
Photophysical and theoretical studies of peripherally halogenated octaphenoxyphthalocyanines†
Saad Makhseed*a,
Basma Ghazala,
Amr Mohamed Abdelmoniema,
Veronika Novakovab and
Petr Zimcikc
aDepartment of Chemistry, Kuwait University, P.O. Box 5969, Safat 13060, Kuwait. E-mail: saad.makhseed@ku.edu.kw
bDepartment of Biophysics and Physical Chemistry, Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Heyrovskeho 1203, Hradec Kralove 50005, Czech Republic
cDepartment of Pharmaceutical Chemistry and Drug Control, Faculty of Pharmacy in Hradec Kralove, Charles University in Prague, Heyrovskeho 1203, Hradec Kralove 50005, Czech Republic
First published on 1st July 2015
Abstract
Peripherally substituted phthalocyanines with halogenated phenoxy groups containing different central metals (In(III), Ga(III), Mg(II) and Zn(II)) were synthesized. X-ray analyses confirmed that the halogen atoms (Cl, Br) at the ortho positions were bulky enough to orient the phenoxy substituents perpendicularly relative to the Pc core and perfectly discouraged cofacial aggregation. The ground state geometry, HOMO–LUMO energies and the electronic properties were theoretically calculated for some complexes and corresponded well with experimental data. Analysis of fluorescence and singlet oxygen quantum yields revealed a strong heavy atom effect provided by the central metal. Irrespective of the present halogen, indium(III) complexes were characterized by high singlet oxygen production and very low fluorescence while reverse data were obtained for Mg(II) complexes.
Introduction
Phthalocyanines (Pcs), a versatile class of organic functional materials, are widely used in a large variety of applications due to their outstanding optical and electronic properties combined with excellent thermal and chemical stability.1–5 However, controlling the Pc bulk material on a molecular level is a crucial requirement to improve the desired photophysical and opto(electronic) properties which can maximize their potential utility in the intended applications.6–10 Therefore, the Pc self-associationrepresents the main limitation barrier of using such materials in optical related applications such as photodynamic therapy (PDT) or optical limiting.11–17 Many strategies have been attempted by several groups over the last decades to overcome the detrimental influence of aggregation by yielding new Pc materials with unique photophysical and optoelectronic properties tailored to the required applications.18–21 Recently, we found that the bulky substituents of 2,3,9,10,16,17,23,24-octakis(2,6-di-iso-propylphenoxy) phthalocyaninato zinc(II) (Pc1-Zn) effectively prohibited self-association of the macrocycle even within solid thin films22 and resulted in the formation of unique nanoporous cubic crystals.23 Introduction of different metals such as zinc, indium and gallium into the central Pc rings could lead to the formation of Pc derivatives with high triplet quantum yield24 which can be considered as a crucial requirement for efficient photosensitizers in PDT application. Similarly, also peripheral substituents may substantially influence photophysical properties. It should be also noted that the halogenated phthalocyanines showed promising photoelectric properties due to the heavy atom effect (i.e. spin–orbit coupling effect).25 Moreover, presence of halogen atoms lowers both the fluorescence quantum yield and the fluorescence lifetime, enhances the triplet quantum yield and decreases the triplet lifetime.26Considerable efforts have been made to explain the electronic structures of MPcs using theoretical calculations. The density functional theory (DFT) calculations were used to investigate the geometric structures and the electronic properties of several MPc complexes. TD-DFT calculations were also used to study the electronic behavior aspects of the Pcs, including the molecular geometries, ground-state and excited-state electronic structures, vibrational spectra and absorption spectra.27–34
Inspired by aforementioned findings, we proposed the synthesis of new Pc derivatives peripherally substituted with phenoxy groups decorated by halogen atoms with different metal ions (Ga, In, Mg, Zn) coordinated in the newly prepared Pc complexes to investigate their impact on photophysical features. In addition theoretical investigation of halogenated phthalocyanines were conducted on selected complexes (Pc2-Zn and Pc3-Zn) to evaluate their electronic properties. The molecular structures of the studied complexes were optimized using the B3LYP/SDD method and compared with a previously reported non-halogenated complex (Pc1-Zn).25
Experimental
General procedures
Among the starting materials, 2,6-dichlorophenol and 2,6-dibromophenol were purchased from Sigma-Aldrich and used directly without further purification. 4,5-Dichlorophthalonitrile (Pn1) was synthesized according to the literature method.35 Anhydrous cesium fluoride and unsubstituted zinc phthalocyanine were purchased from Sigma-Aldrich and used as received. TLC was performed using Polygram sil G/UV 254 TLC plates and visualization was carried out by ultraviolet light at 254 nm and 350 nm. Column chromatography was performed using Merck silica gel 60 of mesh size 0.040–0.063 mm. Anhydrous solvents were either supplied from Sigma-Aldrich and used as they were received or dried as described by Perrin.36 1H and 13C NMR Spectra were recorded using Bruker DPX 400 at 400 MHz and 100 MHz respectively and IR spectra were obtained from Jasco 6300 FTIR. UV-vis studies were done on a Varian Cary 5 spectrometer and Shimadzu UV-2600 spectrophotometer. Elemental analyses were carried out using ElementarVario Micro Cube. High resolution mass analyses (HRMS) were measured on Xevo G2-S QTof (Waters). In addition, clusters of peaks that correspond to the calculated isotope composition of the molecular ion were observed by matrix-assisted laser desorption ionization mass spectroscopy (MALDI-TOF) via ultrafleXtreme (Bruker). The MALDI-TOF mass data for Pcs are presented as the mass of the most intense peak in the cluster instead of exact mass. The calculated exact mass is beyond the detection of the instrument as its abundance is very low (e.g. for brominated Pcs below 0.01%). Melting points were determined via differential scanning calorimetry (DSC) analyses on Shimadzu DSC-50. Single crystal data collections were made on a Rigaku R-Axis Rapid diffractometer using filtered Mo-Kα radiation. The diffraction data were collected at a temperature of −123 °C (Oxford cryosystems). The fluorescence spectra were obtained using an AMINCO Bowman Series 2 luminescence spectrometer and are uncorrected.General method for synthesis of phthalonitriles Pn2 and Pn3
To a stirred solution of 2,6-dihalophenol (2.5 equiv.) and 4,5-dichlorophthalonitrile (1 equiv.) in dry DMF (10 mL) under nitrogen atmosphere, anhydrous cesium fluoride (5 equiv.) was added. The reaction mixture was further heated at 110 °C for 48 h. The reaction mixture, after being cooled, was poured into distilled water and the resulting precipitate was collected by filtration and washed several times with distilled water, then air-dried. Afterwards, the crude product was purified by column chromatography on silica eluting with dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (1:2, v/v) mixture to receive the desired product.
:hexane (1:2, v/v) mixture to receive the desired product.
General methods for the synthesis of metallophthalocyanines (Pc2-M, Pc3-M)
2,3,9,10,16,17,23,24-Octakis(2,6-dichlorophenoxy) phthalocyaninato zinc(II) (Pc2-Zn). Anhydrous Zn(OAc)2 as metal salt. The solvent system used for the column purification was CHCl3
:EtOAc (1:1, v/v) (yield: 93 mg, 18% (method A); 103 mg, 20% (method B)). M.p. over 300 °C; UV-vis (THF) λ/nm (ε/M−1 cm−1): 671 (334720), 642 (44032), 606 (49517), 359 (116350); IR (KBr) ν/cm−1 = 3065, 2916, 2850, 1609, 1570, 1545, 1489, 1477, 1443, 1400, 1340, 1275, 1240, 1205, 1171, 1136, 1092, 1067, 1024, 893, 866, 812, 791, 768, 746, 714; 1H NMR (400 MHz; DMSO-d6) δ/ppm = 7.83 (t, 8H, ArH), 7.88 (d, 16H, ArH), 8.00 (s, 8H, ArH); Anal. calc. for C80H32Cl16N8O8Zn + 2H2O: C, 50.52; H, 1.91; N, 5.89, found C, 50.67; H, 2.17; N, 5.97; MS (MALDI-TOF) (m/z): 1866 [M]+.
2,3,9,10,16,17,23,24-Octakis(2,6-dichlorophenoxy) phthalocyaninato magnesium(II) (Pc2-Mg). Mg(OAc)2·4H2O as the metal salt. The solvent system used for the column purification was CH2Cl2: hexane (2
:1, v/v) followed by CHCl3 (yield: 76 mg, 15% (method A); 96 mg, 19% (method B)). M.p. over 300 °C; UV-vis (THF) λ/nm (ε/M−1 cm−1): 672 (295620), 643 (40805), 609 (43124), 362 (115620); IR (KBr) ν/cm−1 = 3067, 2993, 2924, 2829, 1657, 1639, 1610, 1569, 1545, 1526, 1483, 1443, 1396, 1340, 1277, 1242, 1207, 1169, 1134, 1084, 1022, 895, 866, 791, 771, 752, 715, 536; 1H NMR (400 MHz; CDCl3:DMSO-d6 (5:1)) δ/ppm = 7.60 (t, 8H, ArH), 7.72 (d, 16H, ArH), 8.1 (s, 8H, ArH); Anal. calc. for C80H32Cl16MgN8O8: C, 52.66; H, 1.77; N, 6.14, found C 52.63, H 2.38, N 6.29; MS (MALDI-TOF) (m/z): 1823 [M]+.
2,3,9,10,16,17,23,24-Octakis(2,6-dichlorophenoxy) phthalocyaninato gallium(III) chloride (Pc2-Ga). Anhydrous GaCl3 as the metal salt. The solvent system used for the column purification was CH2Cl2
:hexane (2:1, v/v) followed by CHCl3:DMF (50:1, v/v) (yield: 31 mg, 15% (method A); 29 mg, 14% (method B)). M.p. over 300 °C; UV-vis (THF) λ/nm (ε/M−1 cm−1): 683 (248660), 652 (35933), 616 (38544), 366 (97640); IR (KBr) ν/cm−1 = 3069, 2923, 2851, 1608, 1572, 1487, 1443, 1400, 1342, 1277, 1242, 1205, 1175, 1134, 1092, 1026, 899, 874, 793, 770, 744, 712; 1H NMR (400 MHz; DMSO-d6) δ/ppm = 7.93 (t, 8H, ArH), 8.02 (d, 16H, ArH), 8.14 (s, 8H, ArH); anal. calc. for C80H32Cl17GaN8O8: C, 50.43; H, 1.69; N, 5.88, found C, 50.28; H, 1.52; N, 5.69; MS (MALDI-TOF) (m/z): 1905 [M+].
2,3,9,10,16,17,23,24-Octakis(2,6-dichlorophenoxy) phthalocyaninato indium(III) chloride (Pc2-In). Anhydrous InCl3 as the metal salt. The solvent system used for the column purification was CH2Cl2
:hexane (2:1, v/v) followed by EtOAc:DMF (1:1, v/v) (yield: 88 mg, 17% (method A); 103 mg, 20% (method B)). M.p. over 300 °C; UV-vis (THF) λ/nm (ε/M−1 cm−1): 691 (239400), 658 (38033), 623 (40300), 368 (100520); IR (KBr) ν/cm−1 = 3067, 1724, 1671, 1611, 1571, 1486, 1442, 1399, 1340, 1278, 1240, 1205, 1173, 1135, 1091, 1025, 898, 865, 793, 769, 742, 714, 535; 1H NMR (400 MHz; CDCl3) δ/ppm = 7.36 (t, 8H, ArH), 7.83 (d, 16H, ArH), 8.13 (s, 8H, ArH); anal. calc. for C80H32Cl17InN8O8: C, 49.26; H, 1.65; N, 5.74, found C, 49.48; H, 1.78; N, 5.52; MS (MALDI-TOF) (m/z): 1949 [M]+.
2,3,9,10,16,17,23,24-Octakis(2,6-dibromophenoxy) phthalocyaninato zinc(II) (Pc3-Zn). Anhydrous Zn(OAc)2 as the metal salt. The solvent system used for the column purification was CHCl3
:EtOAc (2:1, v/v) (yield: 92 mg, 18% (method A); 107 mg, 21% (method B)). M.p. over 300 °C; UV-vis (THF) λ/nm (ε/M−1 cm−1): 673 (327260), 642 (44031), 609 (45518), 358 (112200); IR (KBr) ν/cm−1 = 3418, 3064, 2953, 1721, 1611, 1576, 1561, 1486, 1470, 1456, 1433, 1399, 1340, 1273, 1230, 1170, 1136, 1092, 1045, 1027, 947, 894, 864, 808, 768, 746, 725, 658; 1H NMR (400 MHz; CDCl3:DMSO-d6 (5:1)) δ/ppm = 7.42 (t, 8H, ArH), 7.87 (d, 16H, ArH), 8.10 (s, 1H, ArH); anal. calc. for C80H32Br16N8O8Zn: 37.29; H, 1.25; N, 4.35, found C 37.58, H 1.19, N 4.76; MS (MALDI-TOF) (m/z): 2577 [M]+.
2,3,9,10,16,17,23,24-Octakis(2,6-dibromophenoxy) phthalocyaninato magnesium(II) (Pc3-Mg). Mg(OAc)2·4H2O as the metal salt. The solvent system used for the column purification was CHCl3
:hexane (2:1, v/v) followed by CHCl3:EtOAc (1:1, v/v) (yield: 80 mg, 16% (method A); 95 mg, 19% (method B)). M.p. over 300 °C; UV-vis (THF) λ/nm (ε/M−1 cm−1): 675 (276210), 642 (40468), 610 (43366), 363 (123360); IR (KBr) ν/cm−1 = 3065, 2868, 2833, 1611, 1560, 1470, 1455, 1433, 1396, 1339, 1273, 1233, 1168, 1133, 1085, 1046, 1025, 946, 894, 864, 807, 768, 752, 724, 521; 1H NMR (400 MHz; CDCl3:DMSO-d6 (5:1)) δ/ppm = 7.29 (t, 8H, ArH), 7.79 (d, 16H, ArH), 8.15 (s, 8H, ArH); anal. calc. for C80H32Br16MgN8O8: C, 37.89; H, 1.27; N, 4.42, found C, 37.55; H, 1.09; N, 4.61; MS (MALDI-TOF) (m/z): 2535 [M]+.
2,3,9,10,16,17,23,24-Octakis(2,6-dibromophenoxy) phthalocyaninato gallium(III) chloride (Pc3-Ga). Anhydrous GaCl3 as the metal salt. The solvent system used for the column purification was CHCl3
:EtOAc (2:1, v/v) followed by CHCl3:DMF (50:1, v/v) (yield: 78 mg, 15% (method A); 62 mg, 12% (method B)). M.p over 300 °C; UV-vis (THF) λ/nm (ε/M−1 cm−1): 684 (296330), 649 (37295), 619 (45577), 368 (118200); IR (KBr) ν/cm−1 = 3067, 2961, 1666, 1611, 1561, 1506, 1458, 1432, 1405, 1343, 1278, 1237, 1173, 1145, 1095, 1072, 1034, 1001, 898, 804, 768, 748, 725, 545, 521; 1H NMR (400 MHz; CDCl3) δ/ppm = 7.39 (t, 8H, ArH), 7.88 (d, 16H, ArH), 8.18 (s, 1H, ArH); anal. calc. for C80H32Br16ClGaN8O8: C, 36.72; H, 1.23; N, 4.28, found C, 36.58; H, 1.09; N, 4.16; MS (MALDI-TOF) (m/z): 2616 [M]+.
2,3,9,10,16,17,23,24-Octakis(2,6-dibromophenoxy) phthalocyaninato indium(III) chloride (Pc3-In). Anhydrous InCl3 as the metal salt. The solvent system used for the column purification was CHCl3
:EtOAc (2:1, v/v) (yield: 84 mg, 16% (method A); 79 mg, 15% (method B)). M.p. over 300 °C; UV-vis (THF) λ/nm (ε/M−1 cm−1): 693 (300590), 664 (47080), 625 (50661), 369 (120750); IR (KBr) ν/cm−1 = 3066, 1724, 1663, 1607, 1560, 1485, 1454, 1431, 1393, 1335, 1276, 1242, 1170, 1135, 1093, 1068, 1044, 1026, 993, 896, 875, 799, 767, 742, 725, 520; 1H NMR (400 MHz; CDCl3) δ/ppm = 7.36 (t, 8H, ArH), 7.83 (d, 16H, ArH), 8.13 (s, 8H, ArH); anal. calc. for C80H32Br16ClInN8O8: C, 36.10; H, 1.21; N, 4.21, found C 36.25, H 1.09, N 3.98; MS (MALDI-TOF) (m/z): 2661 [M]+.
Computational details
DFT have been implemented to obtain the most energetically stable geometry and molecular orbitals. TD-DFT were further carried out on the optimized geometry to generate the electronic absorption spectra of some halogenated phthalocyanine complexes (Pc2-Zn and Pc3-Zn) compared with the previously reported non-halogenated Pc1-Zn. The aforementioned methods were successfully used to calculate the energy minimized structure, molecular orbitals and electronic absorption spectra of the target complexes.30–33 Geometry optimization and frequency calculations of this symmetrical Pc derivatives were calculated at the density functional B3LYP level with the SDD basis set.37,38 Forty states of each compound were considered in the TD-DFT calculations. All of the calculations were performed using Gaussian09W Multiprocessor/core version (64 bit) RevD software package.39 The final optimized structures were visualized using Gaussview version 5.0.9 and Chemcraft software packages.40,41Fluorescence quantum yields
All samples were repurified before photophysical measurements by preparative TLC (Merck Kieselgel 60 F254) to ensure high purity of the samples using the following eluents: toluene:THF 10:1 (for Pc2-Zn, Pc2-Mg, Pc3-Zn and Pc3-Mg), MeOH followed by chloroform:THF 10:1 (for Pc2-In and Pc3-In) or MeOH followed by chloroform:THF 20:1 (for Pc2-Ga and Pc3-Ga). The corresponding green spots were scraped and the pure product was extracted from silica using chloroform:pyridine 1:1 (for Pc2-In and Pc3-In) or THF (the rest of the samples), filtered and evaporated to dryness. Fluorescence quantum yields (ΦF) of the studied Pcs were determined by comparative method in THF and calculated using the following equation (eqn (1)):
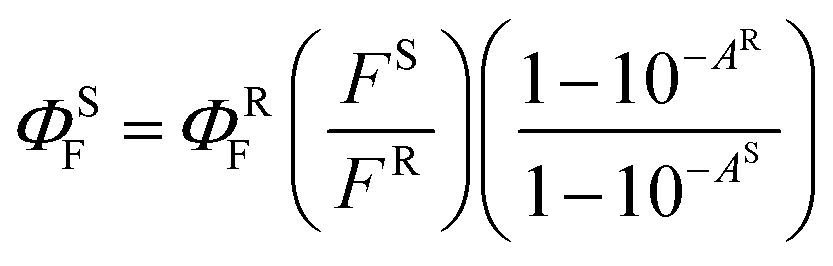 | (1) |
Singlet oxygen quantum yields
The quantum yields of the singlet oxygen (ΦΔ) were determined in THF according to a slightly modified published procedure42 using the decomposition of a chemical trap 1,3-diphenylisobenzofuran (DPBF) with ZnPc as a reference (ΦΔ = 0.53 in THF).43 The detailed procedure was as follows: 2.5 mL of a DPBF stock solution in THF (5 × 10−5 M) was transferred into a 10 × 10 mm quartz optical cell and was saturated with oxygen for 1 min. A stock solution of the tested compound in THF (typically 30 μL) was then added to achieve an absorbance of the final solution in the Q-band maximum of approximately 0.1. The solution was stirred and irradiated using a xenon lamp (100 W, ozone-free XE DC short-arc lamp, Newport). The incident light was filtered through a water filter (6 cm) and an OG530 cut-off filter (Newport) to remove the heat and the light below 523 nm, respectively. Decrease of DPBF in solution with irradiation time was monitored at 414 nm. All experiments were performed in triplicate, and the data presented herein represent the mean of the three experiments (estimated error: ±10%).Results and discussions
Synthesis
The synthetic procedure for preparing the target halogenated phthalocyanine derivatives (Pc2-M, Pc3-M) is depicted in Scheme 1. The starting phthalonitrile precursors Pn2 and Pn3 were prepared in a good yield (∼70%) from the conventional aromatic nucleophilic substitution reaction between 4,5-dichlorophthalonitrile (Pn1) with the 2,6-dihalogenated phenols (2,6-dichlorophenol and 2,6-dibromophenol) using DMF as solvent in the presence of anhydrous CsF. Unfortunately, the fluorinated and iodinated phenols failed to produce the corresponding phthalonitriles and gave cross-linked material and mono-substituted phthalonitrile in very low yield, respectively. Subsequently, the prepared phthalonitriles Pn2 and Pn3 underwent metal-mediated cyclotetramerization in quinoline or pentanol/DBU using the appropriate metal salt: Zn(OAc)2, Mg(OAc)2, GaCl3 and InCl3 to afford the corresponding phthalocyanine derivatives Pc2-M and Pc3-M in reasonable yields. The solubility of the prepared complexes was found to be excellent in most organic solvents (e.g. DCM, chloroform, toluene, ethyl acetate and THF) which facilitated their purification using column chromatography. Structural identifications and purity of the prepared complexes were confirmed by various characterization techniques such as 1H NMR, UV-vis, IR and elemental analysis which gave results consistent with the proposed complex structures.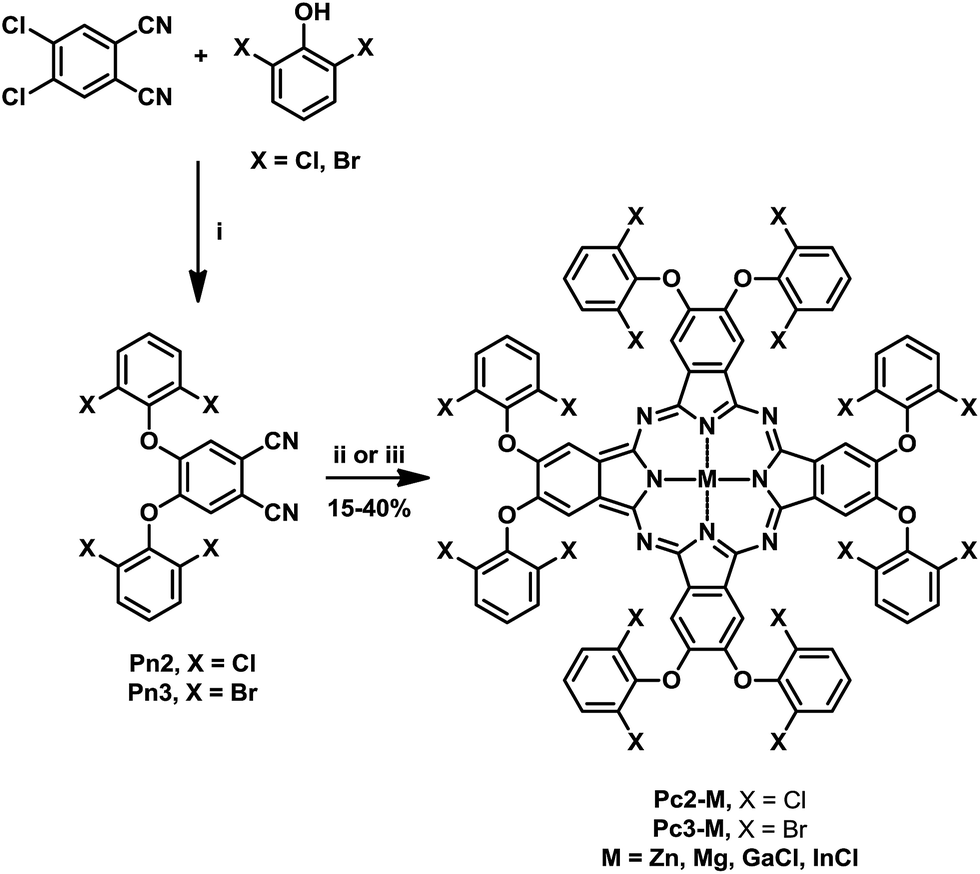 | ||
| Scheme 1 Synthesis of complexes Pc2-M and Pc3-M. Reagents and conditions: (i) DMF, anhydrous CsF, 110 °C, 48 h; (ii) appropriate metal salt, quinoline, 180 °C, 12 h; (iii) n-pentanol, DBU, appropriate metal salt, 140 °C, 24 h. | ||
The complexes were further characterized by MALDI-TOF spectrometry that confirmed the mass corresponding to the expected values. Some of them showed the proposed calculated masses only Fig. S10,† others gave spectra with fragments corresponding to loss of some halogen atoms. For example, the spectrum of Pc3-Zn consists of a cluster of molecular ion peaks centered at 2577 which is assigned to the corresponding mass of Pc complex. However, other clusters also appeared in the spectrum as a consequence of fragmentation. Their m/z matched the complex missing one and more bromine atoms as depicted in Fig. 1. The experimental isotopic pattern perfectly agreed with the theoretical one.
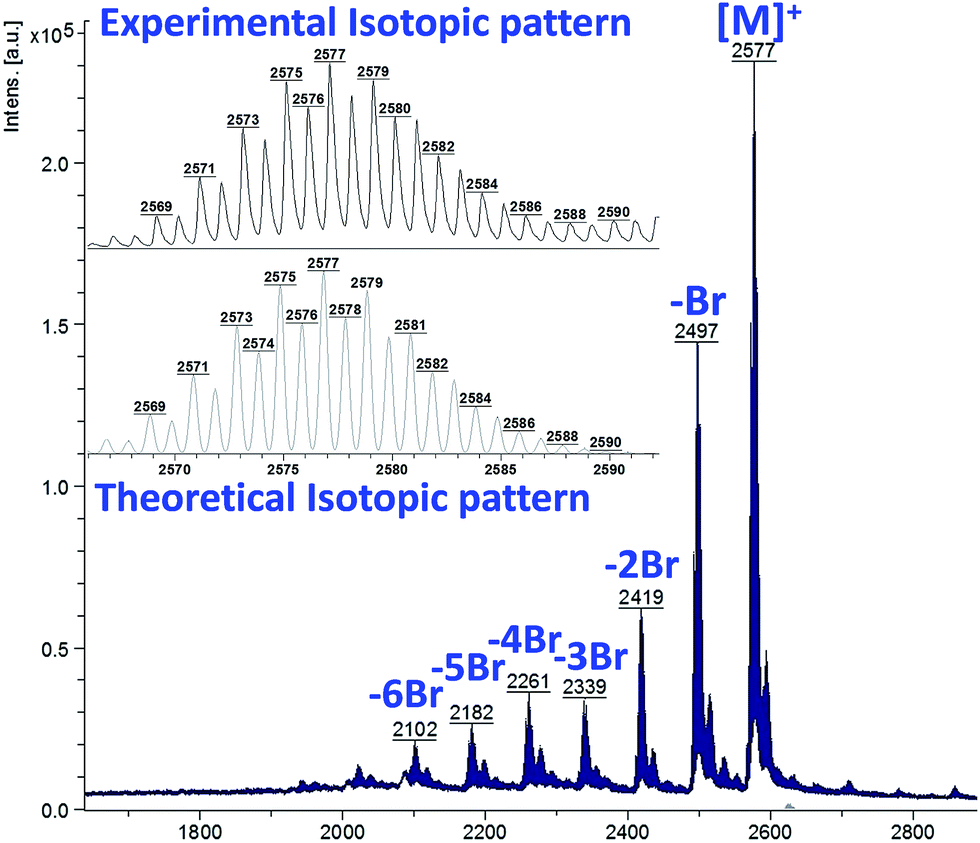 | ||
| Fig. 1 MALDI-TOF (m/z) spectra and isotopic distribution of Pc3-Zn. | ||
Structural and self-association characterization
The intrinsic behavior of disc-like phthalocyanine molecules arising from the π–π stacking can negatively affect their opto-electronic properties and greatly limit their efficiency in many intended applications. Therefore, many structural modification protocols have been adapted to control the degree of molecular self-association. UV-vis and NMR spectroscopic techniques are the best tools to evaluate the aggregation behavior of phthalocyanine molecules. Aggregated Pcs can be detected by 1H NMR spectrum exhibiting characteristic broad peaks. UV-vis spectra can also be used to assess the aggregation from the position and the shape of the corresponding Q-band.The 1H NMR spectra of the prepared complexes Pc2-M and Pc3-M were recorded in either DMSO or CDCl3 and exhibited well resolved spectra. In addition to this, the spectra of these complexes exhibited the same high quality as their corresponding precursors (Pn2 and Pn3) and the previously reported non-aggregating Pc1-Zn.25 As shown in Fig. 2, the NMR spectra of Pc2-Zn and its precursor Pn2 show three distinct sharp signals in the aromatic region (triplet, doublet and singlet). The good quality of the obtained spectra was found to be unaffected by the concentration and remained unchanged with well resolved signals even at high concentration (2 × 10−2 M). Such behavior can be clearly attributed to the steric hindrance exerted by the peripheral phenoxy substituents bearing halogen atoms that lie almost perpendicular to the plane of the Pc core and consequently prevent the efficient π–π stacking of the macrocycle units. This conformation adapted by the bulky phenoxy groups was evidently confirmed by the X-ray analyses as shown in the ESI† and discussed below.
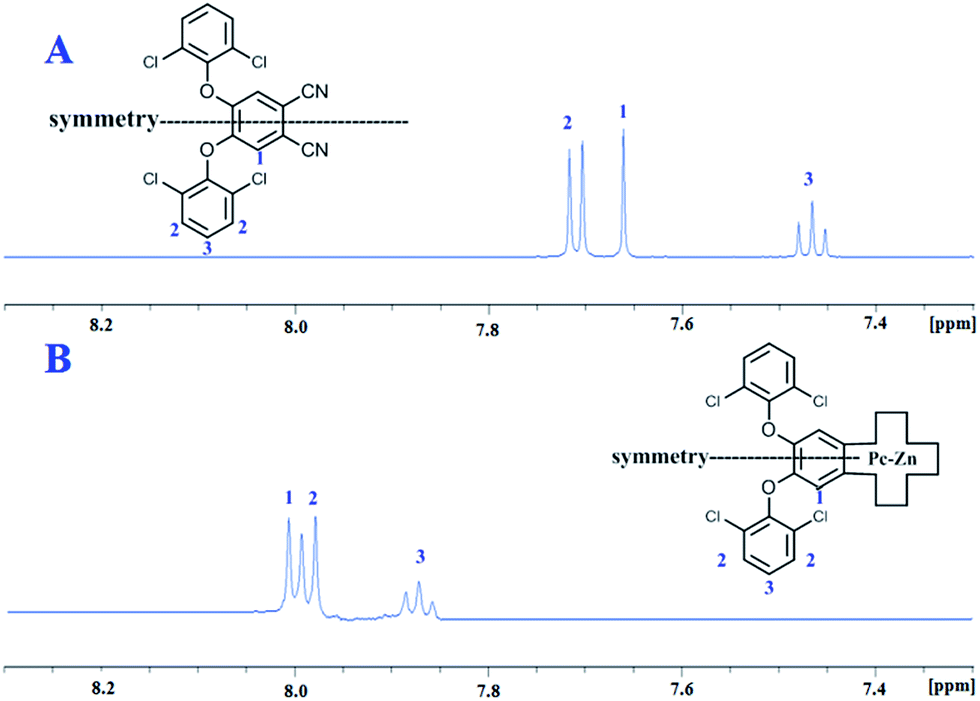 | ||
| Fig. 2 (A) 1H NMR of phthalonitrile Pn2 in DMSO-d6; (B) 1H NMR of Pc2-Zn in DMSO-d6. | ||
The complex Pc2-Zn was examined using the NMR technique at variable temperatures (25–95 °C) (Fig. S11†). The three signals in the Pc2-Zn spectrum moved apart from each other at higher temperature. However, the good quality of the spectrum remained unchanged. The doublet and triplet which belong to the phenoxy substituents shifted slightly into up field region whereas the singlet which belongs to the Pc core shifted into the down field area. This can be attributed to the increased free rotation around aryl ether linkage at high temperature which can decrease the ring current effect of the Pc ring. Conversely, such rotations move the Pc core proton within the proximity effect of the halogen atoms, therefore, the downfield shift of the singlet signal was recognized at high temperature due to the current effects exerted by the halogen atom.
Further assessment of the aggregation behavior of the prepared complexes was performed using UV-vis spectroscopy technique. As anticipated, the sharp unperturbed single Q-band, typical of monomeric species, was the characteristic feature of all the complexes spectra in different organic solvents (DCM, CHCl3, THF and DMF). For example, the absorption spectrum of Pc2-Zn showed a single sharp Q-band at λmax = 671 nm (Fig. 3 and Table 1) which is typical for non-aggregated species belonging to D4h symmetry. Further aggregation evaluation was performed by tracing the Q-band appearance and its position at different concentrations ranging from 10 × 10−6 M to 100 × 10−6 M. There were no spectral changes as the concentration varied within the studied concentration range, confirming that these complexes remained in truly monomeric form. As depicted in (Fig. 3), the Q-band absorption maximum of Pc2-Zn at 671 nm, perfectly follows the Lambert–Beer law in the studied concentration range and its molar extinction coefficient remained constant indicating a purely monomeric form. Another confirmation of non-aggregated character of the spectra is a perfect accordance of excitation spectra with the absorption spectra (see below). According to the aforementioned results based on the NMR and UV-visible spectroscopic analysis, the non-aggregating nature of the prepared Pc systems is well established even at higher solution concentrations. This achievement emphasized that the steric factor of halogen atoms in ortho position (e.g. chlorine and bromine) is sufficiently enough to keep the orthogonal conformation of phenoxy substituents relative to the Pc core (Table 2).
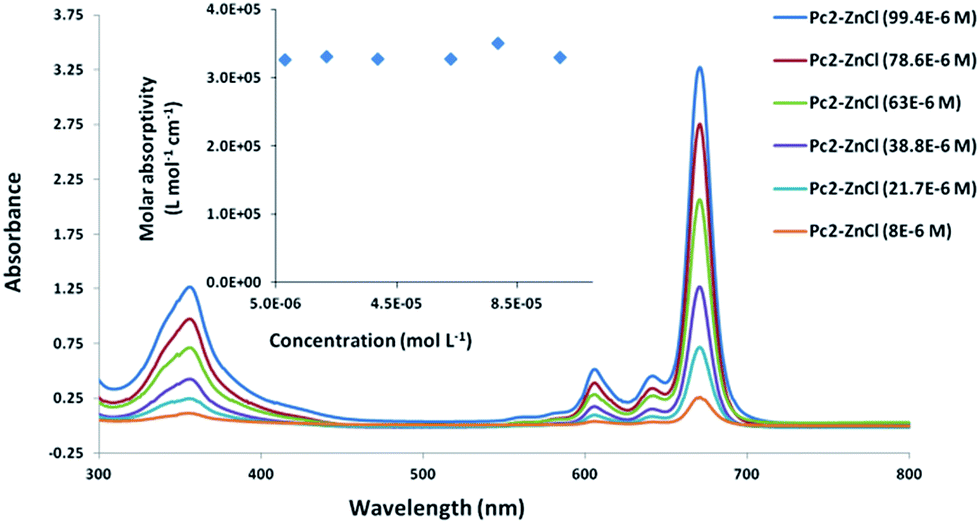 | ||
| Fig. 3 UV-vis spectra of Pc2-Zn in DMF at different concentrations ranging from 10 to 100 μM measured in cuvette with 1 mm path length, inset: dependence of the extinction coefficient at λmax on the concentration of Pc2-Zn. | ||
| Compound | ΔE/eV | f | Main configuration | λmax (nm) | Coefficient | Assignment |
|---|---|---|---|---|---|---|
| Pc1-Zn | 2.0027 | 0.4783 | H → L+1 | 619 | 0.67173 | π–π* |
| 2.7754 | 0.3707 | H−4 → L | 446 | 0.62514 | π–π* | |
| 2.7817 | 0.4405 | H−4 → L+1 | 445 | 0.47044 | π–π* | |
| 3.3330 | 0.0231 | H−8 → L | 371 | 0.39858 | π–π* | |
| Pc2-Zn | 2.0474 | 0.4371 | H → L | 605 | 0.68022 | π–π* |
| 2.0480 | 0.4364 | H → L+1 | 606 | 0.68013 | π–π* | |
| 2.9401 | 0.4375 | H−4 → L | 421 | 0.66382 | π–π* | |
| 2.9409 | 0.4368 | H−4 → L+1 | 421 | 0.66441 | π–π* | |
| 3.5939 | 0.4397 | H−10 → L | 344 | 0.66161 | π–π* | |
| 3.5948 | 0.4414 | H−10 → L+1 | 344 | 0.66032 | π–π* | |
| Pc3-Zn | 2.0428 | 0.4413 | H → L | 606 | 0.67219 | π–π* |
| 2.0429 | 0.4411 | H → L+1 | 606 | 0.67217 | π–π* | |
| 2.9332 | 0.4351 | H−4 → L | 422 | 0.53639 | π–π* | |
| 2.9333 | 0.4353 | H−4 → L+1 | 422 | 0.53664 | π–π* | |
| 3.5699 | 0.3765 | H−8 → L | 347 | 0.55731 | π–π* | |
| 3.5701 | 0.3759 | H−8 → L | 347 | 0.55593 | π–π* |
| HOMO | HOMO−1 | HOMO−2 | HOMO−3 | HOMO−4 | LUMO | LUMO+1 | LUMO+2 | LUMO+3 | LUMO+4 | ΔE (eV) LUMO−HOMO | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pc1-Zn | −5.01 | −6.00 | −6.04 | −6.04 | −6.04 | −2.88 | −2.87 | −1.14 | −0.970 | −0.821 | 2.19 |
| Pc2-Zn | −5.315 | −6.361 | −6.396 | −6.396 | −6.414 | −3.098 | −3.098 | −1.413 | −1.272 | −1.246 | 2.22 |
| Pc3-Zn | −5.309 | −6.352 | −6.387 | −6.387 | −6.405 | −3.095 | −3.095 | −1.427 | −1.393 | −1.376 | 2.21 |
For comparison, octasubstituted zinc Pcs with phenoxy groups bearing a variety of electron-withdrawing and -donating groups at the para position were found to be in aggregated form at concentration over 1 × 10−5 M in DMSO or CHCl3 even bearing bulky tBu substituent.44 Substituents placed in meta positions on aryloxy substituents typically inhibit the aggregation well45,46 but examples of aggregated derivatives may be found47,48 in literature. Indeed, the tendency to aggregations decreases in order of para > meta > ortho substitution49 of the peripheral aryloxy substituent as suggested also by a study of mesomorphic properties of different Pcs. For this reason, substitution of phenoxy substituents in ortho positions seems to be crucial for efficient inhibition of aggregation.
The UV-vis analysis was extended to evaluate the effect of halogen atoms by comparing the spectra of the prepared complexes with the previously reported non-halogenated Pc1-Zn. The Q-band of the Pc2-Zn containing 16 chlorine atoms is slightly blue-shifted in comparison to Pc1-Zn (ca. 4 nm) due to the inductive effect exerted by the chlorine atoms which reduce the contribution of oxygen lone pairs of the aryloxy substituents to Pc core and consequently increase the HUMO–LUMO energy gap. This is supported by the slight red shifted Q-band position of Pc3-Zn (containing 16 bromine atoms) relative to that of Pc2-Zn which could be explained by the virtue of higher electronegativity of chlorine over that of bromine atoms.
The absorption spectra of the prepared complexes containing different metals were also examined to confirm the impact of size and electronic effect of the inserted metals. We found that the insertion of different transition metals within the Pc cores can tune their Q-band position due to the destabilization of the Pc π-system. As shown in (Table 3), the indium central metal produced the largest red shift followed by the gallium metal ion as compared to the corresponding zinc derivatives. This can be attributed either to the distortion of D4h symmetry due to the displacement of indium out of the π-plane structure creating non-planar system or to the weakness of the coordination bond arising from the decreased electronegativity of the inserted metal.
| Complex | λmax (nm) | λem (nm) | εmax (M−1 cm−1) | ΦFb,c | ΦΔb,d |
|---|---|---|---|---|---|
| a Absorption maximum at the Q-band (λmax), emission maximum (λem), extinction coefficient (εmax), fluorescence quantum yield (ΦF) and singlet oxygen quantum yield (ΦΔ).b Mean of three independent measurements, estimated error: ±10%.c With ZnPc as reference (ΦF in THF = 0.32).d With ZnPc as reference (ΦΔ in THF = 0.53). | |||||
| Pc2-Mg | 672 | 676 | 2.96 × 105 | 0.62 | 0.21 |
| Pc2-Zn | 671 | 675 | 3.35 × 105 | 0.33 | 0.53 |
| Pc2-Ga | 683 | 688 | 2.49 × 105 | 0.23 | 0.52 |
| Pc2-In | 691 | 695 | 2.40 × 105 | 0.030 | 0.83 |
| Pc3-Mg | 675 | 679 | 2.76 × 105 | 0.50 | 0.24 |
| Pc3-Zn | 673 | 676 | 3.27 × 105 | 0.32 | 0.53 |
| Pc3-Ga | 684 | 692 | 2.96 × 105 | 0.19 | 0.56 |
| Pc3-In | 693 | 698 | 3.01 × 105 | 0.028 | 0.82 |
Single crystal X-ray diffraction analysis
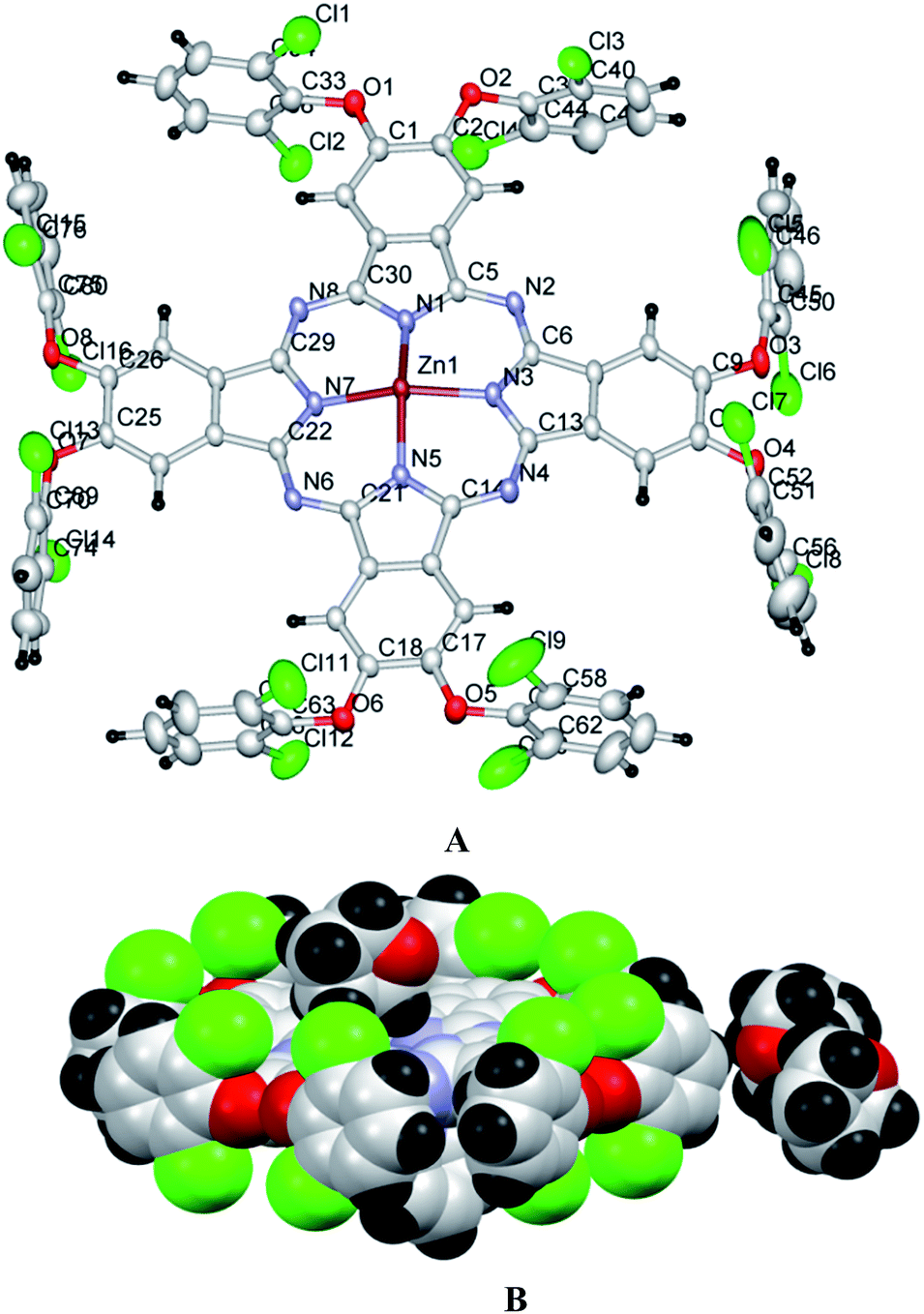 | ||
| Fig. 4 Crystal structure of Pc2-Zn; (A) thermal ellipsoid representation, (B) space filling illustration showing the perpendicular orientation of chlorine atoms with respect to the Pc core and the occupancy of one of the THF solvent within the molecular cage. A molecule of pyridine is coordinated from below (A few drops of pyridine were added to develop good crystal). Color code: red – oxygen; blue – nitrogen; gray – carbon; green – chlorine; black – hydrogen. | ||
The structural features of phthalocyanine precursors and Pc2-Zn obtained from X-ray diffraction analysis can be considered to be similar under solution conditions as well. Therefore, the monomeric nature of the various Pc-M systems confirmed by NMR and UV-vis spectral analysis can be attributed to the steric constraints imposed by the bulky halogen-containing phenoxy substituents as discussed before.
Theoretical calculations
All geometry optimizations were performed using D4h symmetry50 in gas phase. The most important bond distances and angles listed in Table S6,† and the labeling scheme is presented in Fig. 5. The optimized structure of Pc2-Zn (Fig. 5 and Table S4†) is in good agreement with the crystal structure determined by X-ray analysis Fig. 4, which confirms the perpendicular orientation of phenoxy groups in respect to Pc core of the studied complexes (Pc1-Zn, Pc2-Zn and Pc3-Zn). In general, the unsubstituted ZnPc is reported to have perfectly planar structure.51 However, it was noted that the halogen atoms directly attached to porphyrin core at both α and β positions caused a structural deformation.52 This deformation led to significant changes in the energy levels and consequently the position of the Q and B-bands.52 Our results showed that the halogenated phenoxy groups on Pc have a smaller distortion effect than the halogens directly attached to porphyrin core.52 The out of plane deformations are negligibly small and therefore, all complexes retained D4h symmetry. Accordingly, their UV-visible spectra were not affected.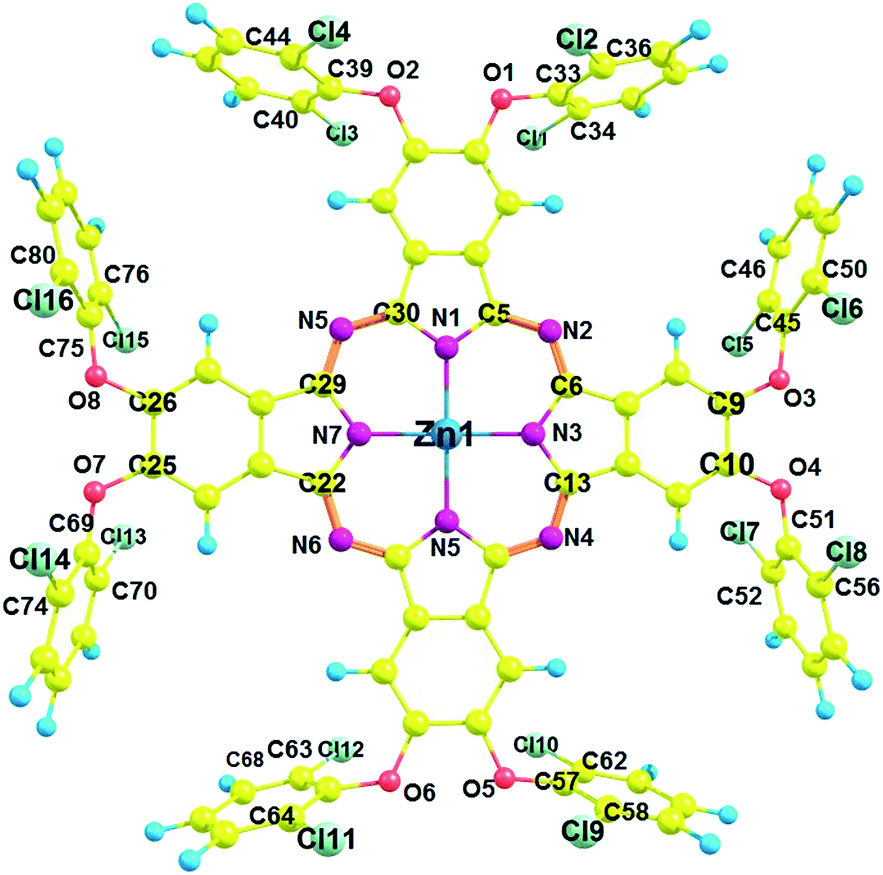 | ||
| Fig. 5 Optimized structure of Pc2-Zn using B3LYP/SDD showing the perpendicular orientation of chlorine atoms with respect to Pc core. Color code: red – oxygen; magenta – nitrogen; yellow – carbon; green – chlorine; blue – hydrogen; dark blue – zinc. | ||
The electronic structure and vertical excitation energy were calculated to gain further insight into the optical and electronic properties for Pc2-Zn and Pc3-Zn along with Pc1-Zn using DFT approach, which can deduce accurate structures and spectroscopic properties of organometallic porphyrins and phthalocyanines.26–33 Such calculations can rationalize the observed spectral features based on the molecular structures of the studied complexes. Time-dependent density functional theory (TD-DFT) calculations for Pc1-Zn, Pc2-Zn and Pc3-Zn were performed to study the influence of the substituents on the electronic spectra, based on the geometries optimized at the B3LYP/SDD level. The results of TD-DFT calculations on selected electronic transitions with the corresponding oscillator strengths and excitation configurations of the target complexes are listed in Table 1. The molecular orbital energy diagrams calculated by a DFT approach are shown in Fig. 6, and the molecular orbital surfaces are depicted in Fig. 7 and Fig. S12.†
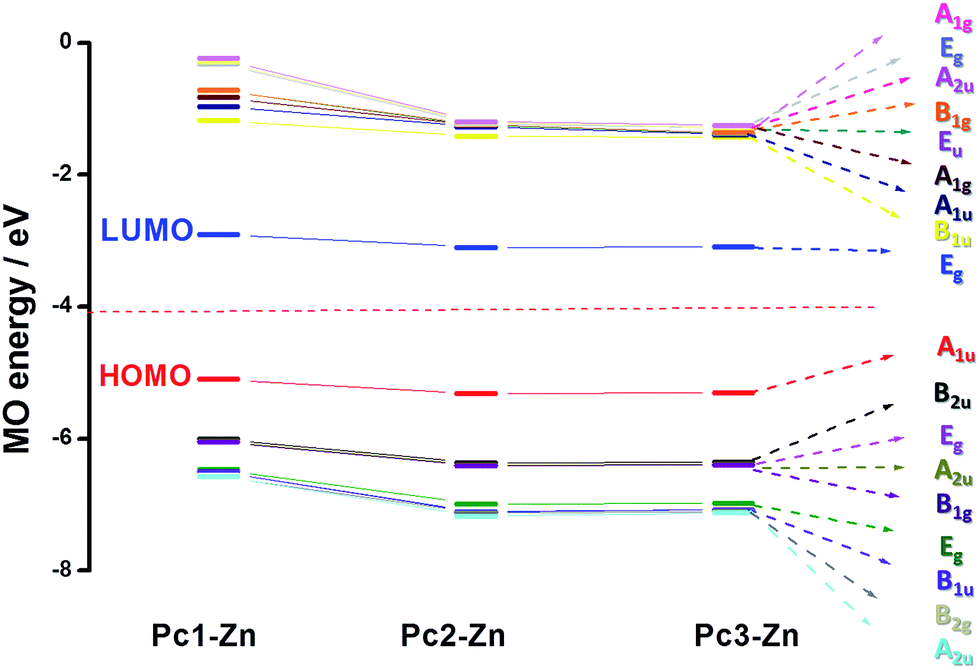 | ||
| Fig. 6 Molecular energy level predicted for (Pc1-Zn, Pc2-Zn and Pc3-Zn). | ||
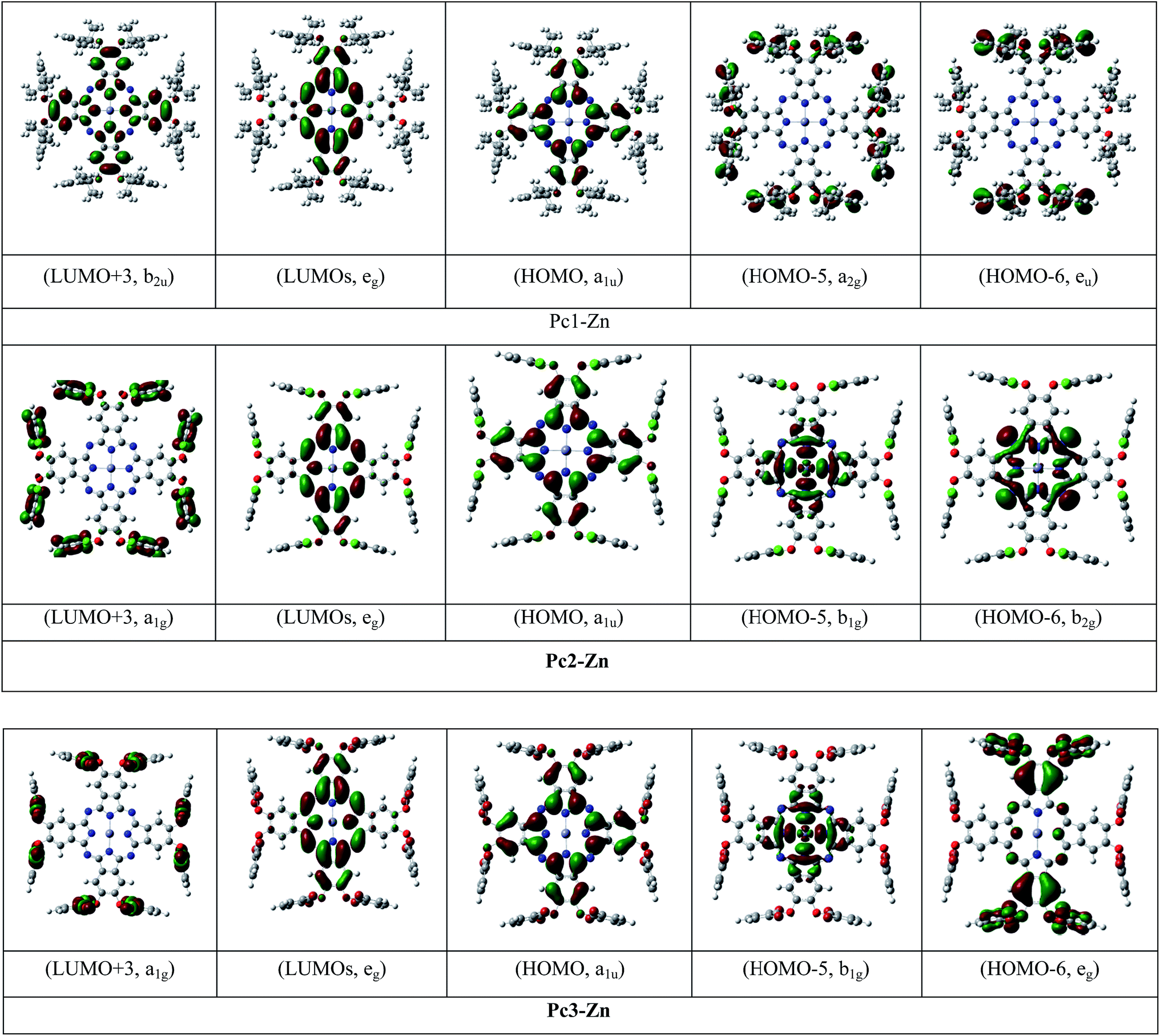 | ||
| Fig. 7 Molecular orbital surfaces of some selected HOMOs and LUMOs for Pc1-Zn, Pc2-Zn and Pc3-Zn. | ||
The UV-vis absorption spectrum is based mainly on the vertical transition between molecular energy levels, which determine the position of the B and Q bands. Thus, we investigated the molecular energy levels and the vertical transition energy to explore the halogenation effects on photophysical properties. From Table 1, the absorption spectra of the non-halogenated Pc1-Zn consists of four main transitions. The first electron transition related to the Q-band (HOMO → LUMO+1) observed at (λmax = 619 nm), which corresponds to the experimentally obtained band at 675 nm in the UV-vis spectra shown in Fig. 8. The B-band spectra consists of 3 main transitions observed at 446, 445 and 371 nm. The calculated band around 371 nm corresponded to the experimentally measured peak at 359 nm shown in Fig. 8. The halogenated complexes Pc2-Zn (λmax = 605 nm) and Pc3-Zn (λmax = 606 nm) exhibited ca. 14 nm hypsochromic shift of the Q-band as compared to Pc1-Zn (619 nm) at the same level of theory (Fig. 8). Both Pc2-Zn and Pc3-Zn complexes were composed of six main transitions. The Q-band region consists of two transitions (HOMO → LUMO, LUMO+1) observed at ca. 606 nm. The other transitions observed at 421 and 334 nm corresponded to B-band (Table 1). Moreover other weak transitions appeared at 466 and 396 nm and were attributed to n → π* transitions in case of Pc1-Zn. Such weak transitions were observed as a small shoulder around (430 nm) in the experimentally determined UV-vis spectra (Fig. 8) and calculated at (421 nm) as shown in Fig. 8 and depicted in Table 1. This shoulder were arises from n → π* transitions of lone pair of electrons of the ether oxygen.53 The band around 421 nm were reported as a ligand–metal charge transfer (LMCT) absorption band shown in Fig. 7 and Fig S12.†54 These n → π* transitions were also observed for both Pc2-Zn and Pc3-Zn as ligand to metal charge transfer (LMCT) shown in Fig. 7 and Fig S12.† The MLCT and LMCT can be represented by b1g, b2g, eu, a1g and eu molecular orbitals as shown in Fig. 7 and Fig S12.† The HOMO is represented by a a1u π-orbital, while the LUMO consists of degenerate eg π-orbitals, as supposed by the classic Gouterman's four-orbital orbital scheme description.55 The DFT calculations show a significant stabilization of the a2u (HOMO−4) π-orbital in comparison to the b2u (HOMO−1) in the studied complexes. However, in case of both halogenated complexes (Pc2-Zn and Pc3-Zn) a significant stabilization on the b1g (HOMO−5) and b2g (HOMO−6) (i.e. nitrogen lone-pair-based n-orbitals) occurs. Therefore, the metal-centred dx2−y2 orbital for Pc2-Zn and Pc3-Zn has a significantly more contribution in the pyrrolic nitrogen-centred nonbonding b1g molecular orbital than in the non-halogenated Pc1-Zn. Moreover, the LUMO+3 of the halogenated complexes (Pc2-Zn and Pc3-Zn) is mainly localized over the phenoxy groups shown in Fig.8 and Fig S12.†
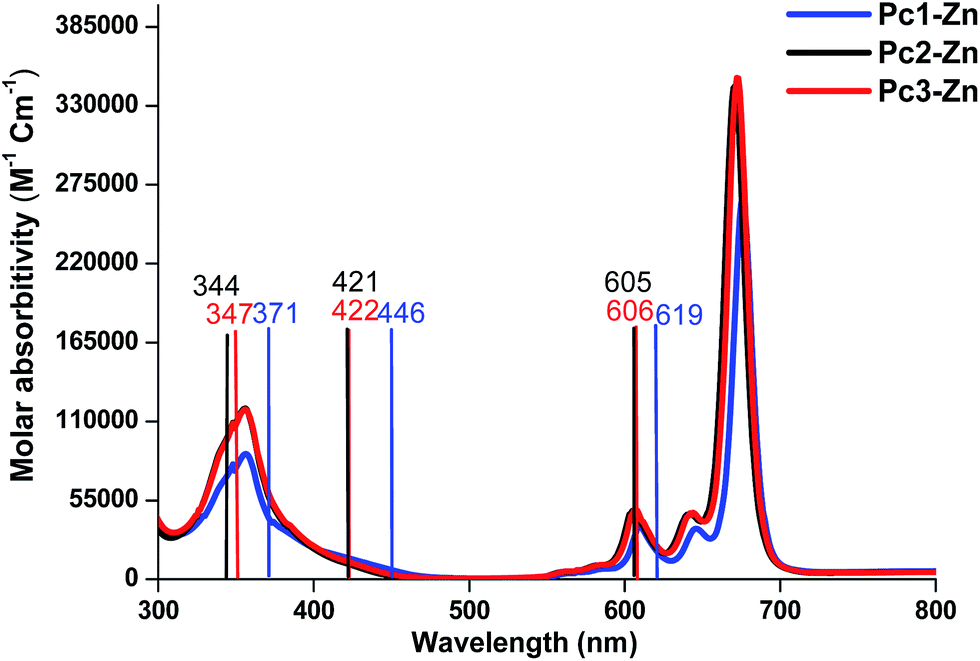 | ||
| Fig. 8 Comparison between gas phase (bars) and the experimentally observed UV-vis spectra of Pc1-Zn, Pc2-Zn and Pc3-Zn. | ||
Photophysics
Fluorescence emission spectra were collected in THF. They had a typical shape for this type of complexes and they mirrored the absorption spectra in the Q-band area (Fig. 9 and Fig. S14†). The excitation spectra perfectly overlapped the absorption spectra Fig. S14† that further confirmed that the complexes were present exclusively in monomeric form and that the aggregates did not influence determination of photophysical parameters.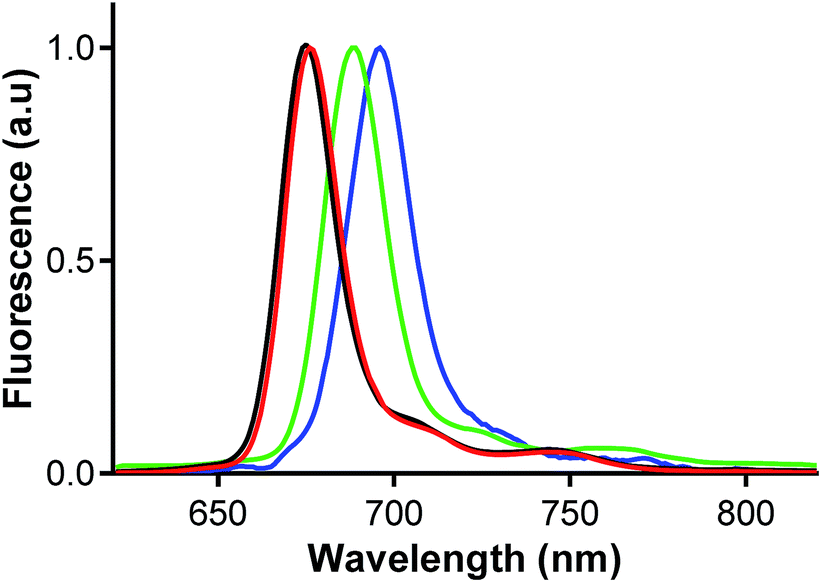 | ||
| Fig. 9 Emission spectra of Pc2-Mg (red), Pc2-Zn (black), Pc2-Ga (green) and Pc2-In (blue) in THF, λexc = 610 nm. | ||
Fluorescence quantum yields (ΦF) were determined in THF with ZnPc as the reference (ΦF in THF = 0.32). The values of ΦF were substantially influenced by presence of the central metal (Table 3). The strongest fluorescence was obtained for Mg derivatives followed by GaCl and Zn complexes. Fluorescence of InCl complexes was always very weak. Reversely to the central cation, effect of peripheral halogen can be considered as negligible (if any) and the observed difference between Pc2-M and Pc3-M series for the same central metal was within the experimental error.
Singlet oxygen quantum yields (ΦΔ) were determined in THF by method of chemical trap (1,3-diphenylisobenzofuran) with ZnPc as reference (ΦΔ in THF = 0.53). The order of the ΦΔ values for complexes with different central metal was reversed to one observed for fluorescence (Table 3), i.e. InCl > GaCl ∼ Zn > Mg. Similarly to fluorescence, the differences between chlorinated (Pc2-M) and brominated (Pc3-M) series felt within the experimental error.
The results from photophysical measurements can be easily interpreted as heavy atom effect of the central cation. The heavy atom effect, described for the first time by McClure,56 shifts the probability of the singlet excited state relaxation toward intersystem crossing (here represented by ΦΔ values) on the account of photon emission. The influence of the metal cation in the current series was very strong as can be easily seen from steep dependences of the quantum yields on atomic weight of the central metal Fig. 10. On the other hand, the effect of halogen substituents was very weak. This fact was further confirmed by comparison of the photophysical data of Pc2-Zn and Pc3-Zn with unsubstituted ZnPc that was used as reference. Both phenoxy substituted zinc complexes reached the same ΦF and ΦΔ values as the reference compound (Table 3). This fact indicates that not only the halogen but actually the whole halogenated phenoxy substituent did not affect the photophysics at all and the changes were induced by central metal only.
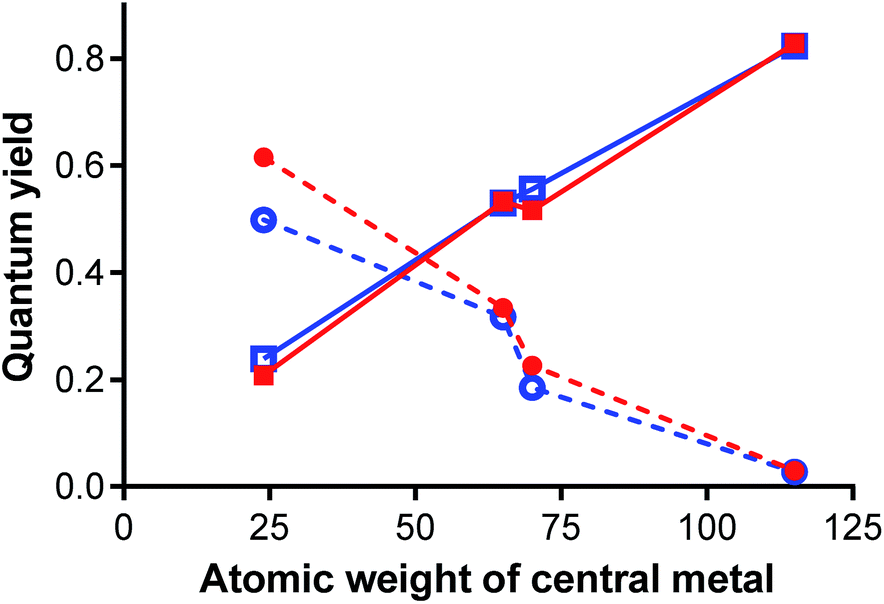 | ||
| Fig. 10 ΦF (circles, dashed lines) and ΦΔ (squares, full lines) values of chlorinated Pc2-M (red, full symbols) and brominated Pc3-M (blue, open symbols) in THF in dependence on the atomic weight of the central metal. | ||
Conclusions
Eight new Pc derivatives with halogenated phenoxy peripheral substituents were synthesized and characterized. Presence of bulky halogens in ortho positions on phenoxy substituents forced them to keep perpendicular orientation towards the Pc plane as predicted by theoretical calculations and unequivocally confirmed by single crystal X-ray diffraction. NMR and UV-vis studies confirmed that such spatial arrangement inhibits fully aggregation in organic solvents and keeps the molecules of Pc separated in solution as monomers even at high concentrations. DFT calculations were used to discuss the differences in absorption spectra of halogenated and non-halogenated Pcs. Strong influence of the central metal on photophysical properties (ΦF and ΦΔ values) was observed and attributed to heavy atom effect.Acknowledgements
The authors acknowledge the support of this work by the Research Administration of Kuwait University, under the Grant Number SC01/09, the facilities of GFS (GS01/01, GS03/01.GS03/08). Chemistry department at Kuwait University is highly acknowledged for using MALDI-TOF. Also we appreciate the useful discussions with Dr Walid M. I. Hassan from Cairo University.Notes and references
- N. B. McKeown, Phthalocyanine materials: synthesis, structure and function, Cambridge University Press, Cambridge, 1998 Search PubMed.
- P. Gregory, J. Porphyrins Phthalocyanines, 2000, 4, 432–437 CrossRef CAS.
- C. G. Claessens, U. Hahn and T. Torres, Chem. Rec., 2008, 8, 75–97 CrossRef CAS PubMed.
- C. C. Leznoff and A. B. P. Lever, Phthalocyanines, Properties and Applications, VCH, NewYork, 1989, vol. 1–4 Search PubMed.
- V. N. Nemykin and E. A. Lukyanets, ARKIVOC, 2010, 136 CrossRef CAS.
- G. de la Torre, P. Vázquez, F. Agulló-López and T. Torres, Chem. Rev., 2004, 104, 3723–3750 CrossRef CAS PubMed.
- D. Dini, M. Hanack and M. Meneghetti, J. Phys. Chem. B, 2005, 109, 12691–12696 CrossRef CAS PubMed.
- S. Vagin, M. Barthel, D. Dini and M. Hanack, Inorg. Chem., 2003, 42, 2683–2694 CrossRef CAS PubMed.
- P. Zimcik, M. Miletin, J. Ponec, M. Kostka and Z. Fiedler, J. Photochem. Photobiol., A, 2003, 155, 127–131 CrossRef CAS.
- V. Novakova, R. Z. U. Kobak, R. Kučera, K. Kopecky, M. Miletin, V. Krepsová, J. Ivincová and P. Zimcik, Dalton Trans., 2012, 41, 10596–10604 RSC.
- S. Makhseed, M. Machacek, W. Alfadly, A. Tuhl, M. Vinodh, T. Simunek, V. Novakova, P. Kubat, E. Rudolf and P. Zimcik, Chem. Commun., 2013, 49, 11149–11151 RSC.
- P. Zimcik, V. Novakova, K. Kopecky, M. Miletin, R. Z. U. Kobak, E. Svandrlikova, L. Váchová and K. Lang, Inorg. Chem., 2012, 51, 4215–4223 CrossRef CAS PubMed.
- M. Machacek, A. Cidlina, V. Novakova, J. Svec, E. Rudolf, M. Miletin, R. Kučera, T. Simunek and P. Zimcik, J. Med. Chem., 2015, 58, 1736–1749 CrossRef CAS PubMed.
- M. Brewis, G. J. Clarkson, P. Humberstone, S. Makhseed and N. B. McKeown, Chem.–Eur. J., 1998, 4, 1633–1640 CrossRef CAS.
- M. Brewis, B. M. Hassan, H. Li, S. Makhseed, N. B. McKeown and N. Thompson, J. Porphyrins Phthalocyanines, 2000, 4, 460–464 CrossRef CAS.
- S. Makhseed, M. Al-Sawah, J. Samuel and H. Manaa, Tetrahedron Lett., 2009, 50, 165–168 CrossRef CAS PubMed.
- H. Manaa, A. A. Mulla, S. Makhseed, M. Al-Sawah and J. Samuel, Opt. Mater., 2009, 32, 108–114 CrossRef CAS PubMed.
- X.-J. Jiang, S.-L. Yeung, P.-C. Lo, W.-P. Fong and D. K. P. Ng, J. Med. Chem., 2010, 54, 320–330 CrossRef PubMed.
- J. T. F. Lau, P.-C. Lo, Y.-M. Tsang, W.-P. Fong and D. K. P. Ng, Chem. Commun., 2011, 47, 9657–9659 RSC.
- V. Mantareva, V. Kussovski, I. Angelov, D. Wöhrle, R. Dimitrov, E. Popova and S. Dimitrov, Photochem. Photobiol. Sci., 2011, 10, 91–102 CAS.
- F. C. Rossetti, L. B. Lopes, A. R. H. Carollo, J. A. Thomazini, A. C. Tedesco and M. V. Bentley, J. Controlled Release, 2011, 155, 400–408 CrossRef CAS PubMed.
- S. Makhseed and J. Samuel, Dyes Pigm., 2009, 82, 1–5 CrossRef CAS PubMed.
- N. B. McKeown, S. Makhseed, K. J. Msayib, L. L. Ooi, M. Helliwell and J. E. Warren, Angew. Chem., Int. Ed., 2005, 44, 7546–7549 CrossRef CAS PubMed.
- T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707–1722 CrossRef CAS PubMed.
- A. Tuhl, H. Manaa, S. Makseed, N. Al-Awadi, J. Mathew and H. M. Ibrahim, Opt. Mater., 2012, 34, 1869–1877 CrossRef CAS PubMed.
- X.-F. Zhang and H.-J. Xu, J. Chem. Soc., Faraday Trans., 1993, 89, 3347–3351 RSC.
- J. E. Van Lier, C. M. Allen and W. M. Sharman, J. Porphyrins Phthalocyanines, 2001, 5, 161–169 CrossRef PubMed.
- E. A. Lukyanets and V. N. Nemykin, J. Porphyrins Phthalocyanines, 2010, 14, 1–40 CrossRef CAS.
- E. A. Makarova and E. A. Lukyanets, J. Porphyrins Phthalocyanines, 2009, 13, 188–202 CrossRef CAS.
- S. P. Keizer, J. Mack, B. A. Bench, S. M. Gorun and M. J. Stillman, J. Am. Chem. Soc., 2003, 125, 7067–7085 CrossRef CAS PubMed.
- V. N. Nemykin, R. G. Hadt, R. V. Belosludov, H. Mizuseki and Y. Kawazoe, J. Phys. Chem. A, 2007, 111, 12901–12913 CrossRef CAS PubMed.
- G. Ricciardi, A. Rosa and E. J. Baerends, J. Phys. Chem. A, 2001, 105, 5242–5254 CrossRef CAS.
- Y. Zhang, X. Zhang, Z. Liu, Y. Bian and J. Jiang, J. Phys. Chem. A, 2005, 109, 6363–6370 CrossRef CAS PubMed.
- L. Yang, L. Guo, Q. Chen, H. Sun, J. Liu, X. Zhang, X. Pan and S. Dai, J. Mol. Graphics Modell., 2012, 34, 1–9 CrossRef CAS PubMed.
- D. Wöhrle, M. Eskes, K. Shigehara and A. Yamada, Synthesis, 1993, 194–196 CrossRef.
- D. D. Perrin, W.L.F. Armarego: Purification of laboratory chemicals, 3rd edn, Pergamon Press, Oxford, 1988 Search PubMed.
- M. Dolg and H. Stoll, Theor. Chim. Acta, 1993, 85, 441–450 CrossRef CAS.
- A. Bergner, M. Dolg, W. Kuechle and H. Stoll, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, USA, 2010 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, version 5, Semichem. Inc., Shawnee Mission, KS, 2009 Search PubMed.
- Data Visualization Tools: ChemCraft, http://www.chemcraftprog.com, accessed January 24 2014 Search PubMed.
- V. Novakova, M. Miletin, T. Filandrová, J. Lenčo, A. Růžička and P. Zimcik, J. Org. Chem., 2014, 79, 2082–2093 CrossRef CAS PubMed.
- L. Kaestner, M. Cesson, K. Kassab, T. Christensen, P. D. Edminson, M. J. Cook, I. Chambrier and G. Jori, Photochem. Photobiol. Sci., 2003, 2, 660–667 CAS.
- S. E. Maree and T. Nyokong, J. Porphyrins Phthalocyanines, 2001, 5, 782–792 CrossRef CAS PubMed.
- S. Vagin, A. Frickenschmidt, B. Kammerer and M. Hanack, Eur. J. Org. Chem., 2005, 2005, 3271–3278 CrossRef PubMed.
- E. N. Ovchenkova and T. N. Lomova, Russ. J. Org. Chem., 2011, 47, 1581–1587 CrossRef CAS.
- C.-F. Choi, P.-T. Tsang, J.-D. Huang, E. Y. M. Chan, W.-H. Ko, W.-P. Fong and D. K. P. Ng, Chem. Commun., 2004, 2236–2237 RSC.
- W. Liu, T. J. Jensen, F. R. Fronczek, R. P. Hammer, K. M. Smith and M. G. H. Vicente, J. Med. Chem., 2005, 48, 1033–1041 CrossRef CAS PubMed.
- M. Yoshioka, K. Ohta and M. Yasutake, RSC Adv., 2015, 5, 13828–13839 RSC.
- S. U. Lee, J. C. Kim, H. Mizuseki and Y. Kawazoe, Chem.–Asian J., 2010, 5, 1341–1346 CrossRef CAS PubMed.
- W. R. Scheid and W. J. Dow, J. Am. Chem. Soc., 1977, 99, 1101–1104 CrossRef.
- H. Shahroosvand, S. Zakavi, A. Sousaraei and M. Eskandari, Phys. Chem. Chem. Phys., 2015, 17, 6347–6358 RSC.
- Z. Musil, P. Zimcik, M. Miletin, K. Kopecky, P. Petrik and J. Lenco, J. Photochem. Photobiol., A, 2007, 186, 316–322 CrossRef CAS PubMed.
- Y. Zhang, X. Cai, X. Zhang, H. Xu, Z. Liu and J. Jiang, Int. J. Quantum Chem., 2007, 107, 952–961 CrossRef CAS PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- D. S. McClure, J. Chem. Phys., 1949, 17, 905–913 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 938006–938008. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra09737a |
| This journal is © The Royal Society of Chemistry 2015 |