DOI:
10.1039/C5RA09361A
(Paper)
RSC Adv., 2015,
5, 58843-58853
Polyurethanes prepared from isocyanates containing triphenylamine derivatives: synthesis and optical, electrochemical, electrochromic and memory properties†
Received
19th May 2015
, Accepted 26th June 2015
First published on 1st July 2015
Abstract
Two novel series of triphenylamine (TPA)-containing aromatic polyurethanes (PUs) were synthesized via polymerization of N′,N′-bis(α-naphthyl)-N,N′-bis(4-phenylisocyanate) biphenyl (M1) and N,N′-diphenyl-N,N′-bis(4-phenylisocyanate) biphenyl (M2) with different dihydroxy compounds. The structures of the polymers were characterized by Fourier transform infrared (FT-IR) spectroscopy, hydrogen nuclear magnetic resonance (1H-NMR) and carbon nuclear magnetic resonance (13C-NMR); and the thermostability and morphology were studied by thermogravimetric analysis (TGA) and scanning electron microscopy (SEM). The molecular weights were measured to be from 1.02 × 104 to 1.31 × 104 by gel permeation chromatography (GPC). The polymers were coated onto indium tin oxide (ITO) and the electrochromic properties were investigated by using the ultraviolet visible (UV-vis) absorption spectra and cyclic voltammetry (CV). The energy gaps were in the range of 2.21–2.26 eV. The polymer films revealed good stability of the electrochromic characteristics by continuous cyclic scans, with the color changing from neutral yellowish to a blue doped form at applied potentials. Meanwhile, the PUs film exhibited non-volatile write-once-read-many-times (WORM) memory behavior in the device resulting from the non-isolated donor–acceptor (D–A).
Introduction
Electrochromism is known as the alternation of color by the application of a potential.1 As eletrochromic materials, conducting polymers (CPs) have attracted scientists' attention because of their outstanding advantages over transition metal oxides, such as long-term stability, high contrast ratio and coloration efficiency (CE), fast response time, low switching potential, and the ease of color control through structure modification.2–5 Of the available electrochromic CPs,in particular, poly(3,4-ethylenedioxythiophene) (PEDOT), polyaniline (PANI), polyfluorenes, poly(3,4-ethylenedithiathiophene) (PEDTT) and polyselenophenes, have also been extensively investigated due to their unique electronic, redox properties, stable and multicolor behaviors.3,6–14 However, because of planar backbones and strong interaction of molecules, most CPs are difficult to melt or be soluble to be processed. In order to expand the application of the polymers, the researchers used many methods to increase the solubility of electroactive polymers such as by introducing long alkyl or side groups.15 Besides, introducing propeller-like TPA group into the polymers is one effective approach to improve solubility without sacrificing thermostability.3 TPA and its derivatives, which have good ability of hole-transport, could also be used as electron donor, electrochromic materials and memory device.16 The polymers based on TPA as core exhibit stable, excellent, multicolor, electrochromic and memory characteristic, furthermore the corporation of electron-donating substituents at the para positions of TPA affords stable radical cations. From 2005, Liou and Hsiao's groups have been researching deeply on a series of TPA-based polyimides, polyamide as electro-active layer in the field of electrochromic and memory device.3,16,17 Iwan and Skene group synthesized and investigated a series of aromatic polyazomethines with attractive optic-electronic properties and with low optical band gaps (1.3–2.0 eV) bearing TPA.18–20 Our group has also systematically investigated the electrochemical, optical and electrochromic properties of polymers and composite with TPA derivatives.21–23
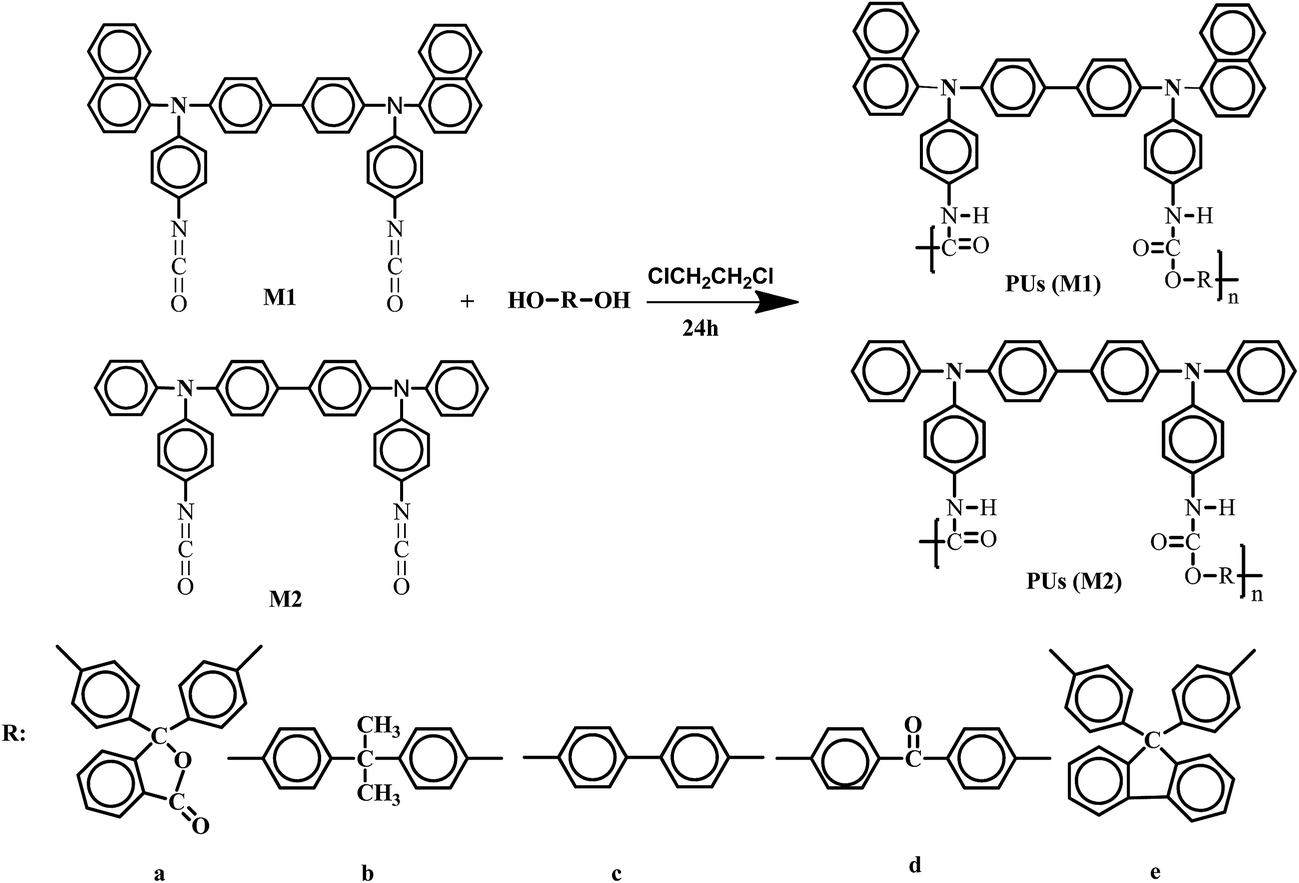
On the other hand, polyurethanes (PUs) form an important family of industrial polymers that have been widely used due to their good properties such as elasticity, flexibility, thermal stability, and excellent chemical resistance.24–29 PU is one of the most versatile polymeric materials with regard to both processing methods and mechanical properties.30 It can be found in almost every aspect of our lives, such as adhesives, in flexible foams, covers, fibers, medical devices and tissue scaffolds.31–34 In recent decades, the research on photoelectric properties of PU has attracted intensively widespread attention. PU has been used in optical devices due to its good optical properties and processibility.35–39 For example, PUs were used as effective hole-transport matrix40–43 as well as light emitting layers.44–46 PUs were reported with better thermal stability, band gap below 2.0 eV, non-linear optical, electrochemical properties, application in solar cell and ion sensor.45,47–49 Zhang reported a new family of PUs with double-ended photo-cross-linkable groups and spindle-type chromophores. The cross-linked films displayed a high electro-optic coefficient up to 41 pm V−1.50 Peng has synthesized successfully electroactive PU containing trimer of amino aniline structure, and found that the electroactive PU had good corrosion resistance performance through a series of electrochemical corrosion test.51 Yang has introduced WO3 into PU materials to improve PU materials electrochromic performance.52 Most investigation is focused on the PU made from toluene-2,4-diisocyanate (TDI) or 4,4′-diphenyl methane-diisocyanate (MDI). In this paper, we used triphosgene to prepare isocyanate which is facile and environmental-friendly to obtain, to the best of our knowledge, there is little research on electrochromic and memory properties study of PU prepared from isocyanate containing TPA.22 In order to distinguish the difference between naphthyl and benzene, and understand the influence of different bisphenol structures on the properties of PUs, we reported two series of TPA-containing aromatic PUs based on the reaction of N,N′-bis(α-naphthyl)-N,N′-bis(4-phenylisocyanate) biphenyl (M1) and N,N′-diphenyl-N,N′-bis(4-phenylisocyanate) biphenyl (M2) with double hydroxyl compounds: phenolphthalein (a), bisphenol A (b), 4,4′-dihydro-xybiphenyl (c), 4,4′-bishydroxybenzophenone (d) and bisphenolfluorene (e), respectively. The characterization of biphenylamine-containing PUs was carried out by means of FT-IR, 1H-NMR, 13C-NMR, TGA and SEM. The optical, electrochemical and electrochromic properties of the PUs films were also tested by UV-vis spectroscopy and CV. The structure and Eg have been calculated by using quantum chemical program. The trend of Egs that was measured by electrochemical method is similar to that of theoretical values which was calculated by quantum chemistry method. The memory devices were prepared with configurating the ITO/polymer/Al, and the memory properties were investigated by I–V measurements. Furthermore, the PUs not only provide a member of new electrochromic materials, but also give theoretical analysis for memory devices.
Experimental
Materials
Dimethyl sulfoxide (DMSO) (analytical reagent) (A.R.), N,N-dimethylformamide (DMF) (A.R.) and dichloroethane were used after standard drying method. 4-Fluoronitrobenzene (A.R.) was distilled before used. Reagent grade aromatic bisphenol monomers that include phenolphthalein (a), bisphenol A (b), 4,4′-dihydroxybiphenyl (c), 4,4′-bishydroxybenzophenone (d), bisphenolfluorene (e) were used as received. Lithium perchlorate (LiClO4) was dried under vacuum at 120 °C for 48 h. All the chemicals were purchased from Shanghai Sinopharm Co and TCI. N,N′-Bis(α-naphthyl)-4,4′-biphenyldiamine (C32H24N2) (1) and N,N′-diphenyl-4,4′-biphenyldiamine (C24H20N2) (1′) were used as received. The intermediate such as N,N′-bis(α-naphthyl)-N,N′-bis(4-aminophenyl)-4,4′-biphenyldiamine (C44H34N4) (3) and N,N′-diphenyl-N,N′-bis(4-aminophenyl)-4,4′-biphenyldiamine (C36H30N4) (3′) and monomers M1, M2 were synthesized according to the modified method of references.22,53
Synthesis of monomers
The synthesis routes of M1 and M2 were outlined in Scheme S1.†
M1 was synthesized by following the procedure. A solution of triphosgene (0.3472 g, 1.170 mmol) and dried dichloroethane (10 ml) were stirred at room temperature under N2, and then was added dropwisely to a stirred solution of compound 3 (0.5450 g, 0.833 mmol). The final solution was heated 100 °C for 8 h under stirring. The obtained compound (M1) was collected by filtration and washed with petroleum ether, subsequently, the solvent was evaporated in vacuum, resulting in 0.4650 g of a light blue solid (yield: 59.4%). The monomer M2 was also synthesized via the same synthesis route of the M1, except for the diamine reactant of N,N′-diphenyl-N,N′-bis(4-aminophenyl)-4,4′-biphenyldiamine and 0.5478 g of a light blue solid product (yield: 57.7%). M1 and M2 were characterized by FT-IR and 1H-NMR and 13C-NMR in ESI.†
Synthesis of PUs
M1 (0.5035 g, 0.513 mmol) and dihydroxy phenolphthalein (0.2532 g, 0.892 mmol) being dissolved in dry dichloroethane (10 ml) were put into a 50 ml, three-neck, round-bottom flask under nitrogen atmosphere. The mixture was stirred at 70 °C for 24 h, yielding a dark viscous solution. The obtained polymer was named of PU (M1)-a and was collected by filtration, resulting in 0.2321 g of a dark solid (yield: 36.3%). Other PUs were also synthesized via the same synthesis route of the PUs (M1)-a as shown in Scheme 1. The formation of PUs was confirmed by FT-IR, 1H NMR and 13C NMR spectroscopy in ESI.†
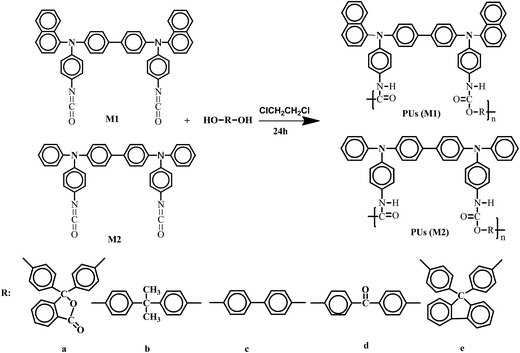 |
| | Scheme 1 Synthesis routes of PUs. | |
Preparation of film electrode
A polymer solution was made by dissolving about 0.02 g of the PU sample in 0.5 ml of DMF. The resulting solution was cast on an ITO-coated glass, which was placed in the oven in vacuum overnight for slow release of the solvent. Then thermally cured films were obtained by programmed increasing temperature to 100 °C at heating rate of 10 °C min−1 and keeping the temperature for 2 h. We controlled the concentration of the solution and casting speed at the same level in order to keep the same thickness of different PUs. The obtained films were used for thermal performance test, optical analyses and electrochemical test.
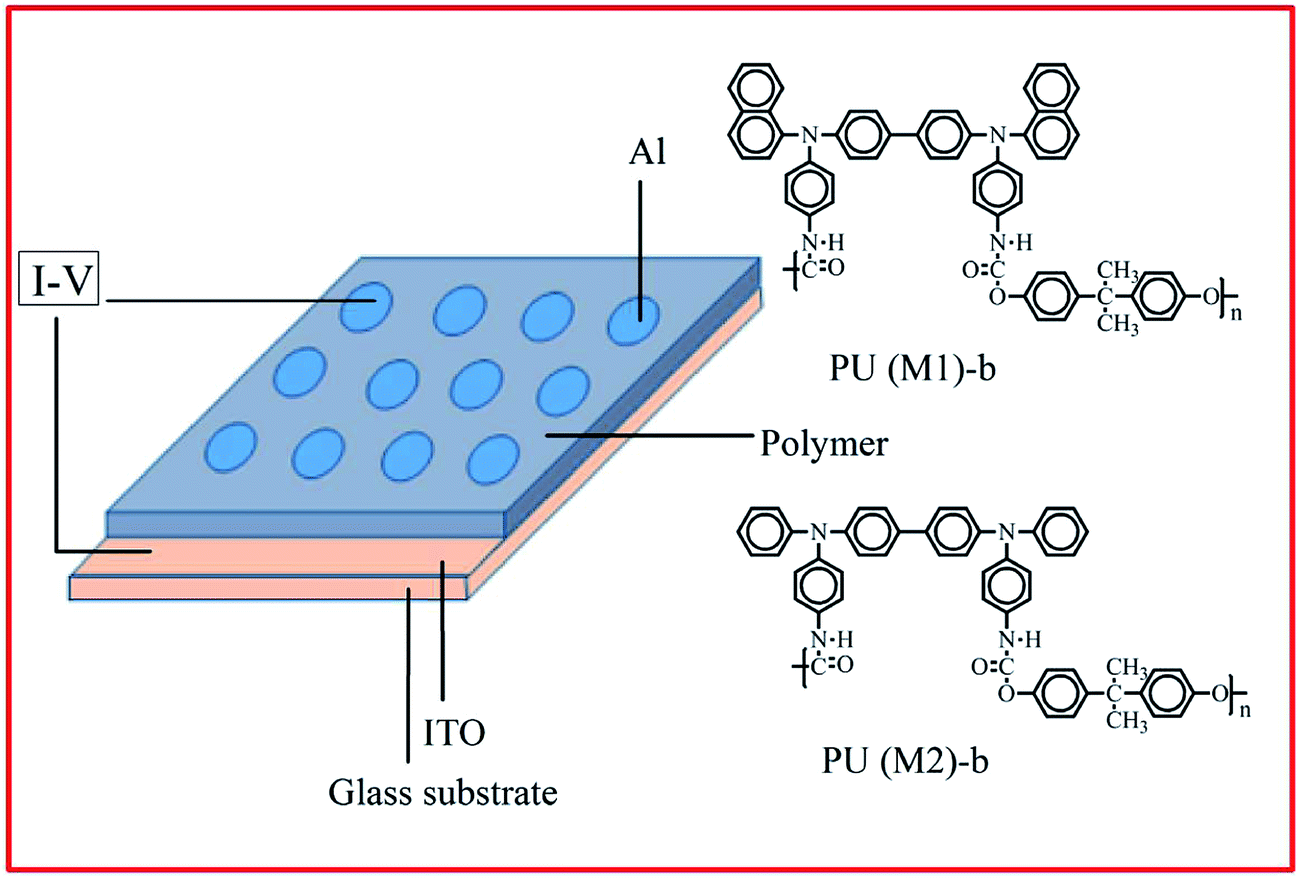
Fabrication and measurement of the memory device
The memory behaviors of these PUs were described by the current–voltage (I–V) characteristics of an ITO/polymer/Al sandwich device as shown in Fig. 1. The ITO glass and polymer film the memory device were the same above film electrode. The film thickness was determined to be around 50 nm. Finally, a 300 nm thick Al top electrode was thermally evaporated through the shadow mask (recorded device units of 0.4 × 0.4 mm2 in size) at a pressure of 10−7 Torr with a depositing rate of 3–5 Å s−1. The electrical characterization of the memory device was performed by a Keithley 4200-SCS semiconductor parameter analyzer. Al was used as the anode during the voltage sweep, and ITO was set as the cathode.
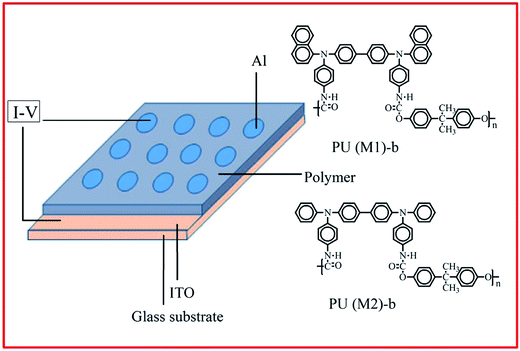 |
| | Fig. 1 Molecular structures of PU (M1)-b and PU (M2)-b and a schematic diagram of the memory device consisting of a polymer thin film sandwiched between an ITO bottom electrode and an Al top electrode. | |
Measurements
FT-IR spectra were recorded on a PerkinElmer Spectrum 100 Model FT-IR spectrometer. 1H and 13C NMR spectra were measured on a Bruker AC-400 and AC-100 MHz spectrometer using DMSO-d6. TGA was conducted on a PerkinElmer Pyris 6. The experiments were carried out by keeping about 6–8 mg powder samples heated in flowing nitrogen (flow rate = 20 cm3 min−1) at a heating rate of 10 °C min−1. SEM measurement was determined on an S4800 instrument with an accelerating voltage of 20 kV. The thickness of PU films (50 nm) coated ITO glass were carried out on profilometer XP-100 (Ambios Technology Inc.). Density-functional theory (DFT) calculations were carried out using the B3LYP functional as implemented in Gaussian 98 program. GPC analysis was performed on a Malvern instrument connected with one refractive index detector (Viscotek-VE3580-RI-DETECTOR) at a flow rate of 0.8 ml min−1 at 35 °C using DMF solution and calibrated with polystyrene standard. UV-vis spectra of the polymer films were determined on a SHIMADZU UV-3600 spectrophotometer whereas the photoluminescence (PL) solution spectra were registered on a Jasco FP-6200 spectrometer. The cyclic voltammograms (CVs) were conducted on a CH Instruments 660A electrochemical analyzer with the use of a three-electrode cell in which a platinum wire was used as an auxiliary electrode against a home-made Ag/AgCl reference electrode and ITO was as a working electrode at a scan rate of 50 mV s−1 with a 0.1 M solution of LiClO4 as an electrolyte under nitrogen atmosphere in dried acetonitrile (CH3CN), and calibrated against the ferrocene/ferrocenium (Fc/Fc+) redox couple. The memory behaviors of polymer were performed by a Keithley 4200-SCS semiconductor parameter analyzer equipped.
Results and discussion
FT-IR analysis and 1H and 13C NMR
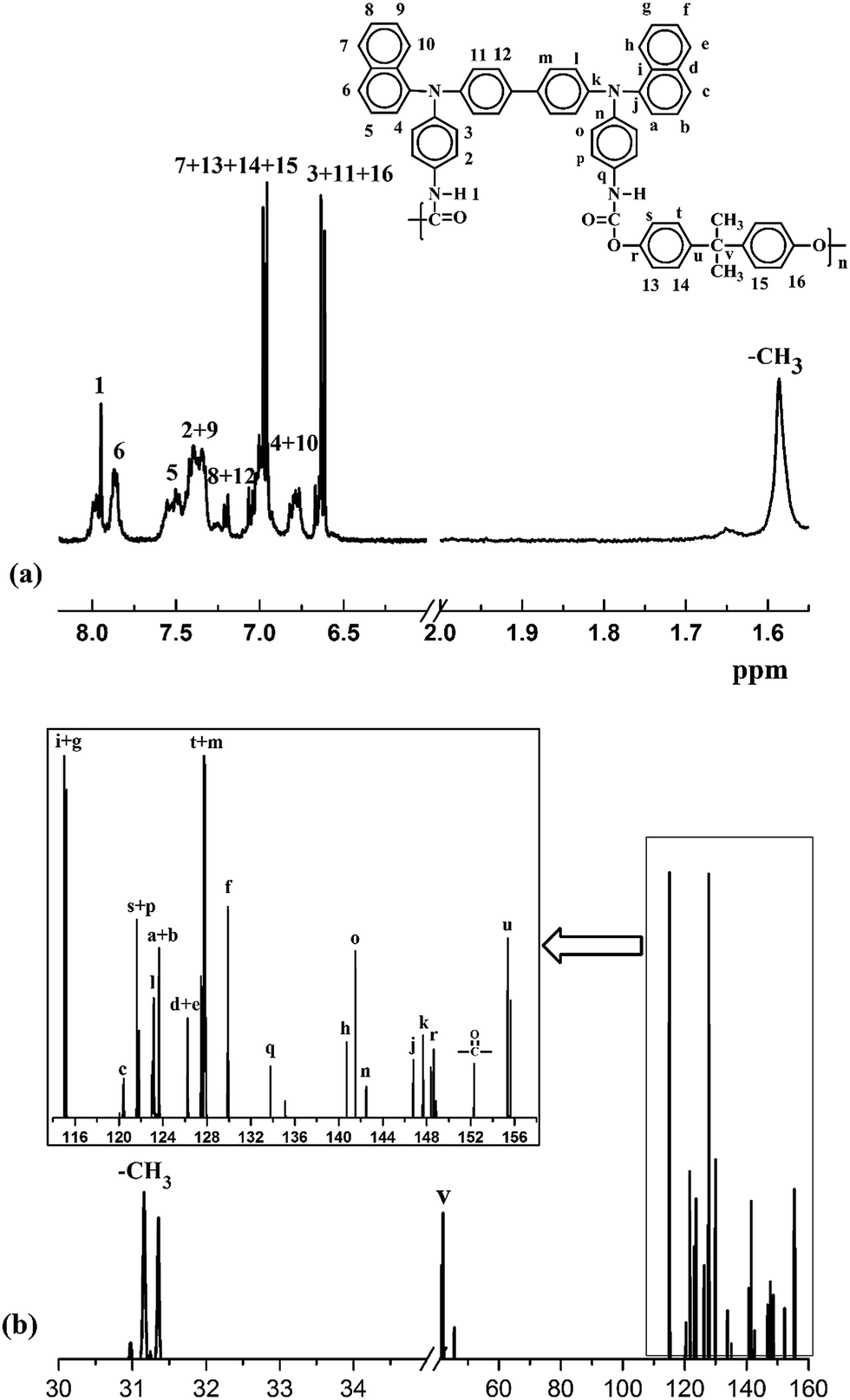
Fig. S1† shows typical FT-IR spectra of M1 and PUs (M1). The distinct characteristic absorption of M1 appears around 2261 cm−1, which confirms the existence of –NCO group. After polymerization, the absorption peak around 2261 cm−1 representing –NCO disappears, which shows the reaction was completed. The characteristic peaks of urethane group of PUs (M1) are observed at 3230 cm−1, 1658 cm−1 and 1275 cm−1 corresponding to stretching vibrations of N–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N–H groups, respectively. FT-IR spectra of M2 and PUs (M2) are also shown in Fig. S1 in ESI.† 1H NMR and 13C NMR spectra of PU (M1)-b in DMSO-d6 are shown in Fig. 2. 1H-NMR spectra of PU (M1)-b confirms that the peak at 7.95 is representing the hydrogen in NH–COO group and peak at 1.67 is representing the hydrogen in –CH3. 13C NMR spectra of PU (M1)-b show the signal at 152.2 corresponding to the ester carbonyl carbon and the singlet at 31.2 due to the –CH3 carbons. All the detected peaks and ratios between the peak areas have been readily assigned to the carbon and hydrogen atoms of the PUs. 1H NMR spectra and 13C NMR spectra of the other PUs in DMSO-d6 are shown in Fig. S2 in ESI.†
O and C–N–H groups, respectively. FT-IR spectra of M2 and PUs (M2) are also shown in Fig. S1 in ESI.† 1H NMR and 13C NMR spectra of PU (M1)-b in DMSO-d6 are shown in Fig. 2. 1H-NMR spectra of PU (M1)-b confirms that the peak at 7.95 is representing the hydrogen in NH–COO group and peak at 1.67 is representing the hydrogen in –CH3. 13C NMR spectra of PU (M1)-b show the signal at 152.2 corresponding to the ester carbonyl carbon and the singlet at 31.2 due to the –CH3 carbons. All the detected peaks and ratios between the peak areas have been readily assigned to the carbon and hydrogen atoms of the PUs. 1H NMR spectra and 13C NMR spectra of the other PUs in DMSO-d6 are shown in Fig. S2 in ESI.†
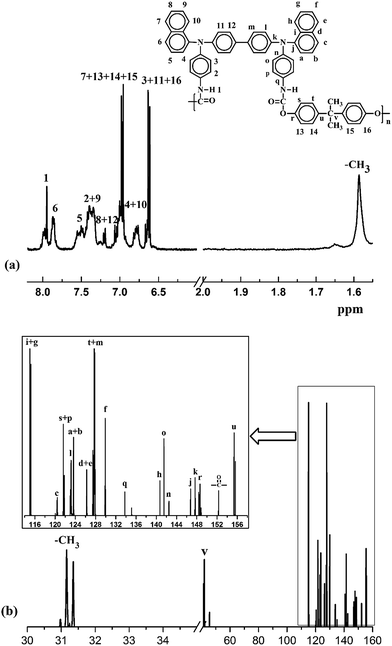 |
| | Fig. 2 1H and 13C NMR spectra of PU (M1)-b in DMSO-d6. | |
Polymer film morphologies, thermal stability and molecular weights
The morphologies of PU (M1)-a, PU (M1)-d and PU (M2)-a, PU (M2)-d are displayed by SEM in Fig. 3 and the morphologies of the other PUs are shown in Fig. S3.† All the original PUs exhibit that the morphologies of the polymers are smooth, which means the morphologies are dependent on the chemical structures of PUs under the same conditions such as concentration, solvent and temperature. The morphology should be controlled to minimize the diffusion distance in PU film in order to achieve sufficient speed of charge. SEM images of PUs suggest the film would provide more ion channel and more reaction surface which makes it facile to transport counter ions.
 |
| | Fig. 3 SEM images of PU (M1)-a, PU (M1)-d and PU (M2)-a, PU (M2)-d. | |
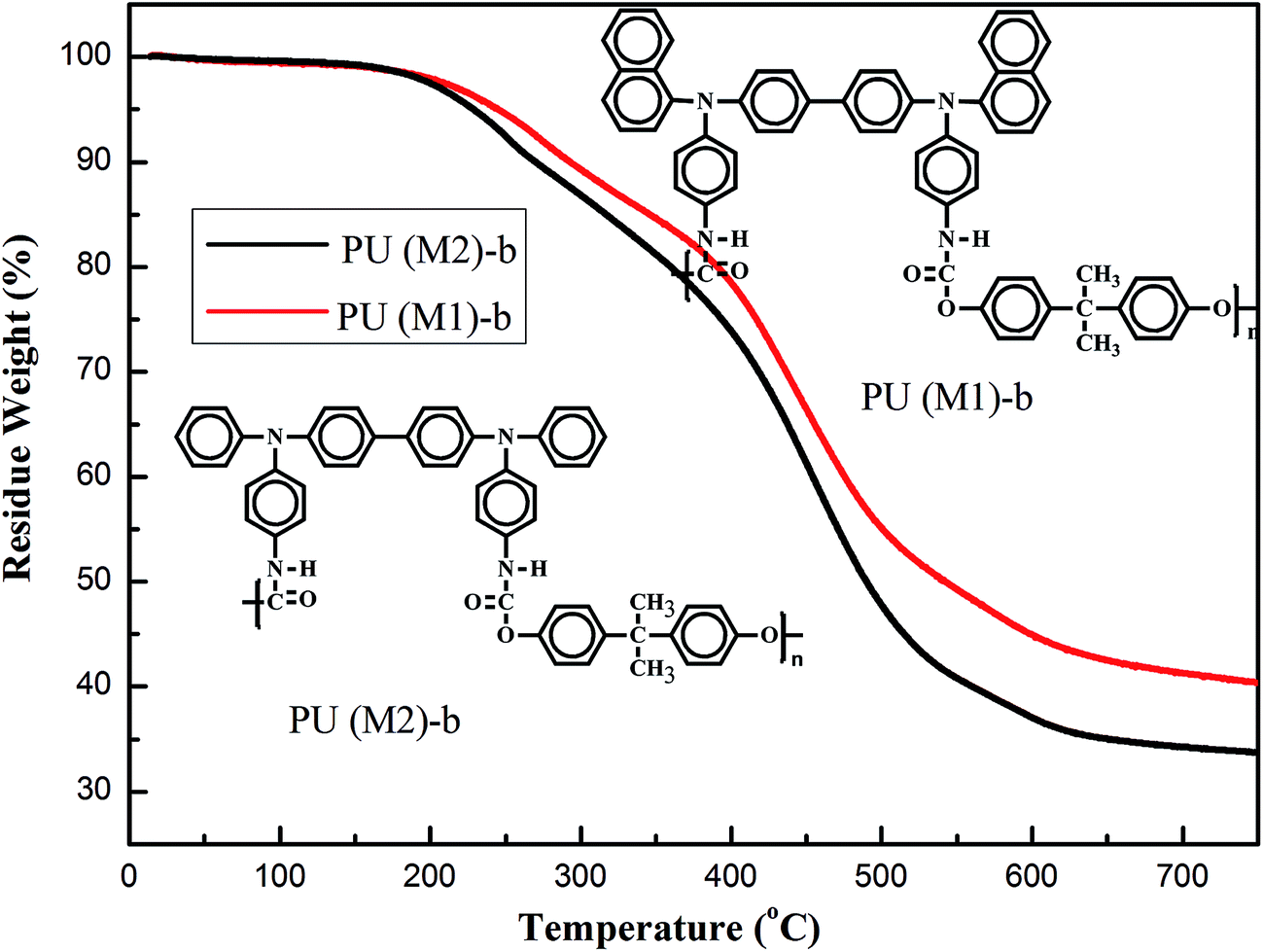
The thermal properties of the PUs were investigated by TGA. Typical TGA curves of representative PU (M1)-b and PU (M2)-b in nitrogen atmospheres are shown in Fig. 4 and the other curves obtained in nitrogen atmospheres are shown in Fig. S4.† The 10% and 60% weight-loss temperatures of these PUs in nitrogen were recorded in the range of 220–270 °C and 317–432 °C, respectively. The amount of carbonized residue (char yield) of these PUs in nitrogen atmosphere was more than 40% at 700 °C. Seen from Fig. 4, and the char yield of PU (M1)-b was superior to PUs (M2)-b. It means that the high char yields of these polymers can be ascribed to their high aromatic content and the bulky naphthylamine-containing chromophores are more effective to preserve the thermal stability in the obtained PUs. Hence, high-temperature stability of the PUs could meet the requirements for optoelectronic thin-layer devices.
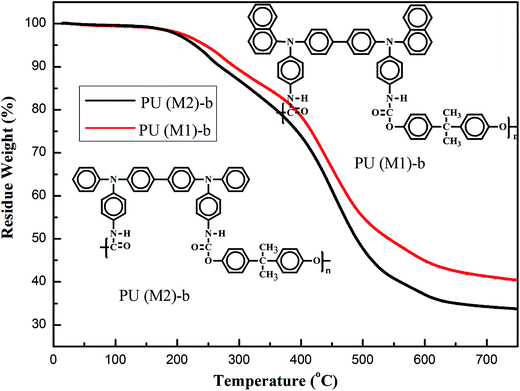 |
| | Fig. 4 TGA thermograms of PU (M1)-b and PU (M2)-b at a scan rate of 10 °C min−1. | |
The number-average molecular weight 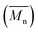 , polydispersity (PDI) and the average degree of polymerization (n) of PUs (M1) and PUs (M2) were determined to be in the range of 10
, polydispersity (PDI) and the average degree of polymerization (n) of PUs (M1) and PUs (M2) were determined to be in the range of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400–12500, 1.06–1.49 and 10–14, 10200–13100, 1.04–1.57 and 11–14, respectively (summarized in Table S1†). The
400–12500, 1.06–1.49 and 10–14, 10200–13100, 1.04–1.57 and 11–14, respectively (summarized in Table S1†). The  and n indicates that the PUs (M1) containing α-naphthyl and PUs (M2) containing biphenyl groups have come into being.
and n indicates that the PUs (M1) containing α-naphthyl and PUs (M2) containing biphenyl groups have come into being.
Optical properties
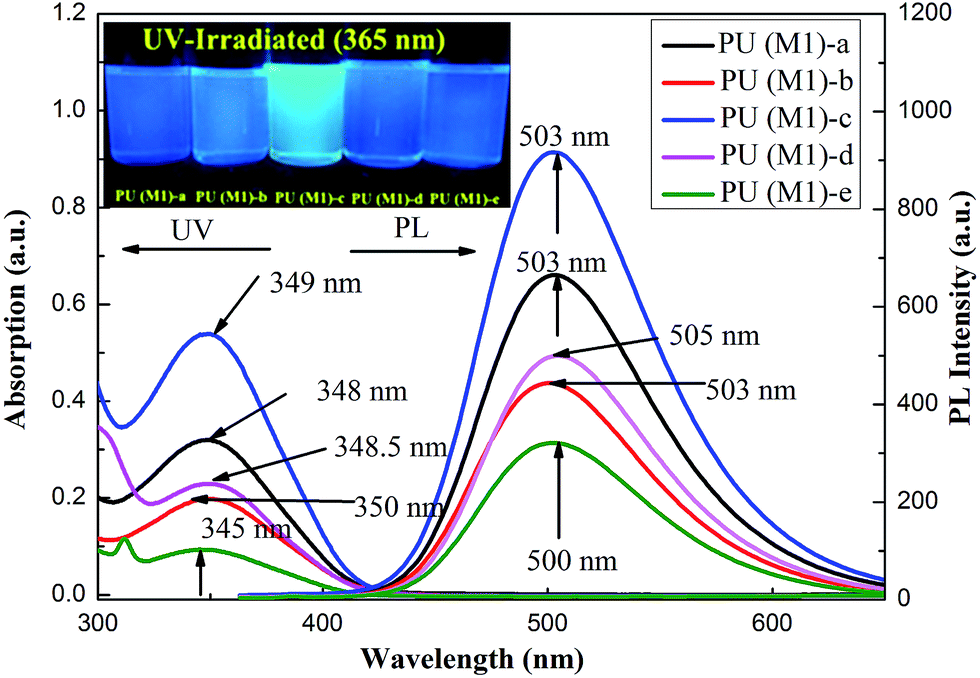
The optical properties of the PUs both in solution and in solid state were investigated by UV-vis spectroscopy and PL spectroscopy. In solid films, these PUs exhibited strong UV-vis absorption bands in the range of 476–480 nm for PU (M1) and 480 nm–484 nm for PU (M2), respectively. Fig. 5 illustrates the strong UV-vis absorption bands PUs (M1) in the range of 345–349 nm in DMSO solution. It can be explained by the π–π* transitions resulting from charge transfer between nitrogen atoms from TPA units and the aromatic rings. The DMSO solution of the PUs (M1) were almost colorless and gave a green PL under irradiation with a general UV lamp (λ = 365 nm). Their PL spectra in dilute DMSO solution (conc.: 10−8 M) showed maximum bands around 500–505 nm with little shift as also shown in Fig. 5. UV-vis absorption and PL spectroscopy of PUs (M2) are displayed in Fig. S5.† PL quantum yields ϕs of the samples in DMSO solvents were measured by using quinine sulfate dissolved in 0.5 M sulfuric acids as reference standard (ϕ = 0.546). The ϕs of these PUs after refractive index correction can be calculated according to the following equation.54
where ϕunk, ϕstd, Iunk, Istd, Aunk, Astd, ηunk and ηstd, are the fluorescent quantum yield, integration of the emission intensity, absorbance at the excitation wavelength, and the refractive indices of the corresponding solutions for the samples and the standard, respectively. The PL quantum yield of PUs in dilute DMSO solution ranges: 3.75% (PU (M1)-d) < 5.08% (PU (M1)-a) < 5.29% (PU (M1)-c) < 5.58% (PU (M1)-e) < 6.23% (PU (M1)-b), 2.37% (PU (M2)-e) < 3.58% (PU (M2)-b) < 3.71% (PU (M2)-d) < 3.84% (PU (M2)-a) < 4.70% (PU (M2)-c). The quantum yield of PU (M1)-b measured in DMSO is the highest among two series of polymers. PUs (M1) containing α-naphthyl show the higher quantum yield than PUs (M2) containing biphenyl groups due to that naphthyl have more conjugated structure. The quantum yield of PU (M1)-b and PU (M2)-c measured in DMSO is the highest among PUs (M1) and PUs (M2) series, respectively. This may be the result of the better electron-donating capability of methyl (–CH3) substituent in the bisphenol A of PU (M1)-b and bisphenolfluorene groups with more planar would contribute more to the emission of PUs than the other groups.
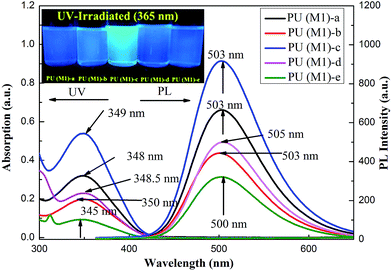 |
| | Fig. 5 UV-vis absorption and PL spectra of PUs (M1) with a concentration of DMSO (conc.: 10−8 M). PL photos of PUs in DMSO solution (10−8 M) by UV irradiation at 365 nm being inset. | |
Electrochemical properties
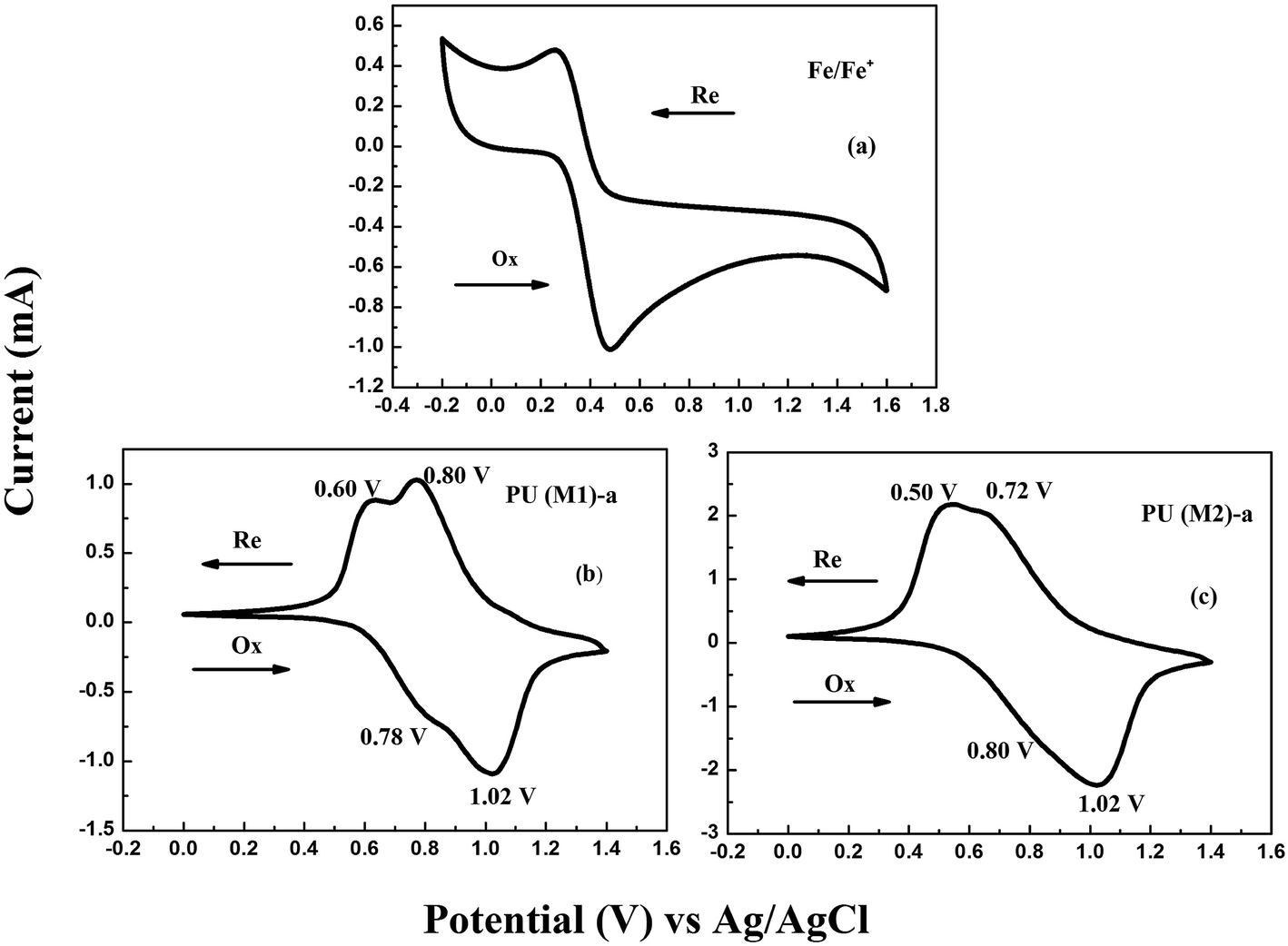
The electrochemical properties of the PUs were investigated by CV. The CVs exhibited two reversible oxidation redox couples for PUs (M1) and one reversible oxidation redox couple for PUs (M2). The color of PUs films changed from yellowish to blue during the electrochemical oxidation of the thin PUs films. The Fc/Fc+ couple was used to calculate the half-wave potentials (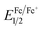 , 0.40 V) vs. Ag/AgCl in CH3CN in order to obtain accurate reoxidation potential. The typical CVs for PU (M1)-a and PU (M2)-a are shown in Fig. 6, and CVs of the other PUs are displayed in Fig. S6.† PU (M1)-a and PU (M2)-a exhibited two reversible oxidation redox couples with E1/21 = 0.69 V vs. Ag/AgCl, E1/22 = 0.91 V vs. Ag/AgCl and with E1/21 = 0.65 V vs. Ag/AgCl, E1/22 = 0.87 V vs. Ag/AgCl, respectively. Each peak is attributed to one electron removal from the nitrogen atom at TPA structure in each repeating unit to yield a stable delocalized radical cation.55 The first peak (at 0.78 and 0.80 V vs. Ag/AgCl) can be attributed to oxidation of the electron-rich nitrogen atom in one of the TPA groups. The second peak (at 1.02 V and 1.02 V vs. Ag/AgCl) can be ascribed to the generation of the dication from radical cation which would cause the radical recombination and formation of a quinoid structure. In addition, the PU (M2)-a had lower reduction potentials than PU (M1)-a due to the latter having a stronger electron withdrawing action of naphthyl group. The energy of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of the investigated PUs can be determined from the onset oxidation potentials and the onset absorption wavelength which are summarized in Table 1. Assuming that the energy level for the Fc/Fc+ standard is 4.80 eV with respect to the zero vacuum level, the HOMO energy values for PUs are calculated using the equation:23
, 0.40 V) vs. Ag/AgCl in CH3CN in order to obtain accurate reoxidation potential. The typical CVs for PU (M1)-a and PU (M2)-a are shown in Fig. 6, and CVs of the other PUs are displayed in Fig. S6.† PU (M1)-a and PU (M2)-a exhibited two reversible oxidation redox couples with E1/21 = 0.69 V vs. Ag/AgCl, E1/22 = 0.91 V vs. Ag/AgCl and with E1/21 = 0.65 V vs. Ag/AgCl, E1/22 = 0.87 V vs. Ag/AgCl, respectively. Each peak is attributed to one electron removal from the nitrogen atom at TPA structure in each repeating unit to yield a stable delocalized radical cation.55 The first peak (at 0.78 and 0.80 V vs. Ag/AgCl) can be attributed to oxidation of the electron-rich nitrogen atom in one of the TPA groups. The second peak (at 1.02 V and 1.02 V vs. Ag/AgCl) can be ascribed to the generation of the dication from radical cation which would cause the radical recombination and formation of a quinoid structure. In addition, the PU (M2)-a had lower reduction potentials than PU (M1)-a due to the latter having a stronger electron withdrawing action of naphthyl group. The energy of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of the investigated PUs can be determined from the onset oxidation potentials and the onset absorption wavelength which are summarized in Table 1. Assuming that the energy level for the Fc/Fc+ standard is 4.80 eV with respect to the zero vacuum level, the HOMO energy values for PUs are calculated using the equation:23
| EHOMO = −(EOX vs. Ag/AgCl + 4.4) eV |
where EOX is the onset oxidation potential. EHOMOs of PUs (M1) and PUs (M2) are calculated to be at in the range from −5.02 to −5.03 eV and from −4.99 to −5.07 eV, respectively. The LUMO energy level values of the PUs are estimated from the HOMO energy level and Eg which could be obtained as following:
ELUMOs and Eg are calculated to be at in the range from at −2.77 eV to −2.78 eV and 2.78 eV to 2.82 eV, respectively. The low-lying HOMO energy level of these polymers suggest that they have potential for use as hole-transporting and blue light-emitting materials in emitting devices. In Table 1, we can see that two series of PUs do not exhibit significant difference in electrochemical properties (EHOMO values, ELUMO values and Eg). It is because the onset oxidation potential is very similar between 0.56 V and 0.64 V due to the influence of similar TPA structures, same experimental conditions and the PDI of molecular weight of PUs. From Table 1, we observed PUs (M2) series had the smaller Eg values because that the pendent naphthyl group is a more conjugated structure and stronger polarity group than benzene group, and the electron transfer of the series of PUs (M1) would need more energy than that of PUs (M2) from the HOMO orbital to LUMO orbital. Seen from the Fig. 6, the peaks of reoxidation are different which is ascribed to difference of polarities of benzene and naphthyl group. Thus, the novel PUs can be used as potential candidates in dynamic electrochromic screen devices due to their suitable thermal stabilities, EHOMO values and reversible electrochemical behaviors.
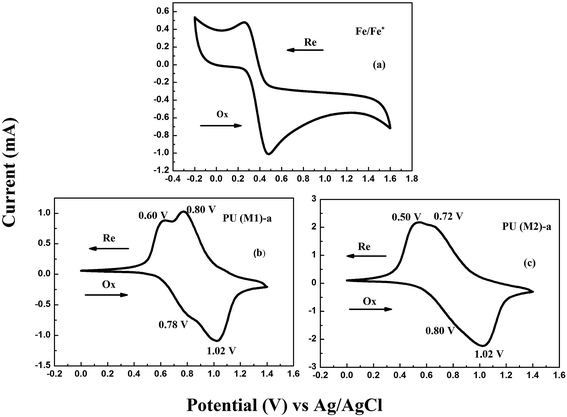 |
| | Fig. 6 CVs of (a) ferrocene (b) PU (M1)-a (c) PU (M2)-a films onto an ITO-coated glass substrate in 0.1 M LiClO4/CH3CN at the scanning rate of 50 mV s−1. | |
Table 1 Optical and electrochemical properties of the PUs
| |
λabsfilma (nm) |
λabsonsetb (nm) |
E1/2 vs. Ag/AgClc |
Epeakonset vs. Ag/AgCl |
EelectroHOMOd (eV) |
EelectroLUMOd (eV) |
Efilmgd (eV) |
EquantumHOMOe (eV) |
EquantumLUMOe (eV) |
Equantumge (eV) |
| UV-vis absorption of the polymer film. λonset of the polymer film. E1/2: average potential of the redox couple peaks. The measured data of the polymers. Theoretical calculation of the polymers. |
| PU(M1)-a |
480 |
552 |
0.69/0.91 |
0.59 |
−5.02 |
−2.77 |
2.25 |
−4.96 |
−1.60 |
3.36 |
| PU(M1)-b |
476 |
554 |
0.70/0.90 |
0.59 |
−5.02 |
−2.78 |
2.24 |
−5.08 |
−1.37 |
3.71 |
| PU(M1)-c |
476 |
552 |
0.71/0.90 |
0.60 |
−5.03 |
−2.78 |
2.25 |
−5.14 |
−1.41 |
3.73 |
| PU(M1)-d |
480 |
554 |
0.71/0.89 |
0.59 |
−5.02 |
−2.78 |
2.24 |
−5.22 |
−1.64 |
3.57 |
| PU(M1)-e |
478 |
549 |
0.69/0.89 |
0.60 |
−5.03 |
−2.77 |
2.26 |
−5.04 |
−1.37 |
3.66 |
| PU(M2)-a |
480 |
561 |
0.65/0.87 |
0.54 |
−4.99 |
−2.78 |
2.21 |
−4.85 |
−1.62 |
3.23 |
| PU(M2)-b |
480 |
554 |
0.72/0.85 |
0.63 |
−5.06 |
−2.82 |
2.24 |
−5.04 |
−1.34 |
3.70 |
| PU(M2)-c |
480 |
554 |
0.70/0.84 |
0.60 |
−5.03 |
−2.79 |
2.24 |
−5.13 |
−1.37 |
3.75 |
| PU(M2)-d |
480 |
552 |
0.73/0.85 |
0.64 |
−5.07 |
−2.82 |
2.25 |
−5.12 |
−1.90 |
3.22 |
| PU(M2)-e |
484 |
553 |
0.68/0.84 |
0.59 |
−5.02 |
−2.78 |
2.24 |
−5.00 |
−1.43 |
3.57 |
Quantum chemical calculation
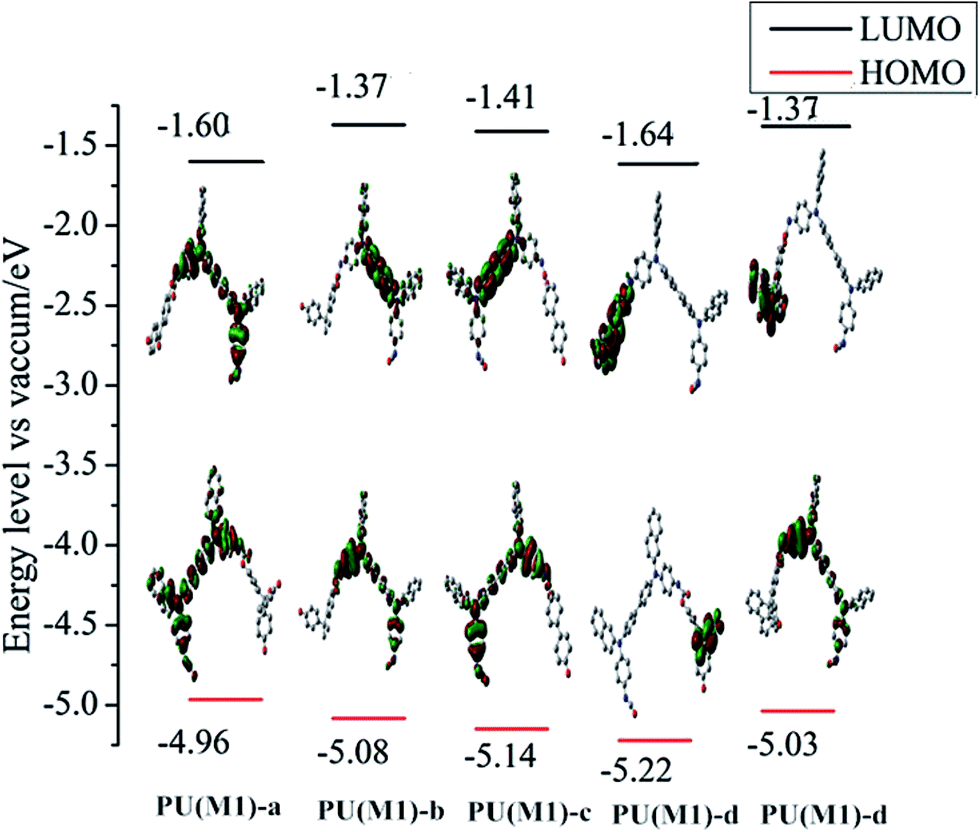
The ground-state geometries of the PUs were optimized by hybrid density functional theory (B3LYP) with 6-31 G basis set, and are presented in Fig. 7 and S7.† EHOMO often expresses the ability of electron donating, and ELUMO often associates with the ability of accepting electrons.56 The HOMO and LUMO energies and their energy gaps (Equantumg) are given in Table 1. The electronic wave function of the HOMO is distributed throughout TPA part of PUs, which is good to get higher hole mobility. The electronic wave function of the LUMO is mainly localized on the dihydroxy portion. The trend of Eg for the series of PUs follows in the order: PUs (M2) < PUs (M1) in Table 1. The measured data have a slight deviation compared with the theoretical data due to the affection of the molecular interaction, molecular structure and solvation process. The results show that the naphthyl group with more polarity and the different bisphenol derivative structures play the key role in regulating the gaps of PUs altogether.
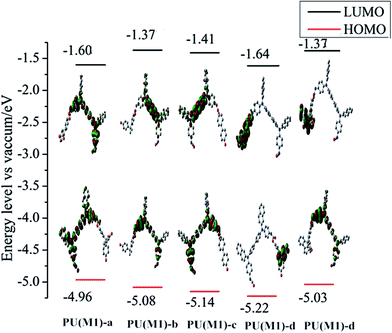 |
| | Fig. 7 Pictorial representations of the electron density in the frontier molecular orbitals of repetition units for PUs (M1). | |
Electrochromic properties
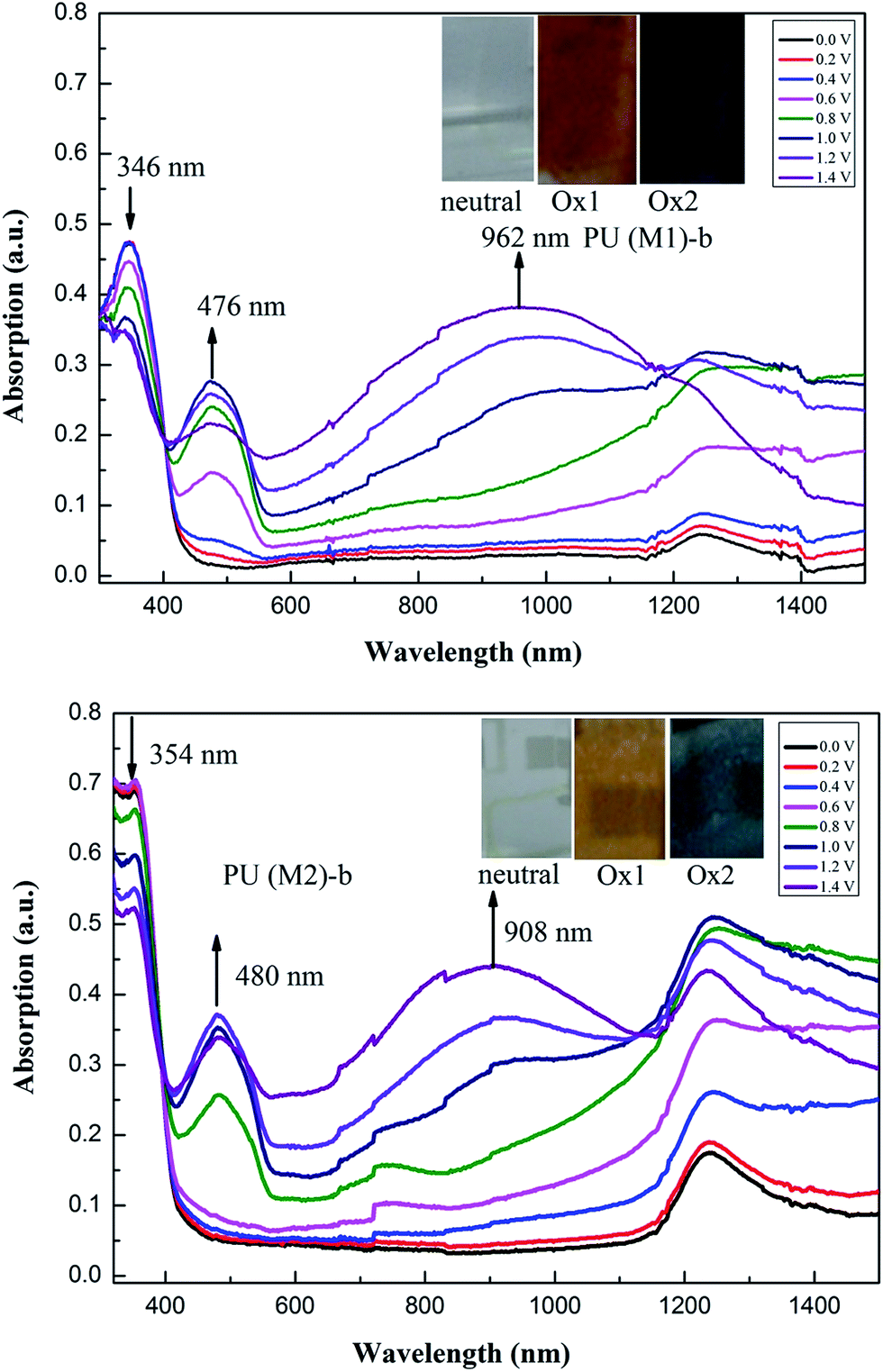
Electrochromic absorption spectra these PUs thin films were examined by a UV-vis spectrometer at different applied potentials, and electrochromism was monitored by casting polymer solution onto an ITO-coated glass substrate. Solution conditions and the electrode preparations were identical to those applied in CV experiments. All these PUs showed similar electrochromic properties, and the typical electrochromic absorbance spectra of PU (M1)-b and PU (M2)-b are illustrated in Fig. 8. In the neutral form, the PU (M1)-b exhibited strong absorption at 346 nm at 0 V due to characteristic of π–π* transitions in TPA. In this oxidized state, PU (M1)-b has two absorption peaks at 346 nm and 476 nm due to the first stage oxidation. When the applied potential increased from 0.6 to 1.4 V, corresponding to the second step oxidation, the peak of absorption at 346 nm decreased gradually, while new band grew up at 962 nm, and the peak of absorption wavelength at 476 nm increased gradually in intensity. For PU (M2)-b, when the applied potentials were adjusted positively from 0.0 to 1.4 V, the peak of absorption at 354 nm decreased gradually for neutral form, while two new bands grew up at 480 and 908 nm, respectively. Meanwhile, the film of PU (M1)-b and PU (M2)-b changed from original colorless to yellow and then to a blue oxidized form. Similar spectral changes for other PUs of PUs (M1) series and the corresponding PUs (M2) series were observed in Fig. S8.† The spectral changes in visible range are assigned to the formation of a stable cationic radical of the TPA center in moiety. Furthermore, the broad absorption in NIR region is the characteristic result due to intervalence-charge transfer (IV-CT) excitation associated with electron transfer (ET) from active neutral nitrogen atom to the cation radical nitrogen center of PUs, which is consistent with the phenomenon classified by Robin and Day.57
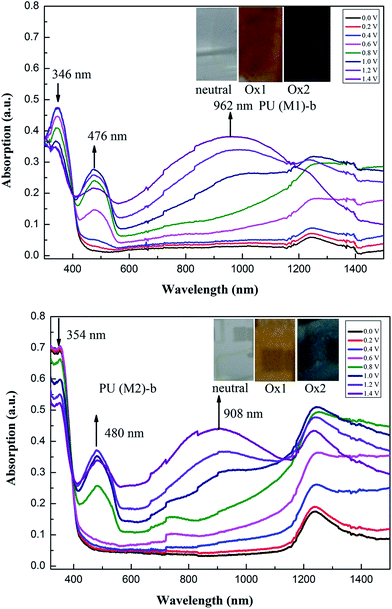 |
| | Fig. 8 Electrochromic behaviors of PU (M1)-b and PU (M2)-b thin film (in CH3CN with 0.1 M LiCO4 as the supporting electrolyte) at 0.0 V to 1.40 V (insets are the pictures of oxidized PU (M1)-b and PU (M2)-b). | |
Electrochromic switching
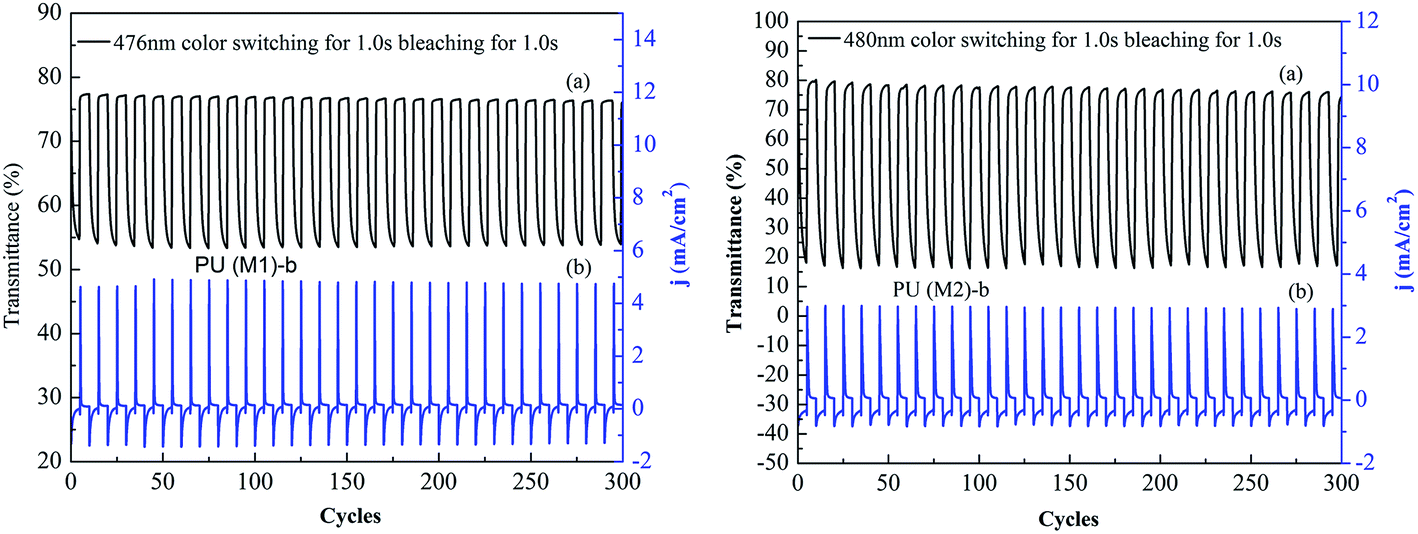
Optical switching processes were further investigated in order to clarify the electrochromic characteristics of these novel PUs. Polymers were cast on ITO-coated glass slides in the same manner as described above, then chronoamperometric and absorbance measurements were performed at the same time. When the voltage was switched, the absorbance at the given wavelength was monitored as a function of time with UV-vis-NIR spectroscopy. The color switching time was estimated by applying a potential step, and the absorbance profile was followed. The switching time was defined as the time required for reach 90% of the full change in absorbance after the switching of the potential. Thin film of PU (M1)-b required 1.0 s at 0.8 V for switching absorbance at 476 nm and 1.0 s for bleaching. The color switching cycles of PU (M1)-b and PU (M2)-b are shown in Fig. 9. After over continuous 300 continuous cyclic switches between 0.0 and 0.8 V in 3000 s, the polymer films still exhibited good stability of electrochromic characteristics. Thin film of PU (M2)-b exhibited same manner as PU (M1)-b. Similar electrochromic switching for the other PUs of PUs (M1) series and the PUs (M2) series were observed in Fig. S9,† and also exhibited good stability of electrochromic characteristics. The electrochromic coloration efficiency (CE; η) is also an important characteristic for the electrochromic materials. CE can be calculated using the equations and given below:58
where Tb and Tc are the transmittances before and after coloration, respectively, δOD is the change of the optical density, which is proportional to the amount of created color centers. η denotes the CE (cm2 C−1) and Q (mC cm−2) is the amount of injected/ejected charge per unit sample area. The electrochromic CEs of PU films are summarized in Table S2.† PUs (M2) containing biphenyl groups show the higher CE than PUs (M1) containing α-naphthyl. The reason is that PU containing biphenyl groups has the more facile CT, which plays the key role in the CEs. PU (M1)-a and PU (M2)-e exhibited the highest CE of the PUs (M1) and PUs (M2), respectively, which were maybe attributed to that phenolphthalein or bisphenolfluorene structures and facile solvation process.
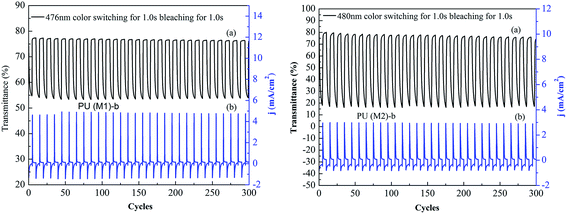 |
| | Fig. 9 Optical switching procedures (0.1 M LiClO4 as the supporting electrolyte with a cycle time of 10 s): (a) potential step transmittance and (b) current consumption of PU (M1)-b and PU (M2)-b at 480 nm. | |
Memory device characteristics
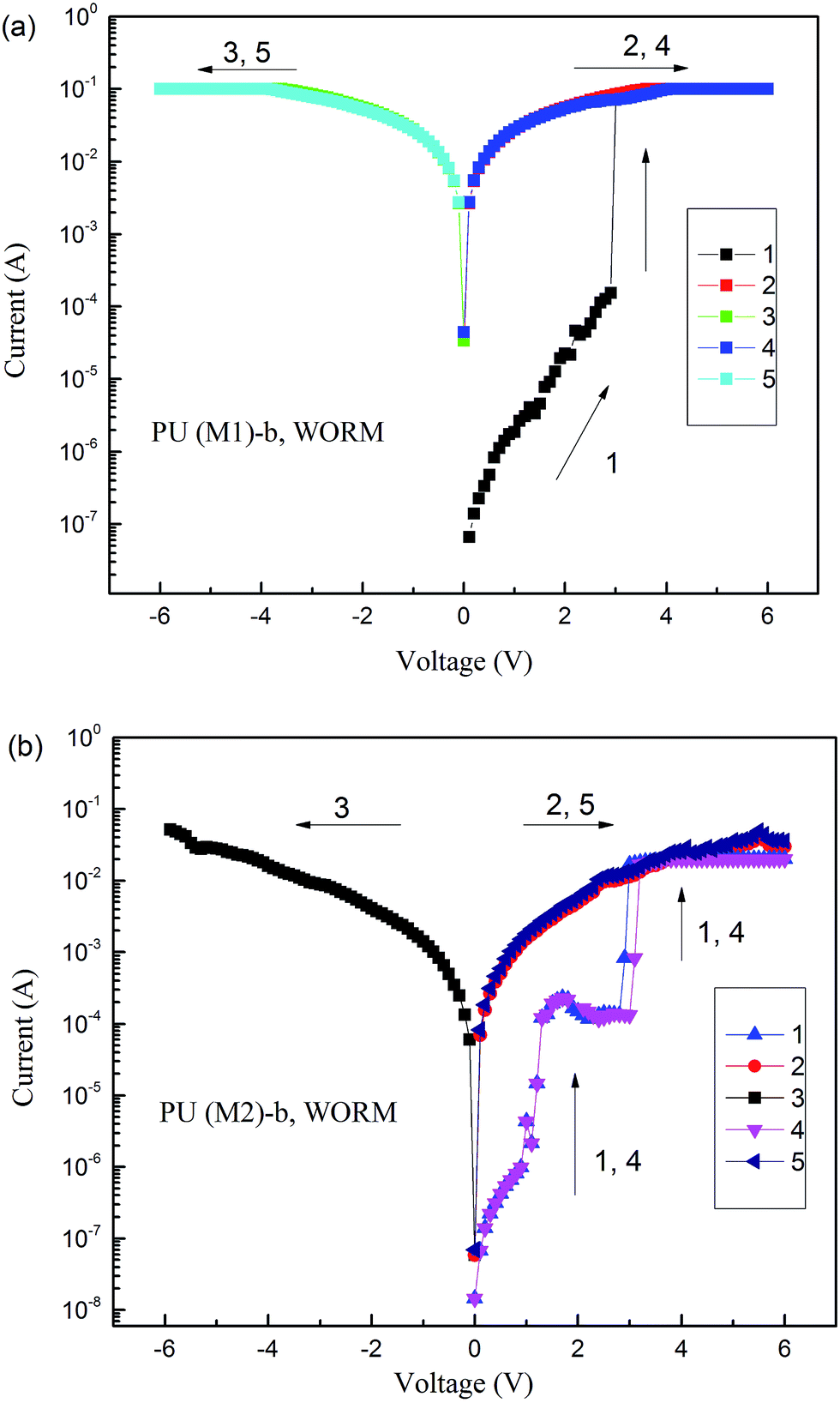
The memory behaviors of these PUs were described by the I–V characteristics of an ITO/polymer/Al sandwich device as shown in Fig. 10. Within the sandwich device, polymer film was used as an active layer between Al and ITO as the top and bottom electrodes. All polymer films standard thickness were coated as 50 nm in order to exclude the effect of the polymer film thickness on memory properties. Fig. 10a depicts the I–V results of PU (M1)-b. During the first positive sweep from 0 V to 3 V, the device based on PU (M1)-b could not be switched to the ON state and stayed in the OFF state with a current range 10−8 to 10−4 A. However, the current increases abruptly from 10−4 to 10−1 A (high-conductivity state) at the threshold voltage of 3.0 V during the positive sweep, indicating that the device undergoes an electrical transition from the OFF state to the ON (high-conductivity) state (writing process). In a memory device, this OFF-to-ON transition can be defined as a “writing” process. The device remained at the ON state during the subsequent positive scan (the second sweep) and then negative scan (the third sweep). The PU (M1)-b memory device also remained in the ON state during the subsequent positive (the fourth sweep) and negative scans (the fifth sweep) after turning off power for 10 min. These results indicate that the PU (M1)-b films exhibit non-volatile write-once-read-many-times (WORM) memory behavior in the device. Fig. 10b reveals the I–V result of PU (M2)-b, the current increasing from 10−8 to 10−5 to 10−4 to 10−1 A (high-conductivity state) instantaneously at the threshold voltage of 2.9 V in the first positive sweep (line 1), indicating the writing process for the transition from the OFF state to the ON state. The device remained in the ON state during the subsequent positive scan (line 2) and the subsequent negative scan (line 3) which could be defined as the reading process. After turning off the power for 10 min, the memory device could be switched on again at threshold voltage 3.2 V (line 4), the PU (M2)-b memory device also remained in the ON state during the subsequent positive scan (the fifth sweep). These results indicate that the PU (M2)-b films also exhibit non-volatile WORM memory behavior in the device. The increased ON/OFF current ratio is advantageous to reduce the probability of misreading between the ON state and the OFF state.16
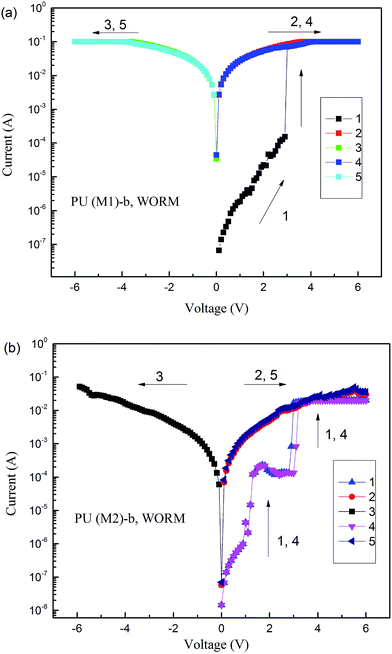 |
| | Fig. 10 Current–voltage (I–V) characteristics of the ITO/polymer (50 nm)/Al memory device. (a) PU (M1)-b, (b) PU (M2)-b. | |
Switching mechanism of the PUs (M1) and PUs (M2)
We could get more understanding of the memory behavior of the PUs (M1) and PUs (M2) devices from Fig. 7 and S7.† These two systems could proceed via a similar charge transfer route from TPA donor to the acceptor due to their similar molecular orbital location. During the positive voltage sweep, both PUs (M1) and PUs (M2), with analogous charge density distribution, proceed via a similar charge transfer pathway from TPA donor to the acceptor as the applied potential reaches the switching-on voltage. Some electrons at the HOMO may accumulate energy then overcome the band gap, thus transit to the LUMO resulting in an excited state. Therefore, charge transfer could occur from the HOMO to the LUMO to form conductive charge transfer complexes. In addition, during the intermolecular charge transfer induced by the applied electric field, the generating holes can be delocalized within TPA moieties resulting in a conducting channel in the polymer chain, which facilitate the migration of charge carriers (holes). The linkage between donor and acceptor in PUs can efficiently prevent the intramolecular backward route, thus stabilizing the CT complex.16
Conclusions
Two series of new TPA-based PUs had been successfully prepared for the first time for memory device applications. PUs displayed a good solubility in common solvents, which is good for polymer film-forming by the solution casting method. The results presented also showed that introducing bulky TPA group into polymer backbone maintained good thermal stability. All obtained PUs revealed good reversible electrochemical behavior, continuous cyclic stability of electrochromic characteristics and memory device characteristics. Thus, these novel TPA-containing PUs have many applications as light-emitting materials, hole-transporting materials, electrochromic materials and memory device application.
Acknowledgements
The authors were grateful to the support of the National Science Foundation of China (Grant No. 51373049, 51372055, 21372067, 21206034, 51473046), Doctoral Fund of Ministry of Education of China (20132301120004, 20132301110001).
References
- C. J. Yao, Y. W. Zhong, H. J. Nie, H. D. Abruna and J. Yao, J. Am. Chem. Soc., 2011, 133, 20720–20723 CrossRef CAS PubMed
 .
. - S. M. Zhang, J. K. Xu, B. Y. Lu, L. Q. Qin, L. Zhang, S. J. Zhen and D. Mo, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1989–1999 CrossRef CAS PubMed .
-
(a) H. M. Wang and S. H. Hsiao, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1172–1184 CrossRef CAS PubMed ;
(b) S. H. Hsiao, P. C. Chang, H. M. Wang, Y. R. Kung and T. M. Lee, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 825–838 CrossRef CAS PubMed ;
(c) S. H. Hsiao, G. S. Liou, Y. C. Kung and T. J. Hsiung, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1013–1023 Search PubMed ;
(d) S. H. Hsiao, G. S. Liou, Y. C. Kung and Y. M. Chang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1013–1023 Search PubMed .
-
(a) H. Y. Qu, H. C. Zhang, N. Li, Z. Q. Tong, J. Wang, J. P. Zhao and Y. Li, RSC Adv., 2015, 5, 803–806 RSC ;
(b) Y. J. Tao, Y. J. Zhou, X. Q. Xu, Z. Y. Zhang, H. F. Cheng and W. W. Zheng, Electrochim. Acta, 2012, 78, 353–358 CrossRef CAS PubMed ;
(c) P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed .
- S. O. Hacioglu, S. Toksabay, M. Sendur and L. Toppare, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 537–544 CrossRef CAS PubMed .
- Y. S. Jeong and K. Akagi, Macromolecules, 2011, 44, 2418–2426 CrossRef CAS .
- A. P. Saxena, M. Deepa, A. G. Joshi, S. Bhandari and A. K. Srivastava, ACS Appl. Mater. Interfaces, 2011, 3, 1115–1126 CAS .
- G. Ćirić-Marjanović, Synth. Met., 2013, 170, 31–56 CrossRef PubMed .
- F. M. Kelly, L. Meunier, C. Cochrane and V. Koncar, Displays, 2013, 34, 1–7 CrossRef CAS PubMed .
- S. X. Xiong, F. Yang, G. Q. Ding, K. Y. Mya, J. Ma and X. H. Lu, Electrochim. Acta, 2012, 67, 194–200 CrossRef CAS PubMed .
- S. M. Zhang, L. Q. Qin, B. Y. Lu and J. K. Xu, Electrochim. Acta, 2013, 90, 452–460 CrossRef CAS PubMed .
- B. Y. Lu, S. M. Zhang, L. Q. Qina, S. Chen, S. J. Zhen and J. K. Xu, Electrochim. Acta, 2013, 106, 201–208 CrossRef CAS PubMed .
- A. Patra and M. Bendikov, J. Mater. Chem., 2010, 20, 422–433 RSC .
- S. S. Zade, N. Zamoshchik and M. Bendikov, Acc. Chem. Res., 2011, 44, 14–24 CrossRef CAS PubMed .
- L. T. Huang, H. J. Yen and G. S. Liou, Macromolecules, 2011, 44, 9595–9610 CrossRef CAS .
-
(a) L. J. Liu, Q. Guo, J. Y. Li, B. Yao and W. J. Tian, Chin. J. Chem., 2013, 31, 456–464 CrossRef CAS PubMed ;
(b) C. J. Chen, Y. C. Hu and G. S. Liou, Polym. Chem., 2013, 26, 06 Search PubMed ;
(c) Y. C. Hu, C. J. Chen, H. J. Yen, K. Y. Lin, J. M. Yeh, W. C. Chen and G. S. Liou, J. Mater. Chem., 2012, 22, 20394–20402 RSC ;
(d) C. J. Chen, H. J. Yen, W. C. Chen and G. S. Liou, J. Mater. Chem., 2012, 22, 14085–14093 RSC ;
(e) Y. Chen, C. L. Tan, H. Zhang and L. Z. Wang, Chem. Soc. Rev., 2015, 44, 2681–2701 RSC ;
(f) Y. Chen, B. Zhang, G. Liu, X. D. Zhuang and E. T. Kang, Chem. Soc. Rev., 2012, 41, 4688–4707 RSC ;
(g) C. L. Tan, Z. D. Liu, W. Huang and H. Zhang, Chem. Soc. Rev., 2015, 44, 2615–2628 RSC .
-
(a) S. H. Cheng, S. H. Hsiao, T. H. Su and G. S. Liou, Macromolecules, 2005, 38, 307–316 CrossRef CAS ;
(b) C. W. Chang, G. S. Liou and S. H. Hsiao, J. Mater. Chem., 2007, 17, 1007–1015 RSC ;
(c) H. J. Yen and G. S. Liou, Chem. Mater., 2009, 21, 4062–4070 CrossRef CAS ;
(d) H. J. Yen, H. Y. Lin and G. S. Liou, Chem. Mater., 2011, 23, 1874–1882 CrossRef CAS ;
(e) L. T. Huang, H. J. Yen and G. S. Liou, Macromolecules, 2011, 44, 9595–9614 CrossRef CAS ;
(f) H. J. Yen and G. S. Liou, Polym. Chem., 2012, 3, 255–264 RSC ;
(g) Y. W. Chuang, H. J. Yen and G. S. Liou, Chem. Commun., 2013, 49, 9812–9814 RSC .
- D. Sek, A. Iwan, B. Jarzabek, B. Kaczmarczyk, J. Kasperczyk, Z. Mazurak, M. Domanski, K. Karon and M. Lapkowski, Macromolecules, 2008, 41, 6653–6663 CrossRef CAS .
- A. Bolduc, S. Dufresne and W. G. Skene, J. Mater. Chem., 2010, 20, 4820–4826 RSC .
- L. Sicard, D. Navarathne, T. Skalski and W. G. Skene, Adv. Funct. Mater., 2013, 23, 3549–3559 CrossRef CAS PubMed .
-
(a) L. N. Ma, H. J. Niu, J. W. Cai, P. Zhao, C. Wang, X. D. Bai, Y. F. Lian and W. Wang, Carbon, 2014, 67, 488–499 CrossRef CAS PubMed ;
(b) Y. Q. Wang, Y. Liang, J. Y. Zhu, X. D. Bai, X. K. Jiang, Q. Zhang and H. J. Niu, RSC Adv., 2015, 5, 11071–11076 RSC .
-
(a) C. Y. Zhang, Y. J. Hou, H. J. Niu, Y. F. Lian, X. D. Bai, C. Wang and W. Wang, J. Electroanal. Chem., 2014, 717, 165–171 CrossRef PubMed ;
(b) V. Bllaert and F. C. de Schryer, Adv. Mater., 1993, 5, 268–270 CrossRef PubMed ;
(c) K. D. Belfield, O. Najjar and S. R. Sriram, polymer, 2000, 41, 5011–5020 CrossRef CAS .
- K. Zhang, H. J. Niu, C. Wang, X. D. Bai, Y. F. Lian and W. Wang, J. Electroanal. Chem., 2012, 682, 101–109 CrossRef CAS PubMed .
- Y. Li, W. Kang, J. O. Stoffer and B. Chu, Macromolecules, 1994, 27, 612–614 CrossRef CAS .
- K. I. Suresh and V. S. Kishanprasad, Ind. Eng. Chem. Res., 2005, 44, 4504–4512 CrossRef CAS .
- R. J. Green, S. Corneillie, J. Davies, M. C. Davies, C. J. Roberts, E. Schacht, S. J. B. Tendler and P. M. Williams, Langmuir, 2000, 16, 2744–2750 CrossRef CAS .
- M. Hasegawa, T. Ikawa, M. Tsuchimori, O. Watanabe and Y. Kawata, Macromolecules, 2001, 34, 7471–7476 CrossRef CAS .
- B. R. Crenshaw and C. Weder, Macromolecules, 2006, 39, 9581–9589 CrossRef CAS .
- J. D. Tovar and C. P. Harvey, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4861–4874 CrossRef PubMed .
- Y. Lu and R. C. Larock, Biomacromolecules, 2008, 9, 3332–3340 CrossRef CAS PubMed .
- O. G. Armagan, B. K. Kayaoglu, H. C. Karakas and F. S. Guner, J. Ind. Text., 2014, 43, 396–414 CrossRef CAS PubMed .
- M. Sponton, N. Casis, P. Mazo, B. Raud, A. Simonetta, L. Rios and D. Estenoz, Int. Biodeterior. Biodegrad., 2013, 85, 85–94 CrossRef CAS PubMed .
- V. Jaso, J. Milic, V. Divjakovic and Z. S. Petrovic, Eur. Polym. J., 2013, 49, 3947–3955 CrossRef CAS PubMed .
- H. Y. Mi, X. Jing, M. R. Salick, T. M. Cordie, X. F. Peng and L. S. Turng, J. Mater. Sci., 2014, 49, 2324–2337 CrossRef CAS .
- M. Špírková, J. Pavličević, A. Strachota, R. Poreba, O. Bera, L. Kaprálková, J. Baldrian, M. Šlouf, N. Lazić and J. Budinski-Simendić, Eur. Polym. J., 2011, 47, 959–972 CrossRef PubMed .
- K. Bagdi, K. Molnár, M. Kállay, P. Schön, J. G. Vancsó and B. Pukánszky, Eur. Polym. J., 2012, 48, 1854–1865 CrossRef CAS PubMed .
- L. Peponi, I. Navarro-Baena, A. Sonseca, E. Gimenez, A. Marcos-Fernandez and J. M. Kenny, Eur. Polym. J., 2013, 49, 893–903 CrossRef CAS PubMed .
- P. Russo, M. Lavorgna, F. Piscitelli, D. Acierno and L. D. Maio, Eur. Polym. J., 2013, 49, 379–388 CrossRef CAS PubMed .
- Z. S. Petrovic, D. P. Hong, I. Javni, N. Erina, F. Zhang and J. Ilavsky, Polymer, 2013, 54, 372–380 CrossRef CAS PubMed .
- C. H. Kuo, K. C. Peng, L. C. Kuo, K. H. Yang, J. H. Lee, M. k. Leung and K. H. Hsieh, Chem. Mater., 2006, 18, 4121–4129 CrossRef CAS .
- C. H. Ku, C. H. Kuo, C. Y. Chen, M. k. Leung and K. H. Hsieh, J. Mater. Chem., 2008, 18, 1296–1301 RSC .
- K. R. Lin, C. H. Kuo, L. C. Kuo, K. H. Yang, M. k. Leung and K. H. Hsieh, Eur. Polym. J., 2007, 43, 4279–4288 CrossRef CAS PubMed .
- C. N. Chuang, C. H. Kuo, Y. S. Cheng, C. K. Huang, M. k. Leung and K. H. Hsieh, Polymer, 2012, 53, 2001–2007 CrossRef CAS PubMed .
- H. Lim, J. Y. Noh, G. H. Lee, S. E. Lee, H. Jeong, K. Lee, M. Cha, H. Suh and C. S. Ha, Thin
Solid Films, 2000, 363, 152–155 CrossRef CAS .
- H. Jeong, D. Zou, T. Tsutsui and C. S. Ha, Thin Solid Films, 2000, 363, 279–281 CrossRef CAS .
- C. H. Ku, C. H. Kuo, M. k. Leung and K. H. Hsieh, Eur. Polym. J., 2009, 45, 1545–1553 CrossRef CAS PubMed .
-
(a) İ. Kaya and M. Kamaci, J. Appl. Polym. Sci., 2012, 125, 876–887 CrossRef PubMed ;
(b) İ. Kaya and M. Kamaci, J. Inorg. Organomet. Polym., 2014, 24, 803–818 CrossRef PubMed ;
(c) İ. Kaya and M. Kamaci, Prog. Org. Coat., 2012, 74, 204–214 CrossRef PubMed ;
(d) İ. Kaya and M. Kamaci, J. Inorg. Organomet. Polym., 2013, 23, 1159–1171 CrossRef ;
(e) İ. Kaya, M. Yildirim and M. Kamaci, Synth. Met., 2011, 161, 2036–2040 CrossRef PubMed .
-
(a) N. P. Wang and T. M. Leslie, Chem. Mater., 1996, 7, 185–191 CrossRef ;
(b) I. Ali, S. M. Al-Zahrani and S. K. Dolui, Polym. Sci., Ser. B, 2012, 54, 342–348 CrossRef CAS .
- A. Shanavas, M. Vanjinathan, A. S. Nasar, S. Amudha and S. A. Suthanthiraraj, High Perform. Polym., 2012, 24, 561–570 CrossRef CAS PubMed .
- X. L. Zhang, M. Li, Z. S. Shi, R. L. Jin, X. B. Wang, Y. F. Yan, M. B. Yi, D. M. Zhang and Z. C. Cui, J. Mater. Sci., 2011, 46, 4458–4464 CrossRef CAS .
- C. W. Peng, C. Hsu and K. H. Lin, Electrochim. Acta, 2011, 58, 614–620 CrossRef CAS PubMed .
- C. H. Yang, L. W. Chong and L. M. Huang, Mater. Chem. Phys., 2005, 91, 154–160 CrossRef CAS PubMed .
- W. Kuran, M. Sobczak, T. Listos, C. Debek and Z. Florjanczyk, Polymer, 2000, 41, 5011–5020 CrossRef .
- G. S. Liou, S. M. Lin and H. J. Yen, Eur. Polym. J., 2008, 48, 2608–2618 CrossRef PubMed .
- G. S. Liou, N. K. Huang and Y. L. Yang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4095–4097 CrossRef CAS PubMed .
- A. A. Nazeer, N. K. Allam, A. S. Fouda and E. A. Ashour, Mater. Chem. Phys., 2012, 136, 1–9 CrossRef CAS PubMed .
- C. H. Duan, W. Z. Cai, F. Huang, J. Zhang, M. Wang and T. B. Yang, Macromolecules, 2010, 43, 5262–5268 CrossRef CAS .
- S. J. Yoo, J. H. Cho, J. W. Lim, S. H. Park, J. Jang and Y. E. Sung, Electrochem. Commun., 2010, 12, 164–167 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09361a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 

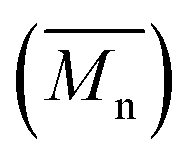 , polydispersity (PDI) and the average degree of polymerization (n) of PUs (M1) and PUs (M2) were determined to be in the range of 10
, polydispersity (PDI) and the average degree of polymerization (n) of PUs (M1) and PUs (M2) were determined to be in the range of 10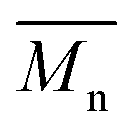 and n indicates that the PUs (M1) containing α-naphthyl and PUs (M2) containing biphenyl groups have come into being.
and n indicates that the PUs (M1) containing α-naphthyl and PUs (M2) containing biphenyl groups have come into being.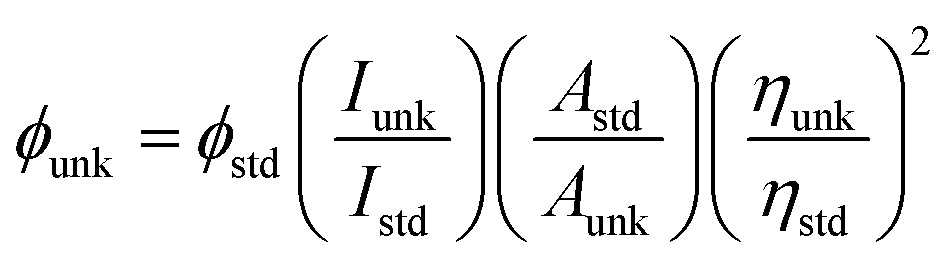
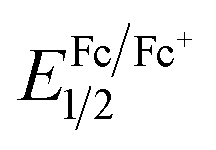 , 0.40 V) vs. Ag/AgCl in CH3CN in order to obtain accurate reoxidation potential. The typical CVs for PU (M1)-a and PU (M2)-a are shown in Fig. 6, and CVs of the other PUs are displayed in Fig. S6.† PU (M1)-a and PU (M2)-a exhibited two reversible oxidation redox couples with E1/21 = 0.69 V vs. Ag/AgCl, E1/22 = 0.91 V vs. Ag/AgCl and with E1/21 = 0.65 V vs. Ag/AgCl, E1/22 = 0.87 V vs. Ag/AgCl, respectively. Each peak is attributed to one electron removal from the nitrogen atom at TPA structure in each repeating unit to yield a stable delocalized radical cation.55 The first peak (at 0.78 and 0.80 V vs. Ag/AgCl) can be attributed to oxidation of the electron-rich nitrogen atom in one of the TPA groups. The second peak (at 1.02 V and 1.02 V vs. Ag/AgCl) can be ascribed to the generation of the dication from radical cation which would cause the radical recombination and formation of a quinoid structure. In addition, the PU (M2)-a had lower reduction potentials than PU (M1)-a due to the latter having a stronger electron withdrawing action of naphthyl group. The energy of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of the investigated PUs can be determined from the onset oxidation potentials and the onset absorption wavelength which are summarized in Table 1. Assuming that the energy level for the Fc/Fc+ standard is 4.80 eV with respect to the zero vacuum level, the HOMO energy values for PUs are calculated using the equation:23
, 0.40 V) vs. Ag/AgCl in CH3CN in order to obtain accurate reoxidation potential. The typical CVs for PU (M1)-a and PU (M2)-a are shown in Fig. 6, and CVs of the other PUs are displayed in Fig. S6.† PU (M1)-a and PU (M2)-a exhibited two reversible oxidation redox couples with E1/21 = 0.69 V vs. Ag/AgCl, E1/22 = 0.91 V vs. Ag/AgCl and with E1/21 = 0.65 V vs. Ag/AgCl, E1/22 = 0.87 V vs. Ag/AgCl, respectively. Each peak is attributed to one electron removal from the nitrogen atom at TPA structure in each repeating unit to yield a stable delocalized radical cation.55 The first peak (at 0.78 and 0.80 V vs. Ag/AgCl) can be attributed to oxidation of the electron-rich nitrogen atom in one of the TPA groups. The second peak (at 1.02 V and 1.02 V vs. Ag/AgCl) can be ascribed to the generation of the dication from radical cation which would cause the radical recombination and formation of a quinoid structure. In addition, the PU (M2)-a had lower reduction potentials than PU (M1)-a due to the latter having a stronger electron withdrawing action of naphthyl group. The energy of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of the investigated PUs can be determined from the onset oxidation potentials and the onset absorption wavelength which are summarized in Table 1. Assuming that the energy level for the Fc/Fc+ standard is 4.80 eV with respect to the zero vacuum level, the HOMO energy values for PUs are calculated using the equation:23