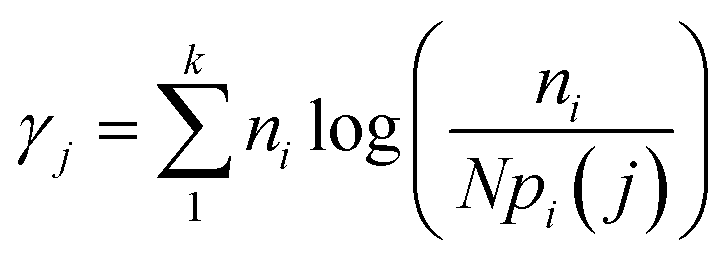
Water contact angle measurements
Water contact angle measurements were performed using an FTA-1000 drop shape instrument; 3 μl water droplets were used and the contact angle of the water droplet was directly observed. The photoactivity of PDMS samples swell encapsulated with titanium dioxide nanoparticles was quantified using dye degradation tests. Resazurin dye was prepared by combining 3 g of a 1.5 wt% aqueous solution of HEC polymer, 0.3 g of glycerol and 4 mg of resazurin dye, this was diluted evenly with ethanol (v/v) to improve spreading across the substrates. 1 mL of this mixture was applied to each of the samples, which had be previously irradiated with UV light (λ = 365 nm, Vilber Lourmat VL-208BLB). The dye degradation was monitored using UV/vis spectroscopy (as above) and optical images as the UV exposure was continued.Results and discussion
QD nanoparticles were synthesised from CdSe@ZnS exhibiting red fluorescence (spectrum included in ESI – S1†) and TiO2 nanoparticles were synthesised hydrothermally to ensure a reasonable size match. From TEM imaging the TiO2 after functionalization with oleic acid had an amorphous aspect and average size 13.1 nm ± 5.6 nm (n = 200). The QDs had a rod like aspect (ratio ∼2.5) with an average length of 12.6 nm ± 2.1 nm (n = 200) (Fig. 2). Nanoparticle dispersions were stable in solution for up to 3 months in the refrigerator. | ||
| Fig. 2 Particle TEM images. (a) QD sample showing rod like CdSe/ZnS nanoparticles. (b) Oleic acid coated TiO2 demonstrating small, but amorphous shapes. | ||
On swelling in organic solvents the samples (originally size – 10 mm × 10 mm × 1 mm) increased to a maximum size of 15 mm × 15 mm × 2 mm after 1 hour, not swelling further after this time, and returned to the original size once dried. The swell encapsulated samples did not appear visually different after swell encapsulation, with no observed discolouration at the longest swelling time. The samples were analysed via UV-vis, however no spectral change was observed in the polymer before and after encapsulation. Examination of the substrates using SEM showed the swelling process for all samples caused wrinkles in the surface material (see ESI – S2†). Analysis of the materials composition was carried out using EDS analysis, which showed an increase in nanoparticle material as swell time was increased (see ESI – S3†). Although EDS analysis shows increased nanoparticulate material (CdSe or TiO2) with longer exposure, both the accuracy and precision of this analysis is unknown, due to the variable detection volume of this technique. Thus not allowing any reasonable estimate of surface coverage.
The swell encapsulated PDMS samples were cross-sectioned, exposing a profile of the particle permeation into the polymer. Fluorescence intensity and lifetime imaging of the cross-sectioned profile of the samples showed a particle concentration gradient which maximised at the edges of the polymer that were exposed to the swelling solution. Fig. 3a–e shows two-dimensional intensity weighted lifetime (τw) maps (photon count × lifetime) of a cross-sectional profile along the middle of PDMS samples that were exposed to swelling solution for up to 48 hours. The samples which endured the longest swell encapsulation time (48 hours) showed both the highest surface concentration of particles, and largest amount of particle permeation into the centre of the polymer samples. Nonetheless, even after 48 hours most of the nanoparticle were encapsulated within 200 μm inside the polymer matrix (for a threshold of 150 photons) as shown in Fig. 3e. Importantly, across samples the lifetime remained the same, with an average value of τ = 3.47 (±0.05 ns), and components (τ1 = 0.96 ns (±0.06 ns) and τ2 = 4.12 ns (±0.09 ns)). Since the PDMS samples had different shape of their cross-section, in order to directly compare between different samples, the intensity weighted lifetime maps were normalised over the imaged sample area. Fig. 3f is a plot of normalised intensity weighted lifetime for samples that were measure after nanoparticle encapsulation up to 48 hours. This indicates that the rate of nanoparticle encapsulation is higher for the initial 6 hours and this rate decreases with longer encapsulation times.
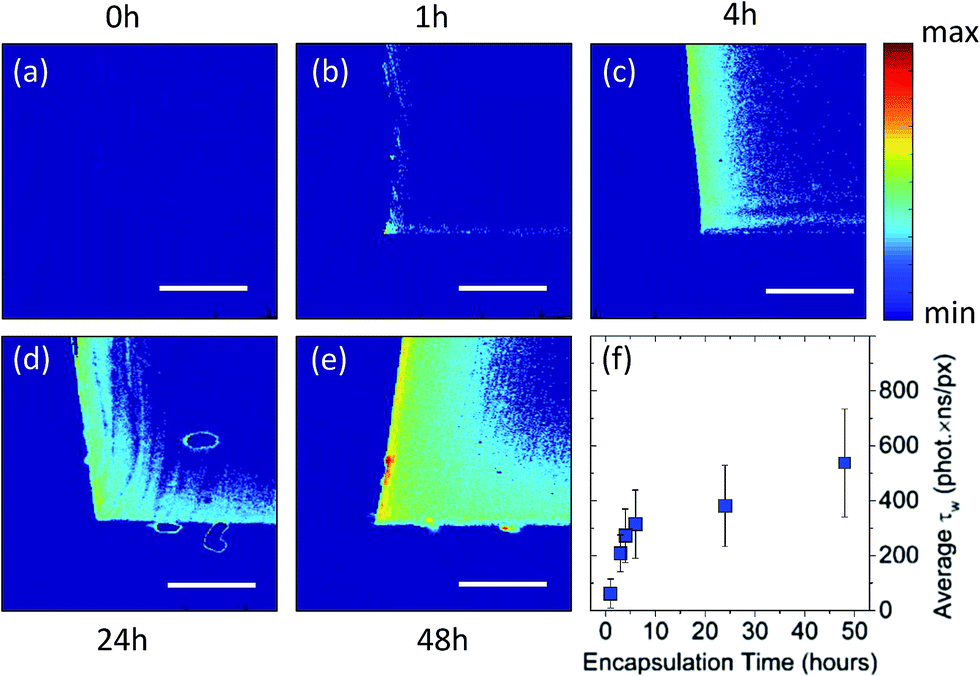 | ||
| Fig. 3 Fluorescence lifetime imaging of swell-encapsulated QDs in PDMS. 2D intensity weighted lifetime maps (photon count × lifetime) showing cross-sectional profile along the middle of PDMS samples prepared after: (a) 0 hours encapsulation (polymer only, no encapsulated of QD), (b) 1 h encapsulation, (c) encapsulation for 4 hours, (d) 24 hours and (e) 48 hours of swell-encapsulation. (f) Intensity weighed lifetime normalized per imaged cross-section area. All scales bars show 100 μm. | ||
Reducing the time of swell encapsulation resulted in a decreased surface particle concentration and a lower number of particles in the centre of the polymer sample. The concentration of particles in the swell encapsulation solution was also varied by dilution, reduced from the original (100%) to 66%, 50% and 33% v/v. The samples, which all underwent the same (48 h) encapsulation time, showed no discernible differences in the fluorescence imaging – indicating little change in particle concentration.
Examination of the fluorescence lifetime images provides further information about rate of QD uptake. The maximum concentration observed at the surface of the samples swell encapsulated for 48 h was noted to be approximately the same as that in the swelling solution [∼0.7 μM] (fluorescence lifetime image included in the ESI – S4†). This concentration is seen to half after penetrating 100 μm into the polymer and falls to approximately zero at 163 μm. This provides an average penetration rate of 3.4 μm per hour for forerunner particles in the samples swell encapsulated for 48 hours. A rate of 28 μm per hour was observed for samples exposed to swell encapsulation for 4 hours, confirming a slowdown of forerunner particles as the encapsulation time is increased (see ESI – S4†). This is much slower when compared to that of the swell encapsulation solvents, which are able to completely saturate the PDMS polymer (1 mm thick) in swelling times between 3–6 hours.
Extended swell encapsulation times provide the highest surface concentration of particles, however they also provide a higher proportion of particles in the polymer's bulk. These nanoparticles, although they are present, would perform limited action in a material where surface interactions are to be considered important. Titanium dioxide nanoparticles were used to examine the surface functionality of polymer–nanoparticle composites, generated by swell encapsulation. The titanium dioxide particle sizes were confirmed as 13.1 nm (±5.6 nm), which are similar to the QD samples which were observed as 12.6 nm (±2.1 nm). Given this similarity, the two particle types were expected to behave similarly during the swell encapsulation process (including the rate/magnitude of penetration), under similar swelling conditions. The series of samples generated were exposed to the swell encapsulation solution for the same time as the QDs used previously (1, 3, 6 and 24 hours).
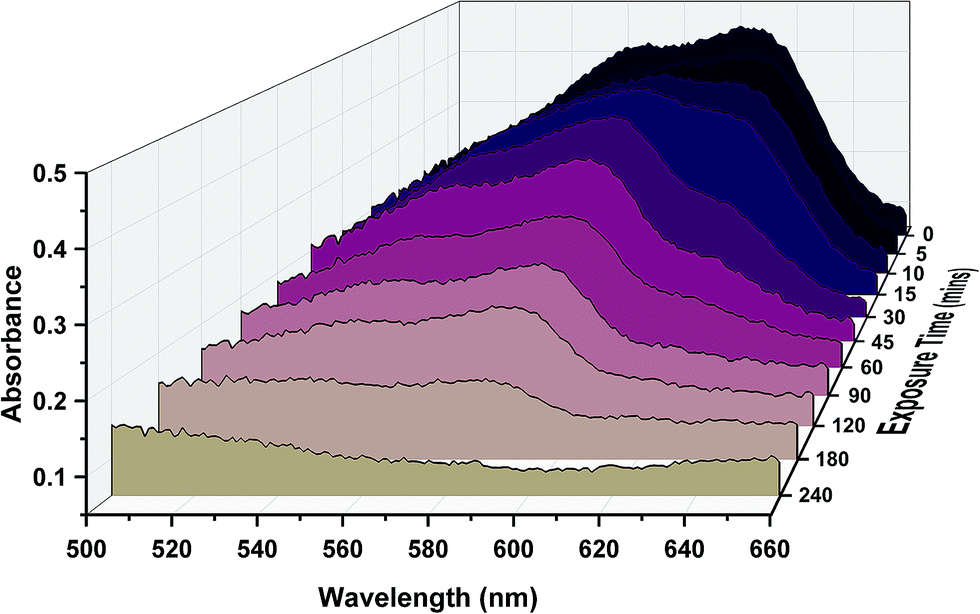
The photoactivity of the titanium dioxide nanoparticle samples was examined using resazurin dye, which upon degradation was converted from originally appearing blue, to pink and then to colourless. Numerous studies have demonstrated this quantifiable degradation. This change was monitored via UV/vis and optical images (Fig. 4). The results show that the samples exposed for the longest swell encapsulation time were the most photoactive. The samples which were swell encapsulated for 1 hour, 3 hours, 6 hours and 24 hours were pre-irradiated with UV-light (λ = 365 nm) for 2 hours before the dye was applied. UV irradiation was continued for a further 4 hours. The amount of dye degradation was estimated from the UV-vis spectra and optical images, and was 1%, 10%, 27%, 56% and 100% for 0 (plain polymer), 1, 3, 6 and 24 h swell encapsulated samples respectively. Some amount of dye degradation was shown for the plain polymer, as resazurin is not completely resistant to UV exposure. It is estimated from this study that the dye degradation would be complete after days of UV exposure under the conditions experienced, this is similar to estimates in other studies.40,41
 | ||
| Fig. 4 Photocatalytic dye degradation of resazurin dye on a PDMS sample swell with embedded titanium dioxide nanoparticles. The particles were embedded by a swell encapsulation process carried out over 24 hours. | ||
The degradation of dye originates from the exposure of the organic components of the dye to the highly active species of the photocatalytic titanium dioxide. Therefore, the speed of dye degradation is directly proportional to the amount of titanium dioxide nanoparticles breeching the surface of the polymer, when passive decomposition of the dye is taken into account. Previous studies using resazurin degradation have been carried out on pure anatase-TiO2, these have demonstrated time of around 20 minutes for complete degradation.31 As the dye degradation is not only dependent on the photo-activity of the sample, but also the amount of dye and sample surface area; only a rough estimate of TiO2 surface coverage of the polymer can be made. The most highly active sample (48 h swell encapsulation) completely degraded the dye in 4 hours, given this, a rough estimate of <10% surface coverage can be made.
The polymers surface coverage with titanium dioxide can also be estimated from monitoring the interaction of water with the samples. The PDMS polymer is inherently hydrophobic, with average water contact angles of 119° (±1°). Any deviation from this can be used to estimate nanoparticle concentration at the surface, as a surface made exclusively from UV-activated titanium dioxide will have a water contact angle approaching zero. Water contact angles for the 1, 3, 6 and 24 h swell encapsulation samples after exposed to UV light (λ = 365 nm) for 2 hours were measures as 118° (±2°), 116° (±3°), 115° (±3°) and 112° (±4°) respectively. The water contact angles can be used to give estimates of the percentage coverage of titanium dioxide nanoparticles, by assuming a relatively flat surface and that the apparent contact angle is an average of the two underlying materials. These rudimentary estimates are 1%, 2.3%, 3.3%, and 5.9%, for the 1, 3, 6 and 24 h samples. A surface coverage of 5.9% also agrees with the estimate gained from the dye degradation experiments (<10%).
The two types of nanoparticles used in this study (CdSe QD and TiO2 nanoparticles) are similar in size and are dispersed in the same swell encapsulation solution. Therefore similar swell encapsulation behaviour is observed for both. The speed and magnitude of QD swell encapsulation process are confirmed by the use of fluorescence imaging. The direct visualisation of the embedded QDs provides an accurate measure of not only the progress of the swell encapsulation process, but also can be used to indicate surface concentration of particles. Using previously reported methods, the surface concentration of titanium dioxide nanoparticles has been estimated, however particle incursion into the bulk was not established.
The quantification of functional nanoparticulate material at the surface of a host matrix is extremely important in many ongoing areas of study. The use of antimicrobial nanoparticles for enhanced bacterial killing is widely reported, however many of these conclusions are based on secondary observations. This includes the enhancement of antimicrobial activity, in addition to concentration approximations through EDS, XPS, Raman, in addition to other techniques. Comprehensive quantification has been carried out in the use of photoactive dyes, however the enhancement brought about by the incorporation of nanoparticles has not been reported previously. The reported method not only allows the visualisation of swell encapsulated nanoparticles, but clearly demonstrates the principles established in a worked example. We propose that this approach can be used to gauge the take-up of various nanoparticles into host matrices. Larger particles, which do not possess inherent fluorescence (as observed for the CdSe QDs) can be fluorescently labelled to provide this method of analysis.
Conclusions
The reported work outlines a method for the quantification of nanoparticles within a host matrix. Swell encapsulated nanoparticles (CdSe@ZnS QDs) were visualised using fluorescence imaging of cross-sectioned PDMS samples. It was found that the highest concentration of nanoparticles is localised toward the edges exposed to the swell encapsulation solution. From these images relative nanoparticle concentrations could be established and the amount of non-functioning particles (those trapped in the bulk of the host material) could also be visualised. The results were validated by using photocatalytic titanium dioxide nanoparticles, these samples demonstrated higher surface activity with longer nanoparticle swell encapsulation times. This was confirmed by examination of the wetting behaviour of the samples, whereby estimates of nanoparticle surface concentrations were established.The quantification of nanoparticle concentration should not only appeal directly to those investigating antimicrobial materials made via swell encapsulation, but also to those interested in designing efficient materials. The localisation of a materials active component at the surface, minimises wasted material which is incorporated but is lost in the bulk.
Acknowledgements
C.R.C. would like to acknowledge the Ramsay memorial trust for their support.Notes and references
- M. Pumera, Chem. Soc. Rev., 2010, 39, 4146–4157 RSC.
- Q. Zhang, E. Uchaker, S. L. Candelaria and G. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 RSC.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- M. S. Olson and P. L. Gurian, J. Nanopart. Res., 2012, 14, 1–7 CrossRef.
- S. Noimark, C. W. Dunnill and I. P. Parkin, Adv. Drug Delivery Rev., 2013, 65, 570–580 CrossRef CAS PubMed.
- E. Lavik and H. von Recum, ACS Nano, 2011, 5, 3419–3424 CrossRef CAS PubMed.
- J. Bear, G. Charron, M. T. Fernández-Argüelles, S. Massadeh, P. McNaughter and T. Nann, in BetaSys: Systems Biology of Regulated Exocytosis in Pancreatic β-Cells, Springer, New York, 2011, vol. 2, pp. 185–220 Search PubMed.
- Z. P. Xu, Q. H. Zeng, G. Q. Lu and A. B. Yu, Chem. Eng. Sci., 2006, 61, 1027–1040 CrossRef CAS PubMed.
- A. Ito, M. Shinkai, H. Honda and T. Kobayashi, J. Biosci. Bioeng., 2005, 100, 1–11 CrossRef CAS PubMed.
- F.-R. Xiu and F.-S. Zhang, J. Hazard. Mater., 2012, 233–234, 200–206 CrossRef CAS PubMed.
- S. Ponja, S. Sathasivam, N. Chadwick, A. Kafizas, S. M. Bawaked, A. Y. Obaid, S. Al-Thabaiti, S. N. Basahel, I. P. Parkin and C. J. Carmalt, J. Mater. Chem. A, 2013, 1, 6271–6278 CAS.
- E. Levashov, V. Kurbatkina and Z. Alexandr, Materials, 2009, 3, 97–109 CrossRef PubMed.
- S. Noimark, E. Allan and I. P. Parkin, Chem. Sci., 2014, 5, 2216–2223 RSC.
- S. Noimark, M. Bovis, A. J. MacRobert, A. Correia, E. Allan, M. Wilson and I. P. Parkin, RSC Adv., 2013, 3, 18383–18394 RSC.
- C. Marambio-Jones and E. M. V. Hoek, J. Nanopart. Res., 2010, 12, 1531–1551 CrossRef CAS.
- K. N. J. Stevens, O. Crespo-Biel, E. E. M. van den Bosch, A. A. Dias, M. L. W. Knetsch, Y. B. J. Aldenhoff, F. H. van der Veen, J. G. Maessen, E. E. Stobberingh and L. H. Koole, Biomaterials, 2009, 30, 3682–3690 CrossRef CAS PubMed.
- X.-G. Li, H. Feng, M.-R. Huang, G.-L. Gu and M. G. Moloney, Anal. Chem., 2012, 84, 134–140 CrossRef CAS PubMed.
- M.-R. Huang, Y.-B. Ding, X.-G. Li, Y. Liu, K. Xi, C.-L. Gao and R. V. Kumar, ACS Appl. Mater. Interfaces, 2014, 6, 22096–22107 CAS.
- X.-G. Li, H. Feng and M.-R. Huang, Chem.–Eur. J., 2009, 15, 4573–4581 CrossRef CAS PubMed.
- J. Stejskal, Prog. Polym. Sci., 2015, 41, 1–31 CrossRef CAS PubMed.
- A. K. Yetisen, Y. Montelongo, M. M. Qasim, H. Butt, T. D. Wilkinson, M. J. Monteiro and S. H. Yun, Anal. Chem., 2015, 87, 5101–5108 CrossRef CAS PubMed.
- M.-R. Huang, H.-J. Lu and X.-G. Li, J. Mater. Chem., 2012, 22, 17685–17699 RSC.
- Q.-F. Lü, M.-R. Huang and X.-G. Li, Chem.–Eur. J., 2007, 13, 6009–6018 CrossRef PubMed.
- C. R. Crick, J. C. Bear, A. Kafizas and I. P. Parkin, Adv. Mater., 2012, 24, 3505–3508 CrossRef CAS PubMed.
- C. R. Crick, J. C. Bear, P. Southern and I. P. Parkin, J. Mater. Chem. A, 2013, 1, 4336–4344 CAS.
- Y. Ma, B. Zhang, M. Gu, S. Huang, X. Liu, B. Liu and C. Ni, J. Appl. Polym. Sci., 2013, 130, 1548–1553 CrossRef CAS PubMed.
- G. C. Jensen, C. E. Krause, G. A. Sotzing and J. F. Rusling, Phys. Chem. Chem. Phys., 2011, 13, 4888–4894 RSC.
- A. Steigerwald and R. Mu, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2008, 26, 1001–1005 CrossRef CAS.
- S. Perni, C. Piccirillo, J. Pratten, P. Prokopovich, W. Chrzanowski, I. P. Parkin and M. Wilson, Biomaterials, 2009, 30, 89–93 CrossRef CAS PubMed.
- S. Perni, C. Piccirillo, A. Kafizas, M. Uppal, J. Pratten, M. Wilson and I. P. Parkin, J. Cluster Sci., 2010, 21, 427–438 CrossRef CAS PubMed.
- S. Noimark, J. Weiner, N. Noor, E. Allan, C. K. Williams, M. S. P. Shaffer and I. P. Parkin, Adv. Funct. Mater., 2015, 25, 1367–1373 CrossRef CAS PubMed.
- D. Gingery and P. Bühlmann, Carbon, 2008, 46, 1966–1972 CrossRef CAS PubMed.
- L. G. Abdelmoti and F. P. Zamborini, Langmuir, 2010, 26, 13511–13521 CrossRef CAS PubMed.
- J. C. Bear, N. Hollingsworth, P. D. McNaughter, A. G. Mayes, M. B. Ward, T. Nann, G. Hogarth and I. P. Parkin, Angew. Chem., Int. Ed., 2014, 53, 1598–1601 CrossRef CAS PubMed.
- J. C. Bear, N. Hollingsworth, A. Roffey, P. D. McNaughter, A. G. Mayes, T. J. Macdonald, T. Nann, W. H. Ng, A. J. Kenyon, G. Hogarth and I. P. Parkin, Adv. Opt. Mater., 2015, 3, 704–712 CrossRef CAS PubMed.
- S. Noimark, K. Page, J. C. Bear, C. Sotelo-Vazquez, R. Quesada-Cabrera, Y. Lu, E. Allan, J. A. Darr and I. P. Parkin, Faraday Discuss., 2015, 175, 273–287 Search PubMed.
- X. C. i. Solvas, V. Turek, T. Prodromakis and J. B. Edel, Lab Chip, 2012, 12, 4049–4054 RSC.
- J. B. Edel, J. S. Eid and A. Meller, J. Phys. Chem. B, 2007, 111, 2986–2990 CrossRef CAS PubMed.
- A. J. D. Monpichar Srisa-Art, Phys. Rev. Lett., 2008, 101, 014502 CrossRef.
- A. Kafizas, D. Adriaens, A. Mills and I. P. Parkin, Phys. Chem. Chem. Phys., 2009, 11, 8367–8375 RSC.
- A. Mills and M. McGrady, J. Photochem. Photobiol., A, 2008, 193, 228–236 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: This includes additional materials characterisation. See DOI: 10.1039/c5ra08788k |
| This journal is © The Royal Society of Chemistry 2015 |