
Diindenothienoacene–tetrathiafulvalene redox systems†
Mikkel A. Christensena,
Gabriel E. Rudebuschb,
Christian R. Parkera,
Cecilie Lindholm Andersena,
Anders Kadziolaa,
Michael M. Haley*b,
Ole Hammerich*a and
Mogens Brøndsted Nielsen*a
aDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark. E-mail: o.hammerich@chem.ku.dk; mbn@kiku.dk
bDepartment of Chemistry & Biochemistry and The Materials Science Institute, University of Oregon, Eugene, Oregon 97403-1253, USA. E-mail: haley@uoregon.edu
First published on 28th May 2015
Abstract
Extended tetrathiafulvalenes with central diindenothienoacene cores were prepared and studied for their redox and spectroelectrochemical properties, which depended strongly on the orientation of the thiophene rings. The cations undergo remarkably strong associations, rendering them attractive as redox-controllable tectons in supramolecular chemistry, and in one case crystals were grown by electrocrystallization.
π-Dimerization of tetrathiafulvalene (TTF) radical cations has become an important tool in supramolecular chemistry owing to the ability to turn on and off this association by reversible oxidation of TTF.1,2 We have recently found3 that combining TTF with indenofluorenes (Fig. 1) causes a remarkable enhancement of the propensity of the radical cations to associate. Association constants for [1·1˙+] and [1˙+·1˙+] of the order of 104 M−1 in CH2Cl2 (0.1 M Bu4NPF6) were estimated, hence much larger than those obtained by Rosokha and Kochi4 for [TTF·TTF˙+] (6.0 M−1) and [TTF˙+·TTF˙+] (0.6 M−1). In addition, 1˙+ is interesting for its optical properties with strong near-infrared absorptions. Frère et al.5 have found that incorporation of thiophene in the π-spacer of extended TTFs also leads to strong association. To tune the redox properties and to enhance association further, we decided to substitute the central benzene ring of 1 with fused thiophene units and here present the synthesis and (spectro)electrochemical properties of four derivatives, which differ by the number and regioisomeric structural arrangements of the thiophene units and hence by the conjugation pathway between the two dithiafulvene units.
 | ||
| Fig. 1 Large π-extended tetrathiafulvalenes. | ||
The synthesis relies on a Horner–Wadsworth–Emmons (HWE) reaction between the phosphonate esters 2a and 2b3,6 and the diketones 3–5 (Scheme 1).
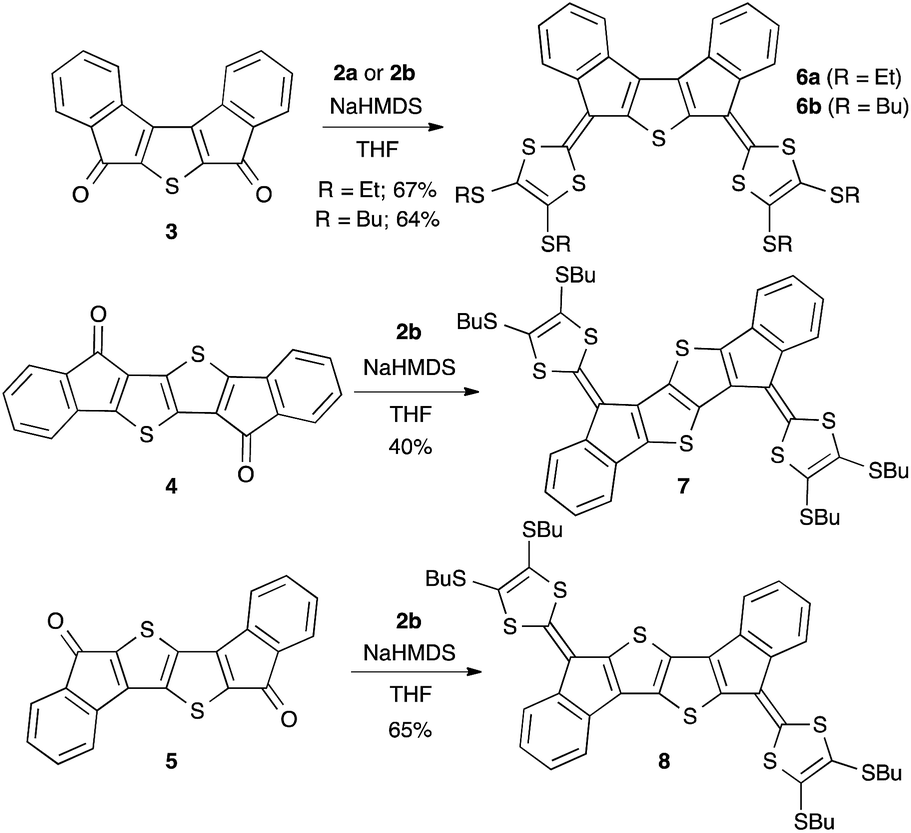 | ||
| Scheme 1 Synthesis of diindenothienoacene–tetrathiafulvalene scaffolds. | ||
Preparation of 4 is described in the ESI† and follows an analogous procedure reported for 3 and 5, used as precursors for quinoidal diindenothienoacenes.7 Treatment of diones 3–5 with 2a or 2b and sodium hexamethyldisilazide (NaHMDS) in THF gave extended TTFs 6a, 6b, 7 and 8 in good yields. Solubility for 6b, 7 and 8 was achieved by using Bu substituents for the solution electrochemistry, while Et substituents in 6a facilitated electrocrystallization.
The structures of 6a, 6b, 7, and 8 were confirmed by X-ray crystallography (Fig. 2 & ESI†).8 The packing is for all four compounds characterized by π–π stacking of closely packed aromatic planes combined with van der Waal contacts from alkyl groups both in and perpendicular to the aromatic planes. No direct S–S contacts are observed.
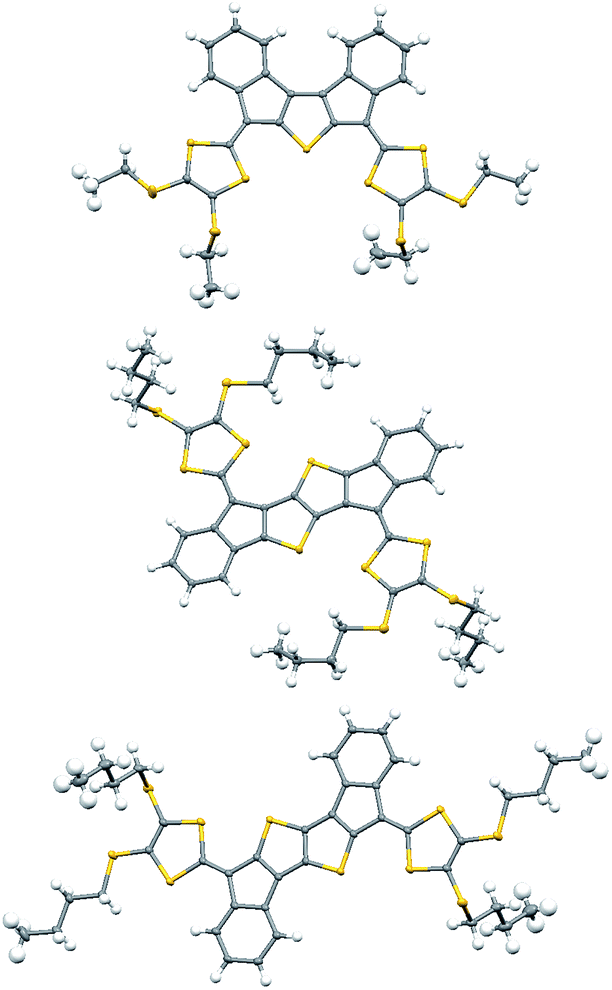 | ||
| Fig. 2 Molecular structures of 6a (top), 7 (middle) and 8 (bottom). All crystals were grown from CS2/heptane. | ||
The electrochemical oxidation of 6b, 7 and 8 was studied by cyclic voltammetry (CV) and differential pulse voltammetry (DPV). The shape of the response curves is in all three cases strongly concentration dependent as illustrated in Fig. 3 and 4. The general trend is that the first wave sharpens up upon dilution, whereas the shape of the second wave essentially is unaffected by the concentration. The same type of response was recently found for 1 and explained by formation of strong [neutral·cation] and [cation·cation] associates during the voltammetric oxidation.3 By digital simulation of the cyclic voltammogram of 6b (see ESI†), we obtain association constants for [6b·6b˙+] and [6b˙+·6b˙+] of 1.8 × 104 M−1 and 2.8 × 103 M−1, very similar to those of 1. Less satisfactory fits were obtained for the associates of 7 and 8 for both of which two clearly distinguishable waves are seen for the first oxidations rather than just a broad one as was the case for 6b. Higher associates now likely play a significant role, which complicate the digital simulations of these CVs. Test simulations seem to indicate that the dimer association constants of [7·7˙+] and [8·8˙+] are indeed one order of magnitude larger for these species.
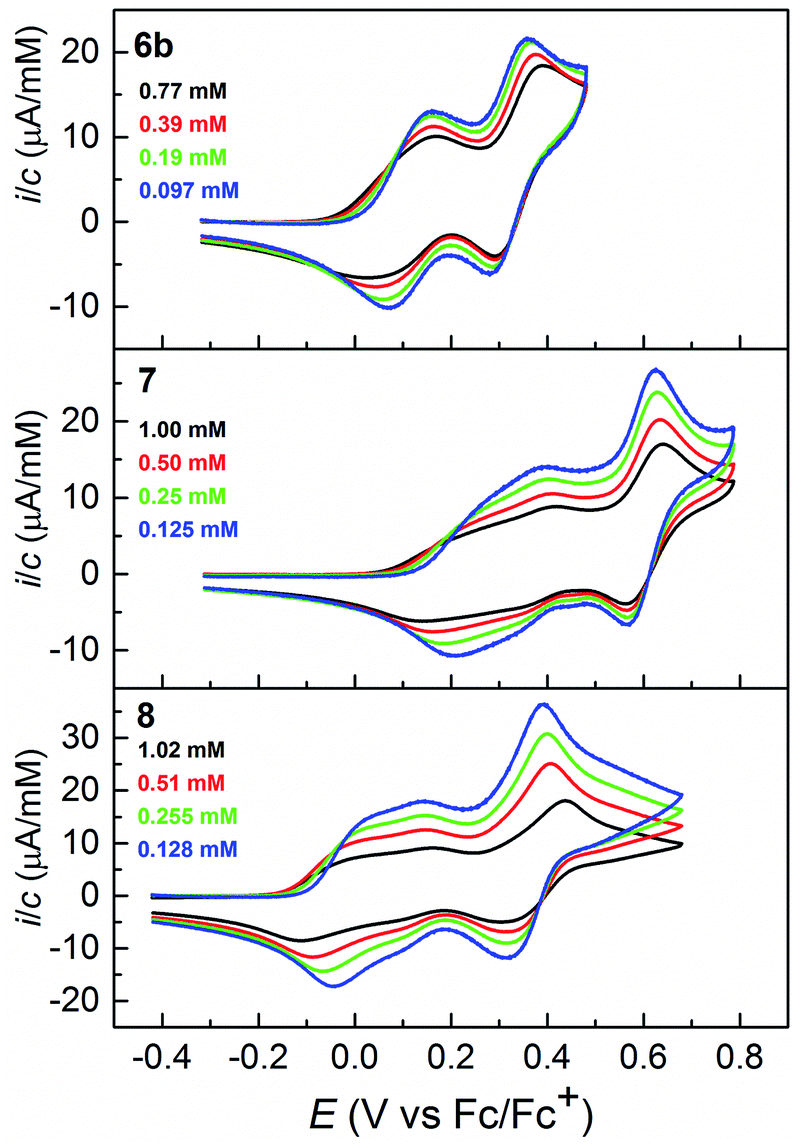 | ||
| Fig. 3 Cyclic voltammograms of 6b, 7 and 8 obtained at a glassy carbon working electrode in CH2Cl2 (0.1 M Bu4NPF6). Voltage scan rate = 0.1 V s−1. The y-axes show concentration–normalized currents to facilitate comparison at different concentrations. In all cases background subtraction was used. | ||
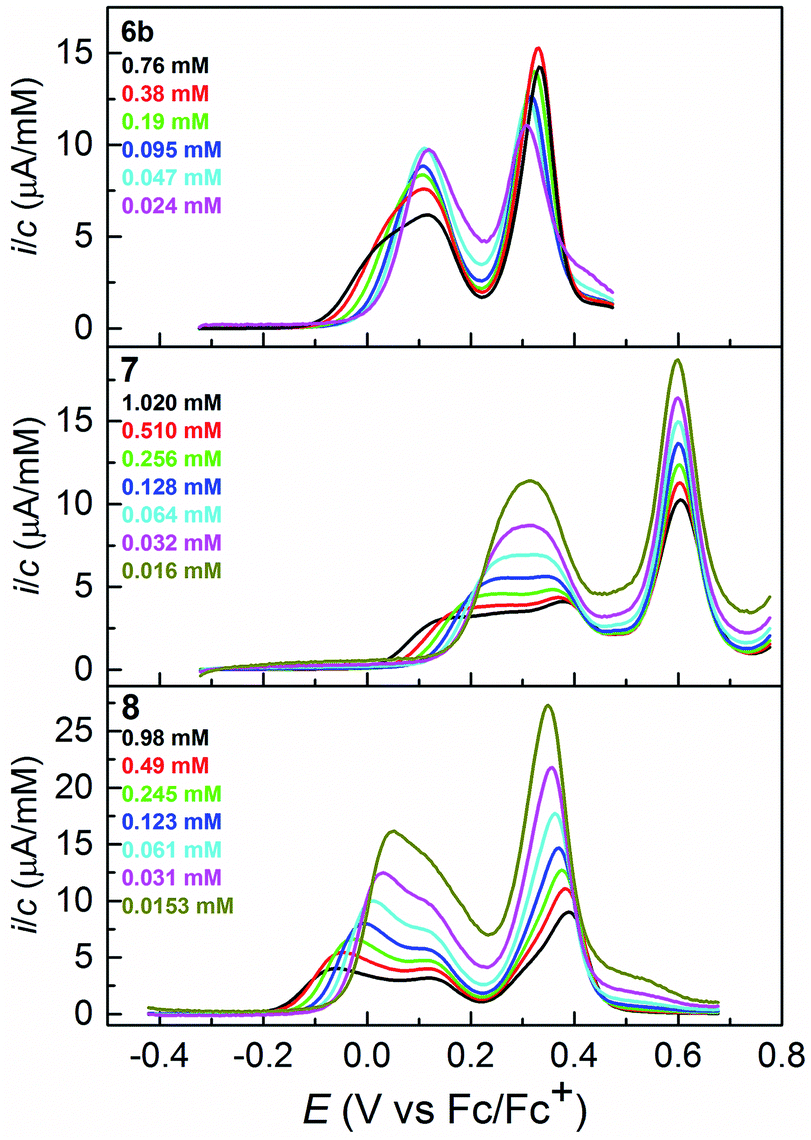 | ||
| Fig. 4 Differential pulse voltammograms of 6b, 7 and 8 obtained at a glassy carbon working electrode in CH2Cl2 (0.1 M Bu4NPF6). Step potential = 0.0045 V and modulation amplitude = 0.025 V. The y-axes show concentration–normalized currents to facilitate comparison at different concentrations. In all cases background subtraction was used. | ||
Dilution to the 2–10 μM range, where the response is almost unaffected by the association phenomena, allowed us to estimate the formal potentials for oxidation, Eo1′ and Eo2′, from the DPV curves (see Fig. S1 in ESI†): +0.14 and +0.31 (6b), +0.32 and +0.60 (7), and +0.09 and +0.33 (8) V vs. Fc/Fc+. It is noteworthy that 6b and 8 are oxidized at essentially the same potentials, whereas 7, an isomer of 8, is more difficult to oxidize by 200–300 mV. Insight into this difference in the ease of oxidation of 7 and 8 was gained by DFT calculations (B3LYP cc-pVDZ, with SEt instead of SBu, 7-SEt/8-SEt; see ESI†). Solution phase calculations predict that 8-SEt is more easily oxidized than 7-SEt by 0.103 V and 0.201 V for the first and second oxidations, respectively. These computed shifts are less than observed experimentally, but still satisfactory considering the modest size of the basis-set used and the application of a continuum solvation model (IEFPCM).
The vacuum phase calculations showed that 8-SEt, independent on the oxidation state, is more stable than 7-SEt, the differences in favor of 8-SEt being 29.9 kJ mol−1 (neutral), 51.4 kJ mol−1 (radical cation) and 64.1 kJ mol−1 (dication). In terms of conjugation, the two dithiafulvene units are connected in a linearly conjugated pathway in 8, but in a cross-conjugated one in 7. We also find from the computations that 72+ and 82+ have open shell singlet ground states with, for 72+, the triplet state being only 2.0 kJ mol−1 less stable. We take this latter result as an indication that 72+ is best described as a biradical; indeed, for this cross-conjugated compound no closed shell valence bond structure can be drawn with all electrons paired as it can with 6b2+ (Fig. 5). For the linearly conjugated 82+, the singlet ground state, but open shell, is indicated by the dashed bonds in Fig. 5.
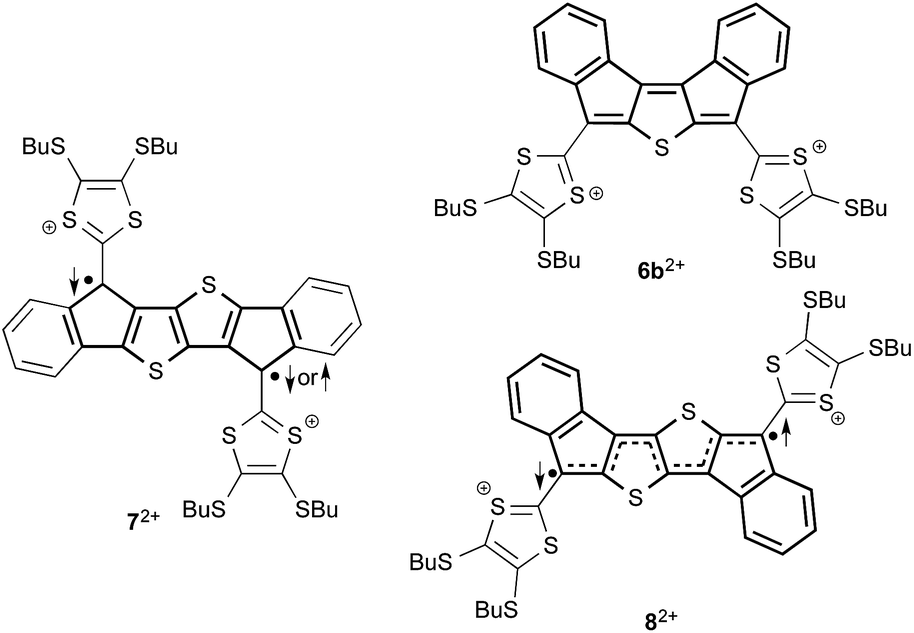 | ||
| Fig. 5 Singlet/triplet (left) vs. singlet (right) ground states of the dications. | ||
Compound 6a with peripheral SEt groups was subjected to electrocrystallization in an H-shaped cell (i = 2 μA) in PhCl/Bu4NPF6 (saturated, <0.02 M). Single crystals were analyzed by X-ray crystallography (Fig. 6),8 revealing the stoichiometry 6a·PF6; all 6a molecules hence exist as monocations. Fig. 7 shows a comparison of selected bond lengths of 6a and 6a˙+. Upon oxidation, the C–S bonds (a and b) in the dithiafulvene rings are reduced in lengths as expected for the generation of 1,3-dithiolium character, while the exocyclic fulvene bond (c) is increased in length. The C–C bonds in the central core (d–g) and the C–S bond (h) are also significantly different in the radical cation relative to in the neutral molecule, signalling that the unpaired electron is spread over the core.
 | ||
| Fig. 6 Molecular structure and packing diagrams of 6a·PF6. | ||
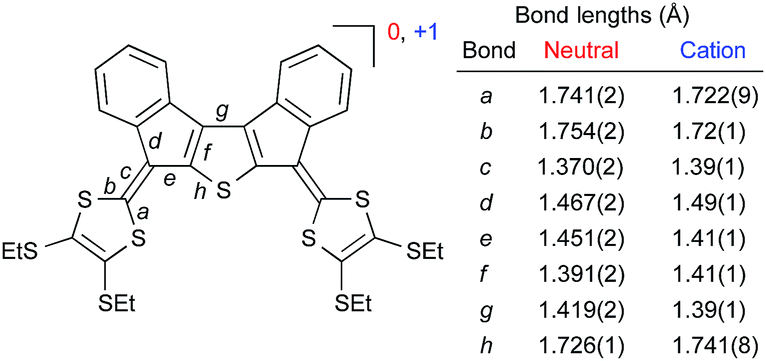 | ||
| Fig. 7 Selected bond lengths from X-ray crystal structures of 6a and 6a·PF6. | ||
Finally, the optical properties of the oxidized species were elucidated by spectroelectrochemistry. Fig. 8 shows the absorption spectra of the neutral, monocation (including associates), and dication species. The absorption spectrum of the neutral compound was not regenerated for 7 after one cycle to 72+ (see ESI†), signalling that the oxidized species are undergoing some chemical reactions, which may reflect the more localized radical character of this compound (vide supra). For all three compounds, new broad and strong near-infrared absorptions emerge for the monocationic species (red curves). These bands are at ca. 1255 and 1565 nm (6b˙+), 1620 nm (7˙+), 1775 and 2450 nm (8˙+). The two absorptions for 6b˙+ and 8˙+ seem to change in relative intensity during the conversion from neutral to monocation, which we take as further evidence for the presence of monomeric as well as [neutral·cation] and [cation·cation] species during the oxidation. Even more significant changes in relative intensities during the first oxidation were observed for the higher energy absorptions, that is, bands at ca. 770 and 665 nm (6b) and at ca. 630/680 and 1140 nm (8) change in relative intensities and also shift in position. For 7, associations are also supported by the spectral evolution in this region. Thus, an initial band at ca. 770 nm shifts in position to ca. 725 nm, when the oxidation to monocation is complete. Thus, the spectroelectrochemical experiments support the associations suggested by the cyclic and differential pulse voltammetry experiments. The dications of 6b and 8 (blue curves) exhibited broad longest-wavelength absorptions at ca. 905/1000 nm (6b2+) and at ca. 905 nm (with a shoulder) (82+). For comparison, a similar absorption was observed for 12+ at 905 nm.3 This characteristic absorption was, however, almost absent for 72+, which again reflects the distinctive properties of this dication with its lack of communication along the cross-conjugated spacer.
 | ||
| Fig. 8 UV-Vis-NIR absorption spectra obtained by spectroelectrochemistry of 6b (top), 7 (middle) and 8 (bottom) in CH2Cl2 + 0.1 M Bu4NPF6. Black curves: neutral species; red curves: monocations (including associates); blue curves: dications. The insets show the spectral evolutions (olive green curves) for selected regions during the oxidation from neutral to monocation. | ||
In conclusion, π-extended TTFs with central diindenothienoacene cores undergo reversible oxidations, generating cationic species, which readily associate in solution and exhibit near-infrared absorptions. The strong association, in particular when the core contains two fused thiophenes (7 and 8), makes these molecules attractive as redox-controllable tectons for generation of self-assembled structures. In addition, the differences in the oxidation potentials and stabilities of all three redox states (0, +1, +2) of 7 and 8 illustrate the need for careful consideration of the regioisomeric structural orientation and hence the nature of the conjugation pathway (linear or cross) between the two dithiafulvene units, depending upon what properties one desires. Future work will examine even more extended π-systems. Such molecules are not only of interest as redox-controllable tectons in supramolecular chemistry, but also as redox-active chromophores for photovoltaics.9
Acknowledgements
The US National Science Foundation (CHE-1301485) and University of Copenhagen are acknowledged for support.Notes and references
- (a) A. Y. Ziganshina, Y. H. Ko, W. S. Jeon and K. Kim, Chem. Commun., 2004, 806 RSC; (b) J. Lyskawa, M. Sallé, J.-Y. Balandier, F. Le Derf, E. Levillain, M. Alain, P. Viel and S. Palacin, Chem. Commun., 2006, 2233 RSC; (c) I. Aprahamian, J.-C. Olsen, A. Trabolsi and J. F. Stoddart, Chem.–Eur. J., 2008, 14, 3889 CrossRef CAS PubMed; (d) P.-T. Chiang, N.-C. Chen, C.-C. Lai and S.-H. Chiu, Chem.–Eur. J., 2008, 14, 6546 CrossRef CAS PubMed; (e) M. Hasegawa, K. Daigoku, K. Hashimoto, H. Nishikawa and M. Iyoda, Bull. Chem. Soc. Jpn., 2012, 85, 51 CrossRef CAS; (f) M. Frasconi, T. Kikuchi, D. Cao, Y. Wu, W.-G. Liu, S. M. Dyar, G. Barin, A. A. Sarjeant, C. L. Stern, R. Carmieli, C. Wang, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 11011 CrossRef CAS PubMed; (g) W.-K. Wang, Y.-Y. Chen, H. Wang, D.-W. Zhang and Z.-T. Li, Chem.–Asian J., 2014, 9, 1039 CrossRef CAS PubMed.
- For a theoretical treatment of TTF π-dimers, see: M. Fumanal, M. Capdevila-Cortada, J. S. Miller and J. J. Novoa, J. Am. Chem. Soc., 2013, 135, 13814 CrossRef CAS PubMed.
- M. A. Christensen, C. R. Parker, T. J. Sørensen, S. de Graaf, T. J. Morsing, T. Brock-Nannestad, J. Bendix, M. M. Haley, P. Rapta, A. Danilov, S. Kubatkin, O. Hammerich and M. B. Nielsen, J. Mater. Chem. C, 2014, 2, 10428 RSC.
- S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2007, 129, 828 CrossRef CAS PubMed.
- (a) P. Frère, M. Allain, E. H. Elandaloussi, E. Levillain, F.-X. Sauvage, A. Riou and J. Roncali, Chem.–Eur. J., 2002, 8, 784 CrossRef; (b) P. Frère and P. J. Skabara, Chem. Soc. Rev., 2005, 34, 69 RSC.
- J. Rybácek, M. Rybácková, M. Høj, M. Belohradsky, P. Holy, K. Kilså and M. B. Nielsen, Tetrahedron, 2007, 63, 8840 CrossRef PubMed.
- G. E. Rudebusch, A. G. Fix, H. A. Henthorn, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Chem. Sci., 2014, 5, 3627 RSC.
- ESI.†.
- Some recent examples: (a) M. Dogru, M. Handloser, F. Auras, T. Kunz, D. Medina, A. Hartschuh, P. Knochel and T. Bein, Angew. Chem., Int. Ed., 2013, 52, 2920 CrossRef CAS PubMed; (b) A. J. Pearson, D. C. Watters, H. Yi, M. S. Sarjadi, L. X. Reynolds, P. P. Marchisio, J. Kingsley, S. A. Haque, A. Iraqi and D. G. Lidzey, RSC Adv., 2014, 4, 43142 RSC; (c) N. Zhou, K. Prabakaran, B. Lee, S. H. Chang, B. Harutyunyan, P. Guo, M. R. Butler, A. Timalsina, M. J. Bedzyk, M. A. Ratner, S. Vegiraju, S. Yau, C.-G. Wu, R. P. H. Chang, A. Facchetti, M.-C. Chen and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 4414 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. CCDC 1056502–1056506. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra09581f |
| This journal is © The Royal Society of Chemistry 2015 |