
Organocatalytic [4+1]-annulation approach for the synthesis of densely functionalized pyrazolidine carboxylates†
Komal G. Lalwani‡
,
Ravindra D. Aher‡ and
Arumugam Sudalai*
Chemical Engineering & Process Development Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pune, Maharashtra, India-411008. E-mail: a.sudalai@ncl.res.in
First published on 27th July 2015
Abstract
A novel one-pot [4+1]-annulation process for the asymmetric synthesis of densely functionalized pyrazolidine carboxylates is described. The in situ generated γ-hydrazino-α,β-unsaturated ester obtained via proline catalysis acts as a four-atom component, and Corey's sulfur ylide or ethyl bromoacetate acts as a one-atom carbon source to construct pyrazolidine carboxylate units in a highly enantio- and diastereoselective fashion.
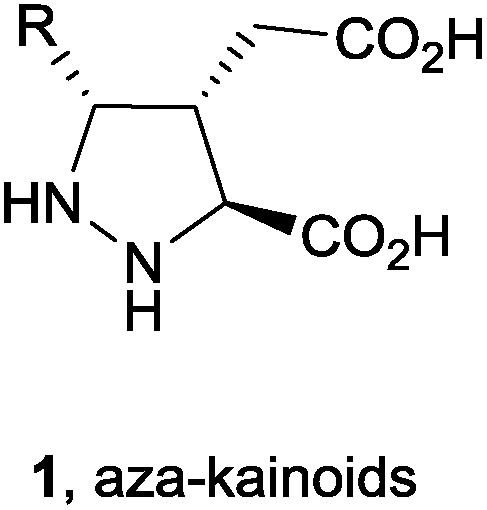
The pyrazolidine and pyrazoline moieties are an interesting class of nitrogen-containing heterocyclic structural units found in many complex natural products1 with significant biological activities (e.g. anticancer,2 antidepressant,3 antibacterial,4 anticonvulsant,5 antiviral,6 etc.) and other uses (as an arthropodicidal agent7 in agriculture or optical brightening agent).8 Furthermore, they can be considered as powerful starting materials for the synthesis of enantiopure azaprolines9 and densely functionalized 1,3-diamino derivatives10 after reductive cleavage of the N–N bond. In particular, recent SAR studies have established that aza-kainic acid derivatives (1) have been proven to exhibit potent neuroexcitatory activity.11
One of the most efficient strategies for the construction of such fused skeletons generally relies on [3+2] cycloadditions of hydrazones to olefins in the presence of Bronsted12/Lewis13 acids or with strong heating.14 Their asymmetric synthesis are also reported employing chiral Zr/BINOL,15 Si-based Lewis acids,16 transition metal (Pd, Ni, Au)-catalyzed intramolecular annulations17 including sequential organocatalysis.18 However, these methods are rather limited due to harsh reaction conditions, complex chiral pool resources, expensive chiral ligands and metal catalysts often involving multistep reaction sequences. To the best of our knowledge, no method has been previously reported for the synthesis of densely substituted pyrazolidine carboxylates in “one-pot” fashion using organocatalysis.
In recent years, proline-catalyzed sequential reactions have gained prominence for the asymmetric synthesis of structurally diverse molecular architectures.19,20 As part of our program directed towards asymmetric synthesis of bioactive molecules employing organocatalytic sequential reactions,21 we envisaged that in situ trapping of γ-hydrazino-α,β-unsaturated ester 6 (ref. 20e) with Corey's sulfur ylide (dimethyloxosulfonium methylide)22 under basic conditions should provide the corresponding highly functionalized cyclopropane carboxylate 3, potent and selective group II metabotropic glutamate receptor (mGluR) antagonists.3e
Surprisingly, the reaction took a different course to afford the corresponding 3,4-disubstituted pyrazolidine carboxylate 4a as a single diastereomer in 68% yield (Scheme 1).
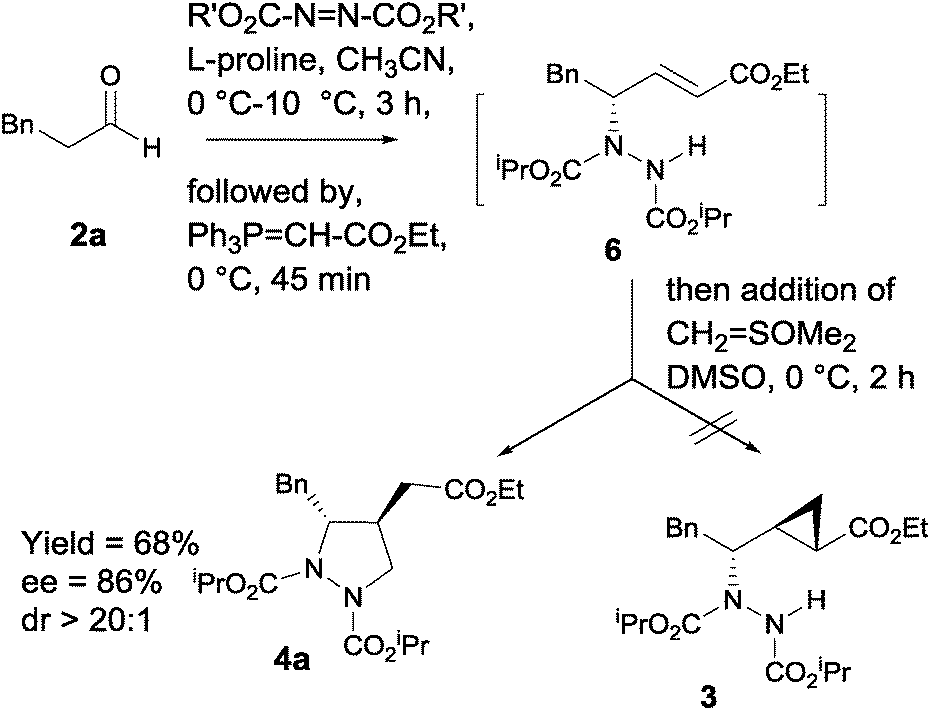 | ||
| Scheme 1 In situ trapping of γ-hydrazino-α,β-unsaturated ester 6 with dimethyloxosulfonium methylide. | ||
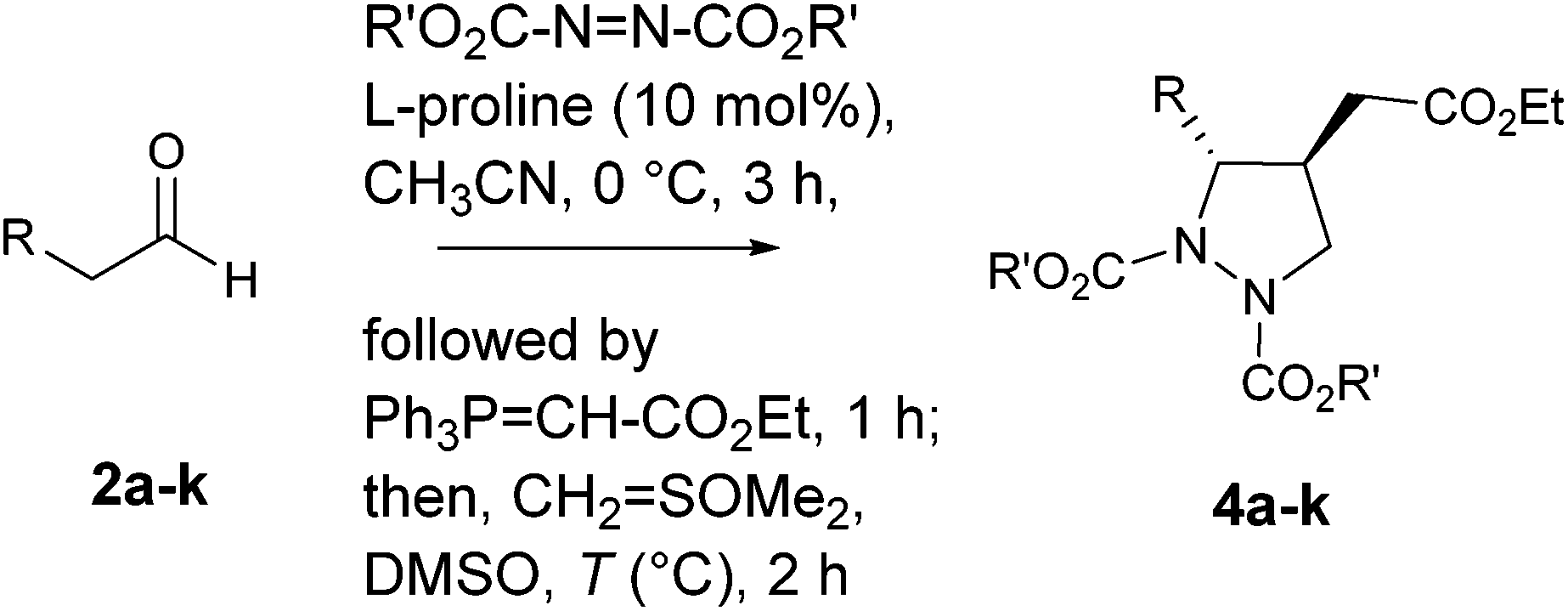
In this communication, we describe a one-pot sequential procedure for a tandem [4+1] annulation reaction of γ-hydrazino-α,β-unsaturated ester 6 generated in situ with Corey's sulfur ylide or ethyl bromoacetate that proceeds to give densely functionalized chiral pyrazolidine carboxylates 4 & 5 in a highly enantio- and diastereoselective manner (Table 1 and 2).
|
|||||
|---|---|---|---|---|---|
| Entry | Aldehyde (2a–k) (R) | Amine (R′) | T (°C) | Products (4a–k) | |
| Yieldb (%) | eec (%) | ||||
a Aldehyde (2.5 mmol), amine (R′O2C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–CO2R′) (2.5 mmol), L-proline (10 mol%), Ph3PCHCO2Et (3.75 mmol), dimethyloxosulfonium methylide (5.0 mmol).b Diastereomeric ratio (dr > 20 N–CO2R′) (2.5 mmol), L-proline (10 mol%), Ph3PCHCO2Et (3.75 mmol), dimethyloxosulfonium methylide (5.0 mmol).b Diastereomeric ratio (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was determined from proton NMR analysis of the crude product.c %ee were determined from chiral HPLC analysis; nd = not determined. :1) was determined from proton NMR analysis of the crude product.c %ee were determined from chiral HPLC analysis; nd = not determined. |
|||||
| 1 | Benzyl (2a) | iPr | 0 | 68 | 86 |
| 2 | iPr | 10 | 75 | 86 | |
| 3 | iPr | 25 | 80 | 86 | |
| 4 | Et | 25 | 79 | 81 | |
| 5 | tBu | 25 | 70 | 86 | |
| 6 | 4-Methylbenzyl (2b) | iPr | 25 | 71 | 84 |
| 7 | 4-Methoxybenzyl (2c) | iPr | 25 | 67 | 87 |
| 8 | 4-Thiomethylbenzyl (2d) | iPr | 25 | 75 | 92 |
| 9 | 2-CN-4,5-methylene-dioxybenzyl (2e) | iPr | 25 | 65 | 81 |
| 10 | 2-NO2-4,5-methylene-dioxybenzyl (2f) | iPr | 25 | 69 | 92 |
| 11 | 2-NO2-4,5-methylene-dioxybenzyl (2f) | tBu | 25 | 66 | 94 |
| 12 | 2-Bromo-3,4,5-trimethoxy-benzyl (2g) | iPr | 25 | 72 | 94 |
| 13 | Naphthalene-1-yl-methyl (2h) | iPr | 25 | 68 | 90 |
| 14 | 3-Benzyloxypropyl (2i) | iPr | 25 | 70 | 92 |
| 15 | Propyl (2j) | iPr | 25 | 79 | nd |
| 16 | Methyl (2k) | iPr | 25 | 80 | nd |
|
|||||
|---|---|---|---|---|---|
| Entry | Aldehyde (2) (R) | Amine (R′) | T (°C) | Products (5a–h) | |
| Yieldb (%) | eec (%) | ||||
| a Aldehyde (2.5 mmol), amine (R′O2C–NN–CO2R′) (2.5 mmol), L-proline (10 mol%), Ph3PCHCO2Et (3.75 mmol), ethyl bromoacetate (3.75 mmol), Cs2CO3 (6.25 mmol).b Isolated yield of products.c %ee were determined from chiral HPLC analysis.d Diastereomeric ratio (dr > 20:1) was determined from proton NMR analysis of the crude product.e dr = 7:3.f dr = 6:1; nd = not determined. |
|||||
| 1 | Benzyl | tBu | 25 | — | — |
| 2 | tBu | 50 | — | — | |
| 3 | tBu | 80 | 50 (5a)d | 86 | |
| 4 | Et | 50 | 72 (5a)d | 96 | |
| 5 | 4-Methoxybenzyl | Et | 50 | 77 (5b)d | 96 |
| 6 | 4-F-benzyl | Et | 50 | 64 (5c)d | 95 |
| 7 | 3,4-Dimethylbenzyl | Et | 50 | 75 (5d)d | 94 |
| 8 | 2-NO2-4,5-methyl-enedioxybenzyl | Et | 50 | 64 (5e)e | 88 |
| 9 | Naphthalene-1-yl-methyl | Et | 50 | 79 (5f)d | 94 |
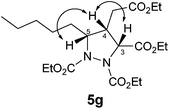
| 10 | Pentyl | Et | 50 | 72 (5g)d | nd |
| 11 | Methyl | Et | 50 | 62 (5h)f | nd |
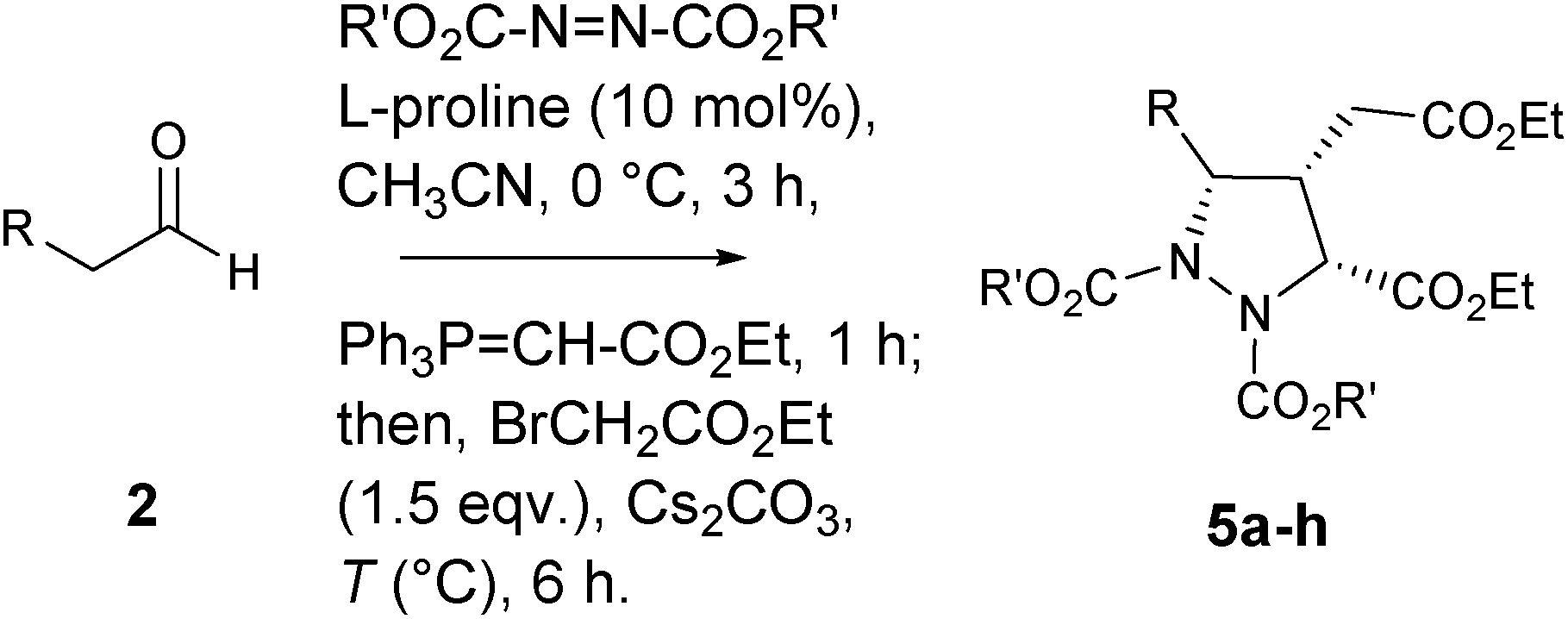
In order to optimize the reaction conditions, initially the amination/Wittig olefination of hydrocinnamaldehyde 2a was carried out following our amination protocol 20e that produced the corresponding γ-hydrazino-α,β-unsaturated ester 6 in situ. This was followed by the addition of a solution of dimethyloxosulfonium methylide in DMSO [sulfur ylide (2.0 equiv.), prepared in situ from OSMe3I/NaH in DMSO] at 0 °C that gave 4a as a single diastereomer in 68% yield with 86% ee (entry 1). A significant improvement in yield (80%) was, however, realized when the reaction was conducted at 25 °C for 2 h. Increase of temperature (50 °C) resulted in complex reaction mixture. Also, use of other solvents such as CH2Cl2 and THF for the tandem protocol resulted in a sluggish reaction with poor yields (<10%). Furthermore, (S)-α,α-diarylprolinol silyl ether as a modified proline catalyst was found to be less effective for the reaction. We then turned our attention to investigate the scope of amine sources, the results of which indicated that diisopropyl was found to be better candidate (Table 1, entry 3). With the optimized reaction condition in hand, we next examined the scope of the reaction. Aldehydes bearing Br, CN, NO2, OMe, SMe and methylenedioxy groups on the aromatic nucleus, and benzyl ether substitutions in aliphatic compounds were found to be well-tolerated under the reaction condition. For all the cases studied, the products 4a–k were indeed obtained in high yields (65–80%) and excellent enantioselectivities (80–94%) with dr > 20:1 (Table 1, entry 6–16).
In order to further extend the scope of [4+1]-annulation strategy, the in situ generated amino ester 6 (R = Bn & R′ = tBu) was treated with ethyl bromoacetate (1.5 equiv.) in presence of K2CO3 as base at 50 °C and found that the annulation was unsuccessful. However, when Cs2CO3 was used as base and heating the mixture at 80 °C, the annulation proceeds smoothly producing the desired pyrazolidine dicarboxylate 5a in 50% yield and 86% ee with dr > 20:1. The best results were obtained when the amine source was changed to R′ = Et (72% yield, 96% ee, and dr = 20:1 at 50 °C) (entry 4, Table 2). With this optimized reaction conditions, other substrates bearing F, NO2, Me and OMe substituents on the aromatic nucleus underwent this [4+1] annulations cascade smoothly, affording the corresponding pyrazolidine dicarboxylates 5a–h in high yields with excellent enantio- and diastereoselectivities (Table 2).
The absolute configuration of the newly generated stereogenic centers was assigned on the basis of the previously established configuration of γ-hydazino-α,β-unsaturated ester.20e The relative stereochemistry in pyrazolidines 4 and 5 is proven unambiguously from X-ray crystallographic analysis23 (Fig. 1, CCDC 1041539†) as well as COSY and NOESY NMR studies.24
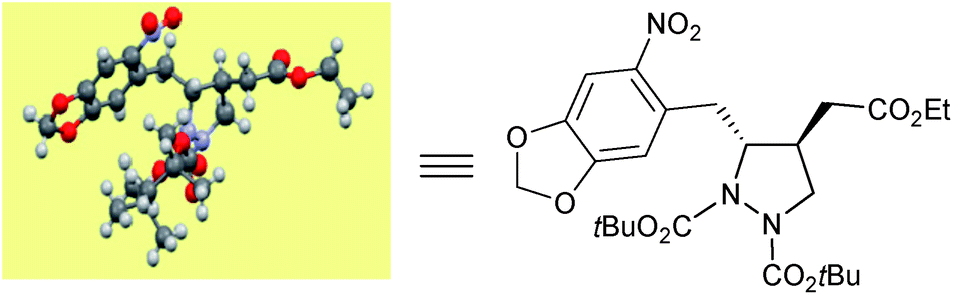 | ||
| Fig. 1 ORTEP diagram of 4f (R′ = tBu). | ||
A probable mechanistic pathway is shown in Scheme 2 in which sulfur ylide adds onto β-carbon of the in situ generated γ-amino-α,β-unsaturated ester 6 to form species A. This in turn is followed by a facile proton exchange25 from the carbamate nitrogen to the basic carbanion A to give the stable species B, which then subsequently undergoes intramolecular cyclization with the removal of DMSO to afford products 4a–k, all occurring sequentially under “one-pot” fashion.
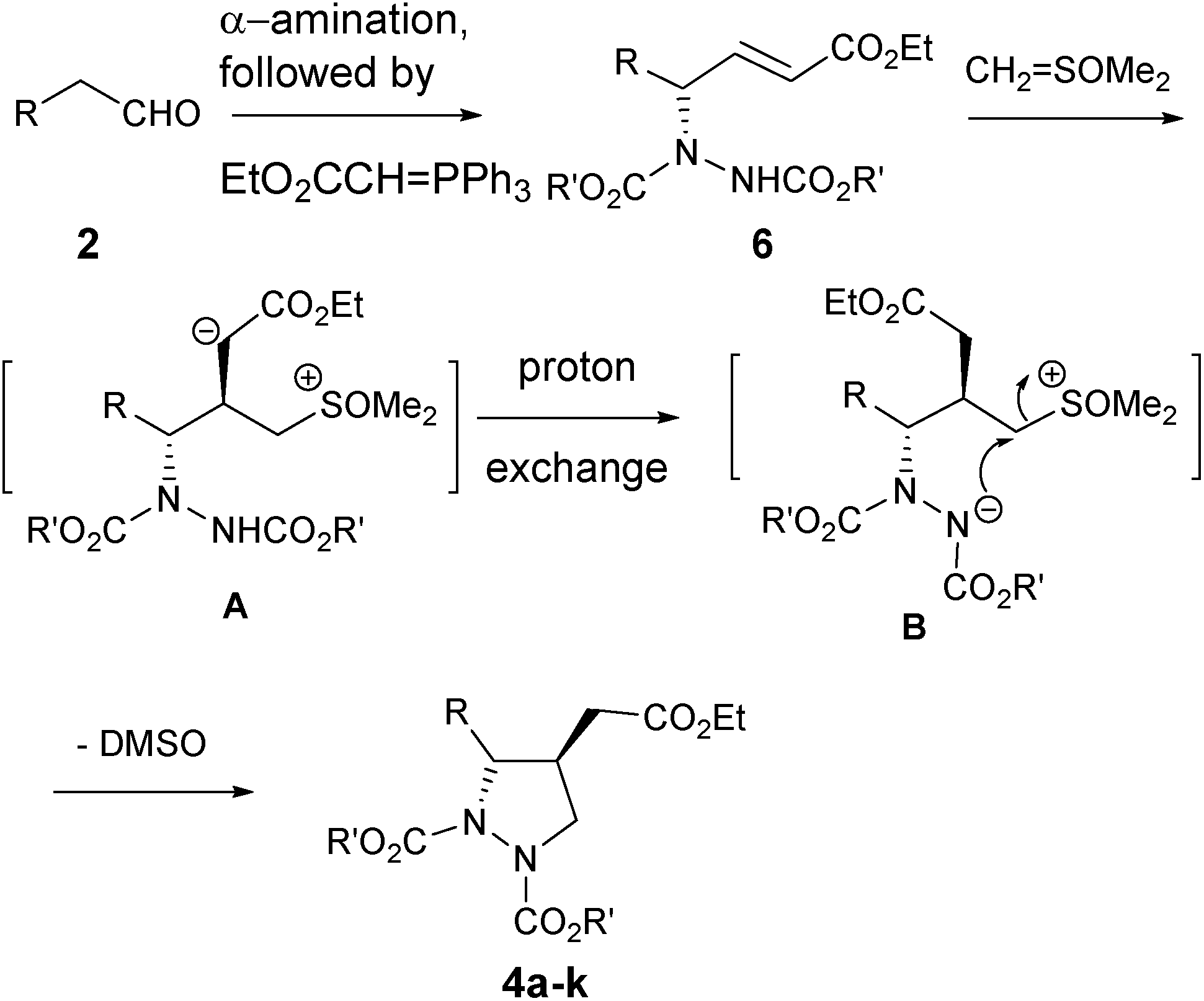 | ||
| Scheme 2 Probable mechanistic pathway for the formation of 4a–k. | ||
To rationalize the observed high ‘anti’ diastereoselectivity between the two substituents in the formed pyrazolidines 4a–k, Felkin Anh model26 has been proposed (Fig. 2). In this model, nucleophilic attack of the sulfur ylide takes place exclusively at the ‘Si’ face of olefin incorporating Bürgi-Dunitz trajectory27 leading to highly diastereoselective pyrazolidines 4a–k, following transition states (TS-I to TS-III).
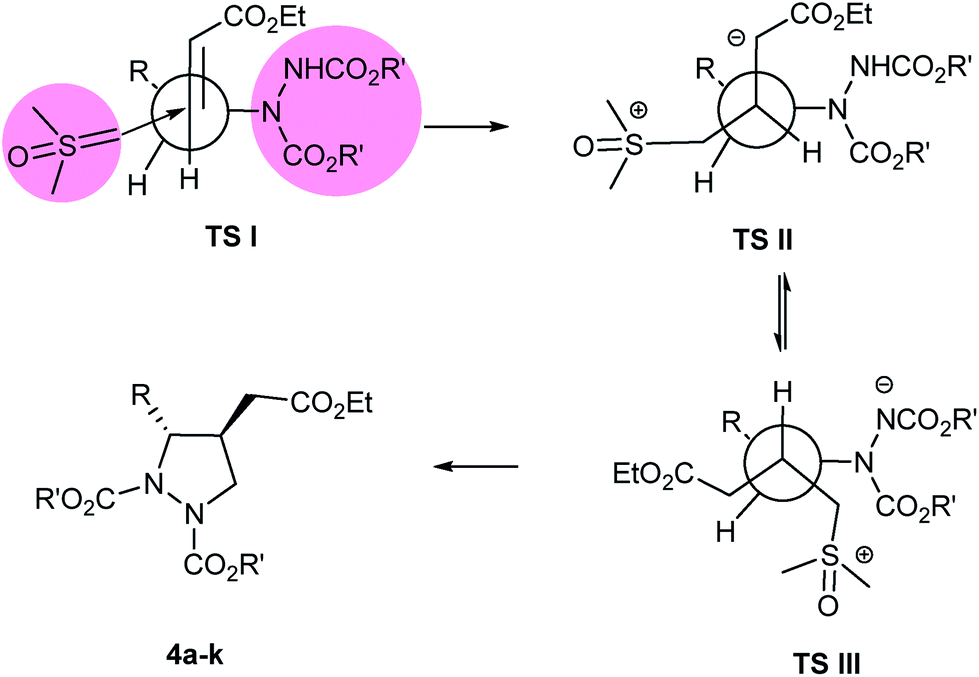 | ||
| Fig. 2 Proposed transition state model (R = alkyl or alkylaryl and R′ = Et, iPr, tBu). | ||
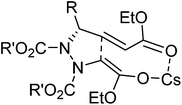
In the case of 5a–h, only one diastereomer was obtained predominantly out of four possible diastereomers during Michael addition. This high diastereoselectivity can be explained on the basis of the chelation controlled favorable transition state model.28
To demonstrate its potential applicability, 4a was subjected to reductive cleavage of N–N bond under metal/NH3 conditions (Na in liquid NH3, −70 to −40 °C, THF, 1 h) to afford the corresponding anti-1,3-diamino acid 7 (60% yield),29 which are common structural subunits present in many natural products and also useful as chiral ligands (Scheme 3). We have carried out several experiments to deprotect carbamate moieties in 4, 5 and 7 to demonstrate the further utility of this methodology. Unfortunately, we have ended up with complex reaction mixtures in the case of benzyl (Cbz) and tert butyl carbamates (Boc). The deprotection of ethyl carbamate under basic condition was also not successful; starting material was recovered, which may be a limitation of this methodology.30
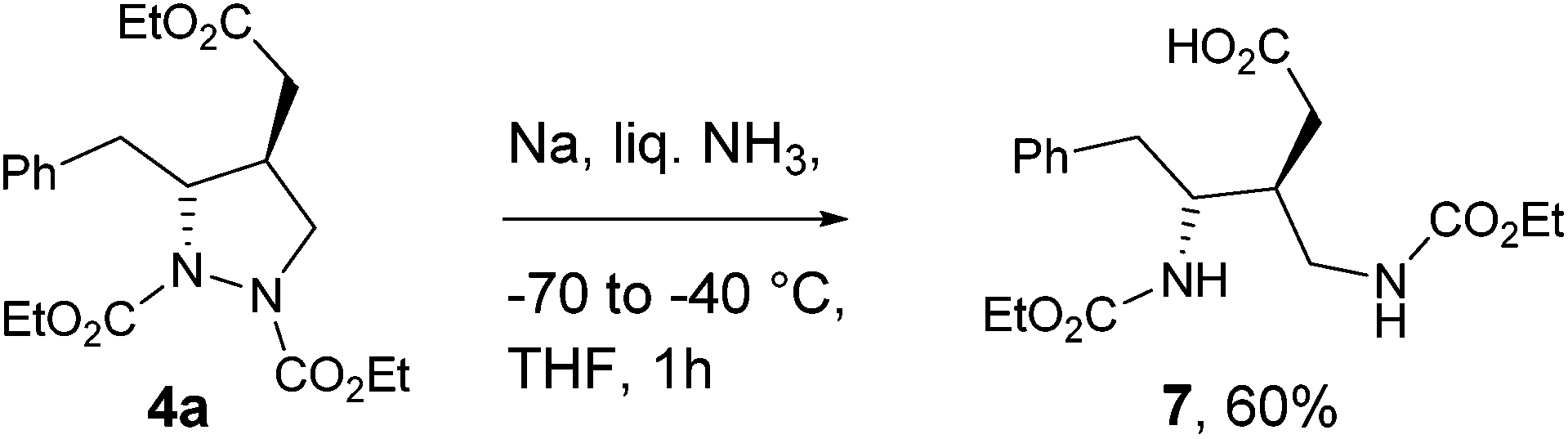 | ||
| Scheme 3 Reductive cleavage of N–N bond. | ||
Conclusions
In conclusion, we have described, for the first time, a novel [4+1]-annulation strategy involving a sequential α-amination/Wittig olefination/Corey–Chaykovsky reaction or intramolecular Michael reaction of aldehydes that leads to the synthesis of densely functionalized pyrazolidine carboxylates 4 & 5 containing two to three stereogenic centers with high yields and excellent enantio- and diastereoselectivities. The reductive cleavage of N–N bond in pyrazolidine afforded optically active 2,3-disubstituted 1,3-diamino acid 7. The ready availability of starting materials, milder reaction conditions and the formation of two to three stereogenic centers under “one-pot, metal-free conditions” makes this protocol quite useful in organic synthesis.Acknowledgements
We sincerely thank CSIR, New Delhi, India (Indus MAGIC project CSC-0123) and DST-SERB (no. SB/S1/OC-42/2014), for financial support. K. G. L and R. D. A thank CSIR, New Delhi, India for senior research fellowships. We thank Dr Rajesh G. Gonnade and Mr Shridhar Thorat (Center for Materials Characterization, CSIR-NCL) for assistance with X-ray crystallography. We also thank Dr B. Senthil Kumar for his initial suggestion and encouragement.Notes and references
- (a) S. M. Sondhi, M. Dinodia, J. Singh and R. Rani, Curr. Bioact. Compd., 2007, 3, 91 CrossRef CAS; (b) J. Elguero in Comprehensive Heterocyclic Chemistry, ed. K. T. Potts, Pergamon Press, Oxford, 1984, vol. 5, p. 167 Search PubMed; (c) J. M. Ellis and S. B. King, Tetrahedron Lett., 2002, 43, 5833 CrossRef CAS; (d) L. C. Behr, R. Fusco and C. H. Jarboe, in The Chemistry of Heterocyclic Compounds, Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles and Condensed Rings, ed. A. Weissberger, Interscience Publishers, New York, 1967 Search PubMed; (e) F. M. Guerra, M. R. Mish and E. M. Carreira, Org. Lett., 2000, 2, 4265 CrossRef CAS PubMed; (f) G. A. Whitlock and E. M. Carreira, Helv. Chim. Acta, 2000, 83, 2007 CrossRef CAS; (g) J. Barluenga, F. Fernández-Marí, A. L. Viado, E. Aguilar, B. Olano, S. García-Granda and C. Moya-Rubiera, Chem.–Eur. J., 1999, 5, 883 CrossRef CAS; (h) B. Stanovnik, B. Jelen, C. Turk, M. Žličar and J. Svete, J. Heterocycl. Chem., 1998, 35, 1187 CrossRef CAS PubMed; (i) M. R. Mish, F. M. Guerra and E. M. Carreira, J. Am. Chem. Soc., 1997, 119, 8379 CrossRef CAS; (j) T. P. Lebold and M. A. Kerr, Org. Lett., 2009, 11, 4354 CrossRef CAS PubMed; (k) A. Chauveau, T. Martens, M. Bonin, L. Micouin and H. P. Husson, Synthesis, 2002, 1885 CAS.
- (a) Z. Brzozowski, F. Sączewski and M. Gdaniec, Eur. J. Med. Chem., 2000, 35, 1053 CrossRef CAS; (b) C. D. Cox, M. J. Breslin, B. J. Mariano, P. J. Coleman, C. A. Buser, E. S. Walsh, K. Hamilton, H. E. Huber, N. E. Kohl, M. Torrent, Y. Yan, L. C. Kuo and G. D. Hartman, Bioorg. Med. Chem. Lett., 2005, 15, 2041 CrossRef CAS PubMed; (c) M. E. Camacho, J. León, A. Entrena, G. Velasco, M. D. Carrión, G. Escames, A. Vivó, D. Acuña-Castroviejo, M. A. Gallo and A. Espinosa, J. Med. Chem., 2004, 47, 5641 CrossRef CAS PubMed.
- (a) Y. Rajendra Prasad, A. Lakshmana Rao, L. Prasoona, K. Murali and P. Ravi Kumar, Bioorg. Med. Chem. Lett., 2005, 15, 5030 CrossRef CAS PubMed; (b) E. Palaska, D. Erol and R. Demirdamar, Eur. J. Med. Chem., 1996, 31, 43 CrossRef CAS; (c) F. Chimenti, A. Bolasco, F. Manna, D. Secci, P. Chimenti, O. Befani, P. Turini, V. Giovannini, B. Mondovì, R. Cirilli and F. La Torre, J. Med. Chem., 2004, 47, 2071 CrossRef CAS PubMed; (d) N. Gökhan-Kelekçi, S. Koyunoğlu, S. Yabanoglu, K. Yelekçi, O. Ozgen, G. Ucar, K. Erol, E. Kendi and A. Yeşilada, Bioorg. Med. Chem., 2009, 17, 675 CrossRef PubMed; (e) P. L. Ornstein, T. J. Bleisch, M. B. Arnold, R. A. Wright, B. G. Johnson and D. D. Schoepp, J. Med. Chem., 1998, 41, 346 CrossRef CAS PubMed.
- F. Chimenti, B. Bizzarri, F. Manna, A. Bolasco, D. Secci, P. Chimenti, A. Granese, D. Rivanera, D. Lilli, M. M. Scaltrito and M. I. Brenciaglia, Bioorg. Med. Chem. Lett., 2005, 15, 603 CrossRef CAS PubMed.
- (a) M. J. Kornet and R. J. Garrett, J. Pharm. Sci., 1979, 68, 377 CrossRef CAS PubMed; (b) Z. Özdemir, H. B. Kandilci, B. Gümüşel, Ü. Çalış and A. A. Bilgin, Eur. J. Med. Chem., 2007, 42, 373 CrossRef PubMed.
- J. R. Goodell, F. Puig-Basagoiti, B. M. Forshey, P. Y. Shi and D. M. Ferguson, J. Med. Chem., 2006, 49, 2127 CrossRef CAS PubMed.
- P. L. Zhao, F. Wang, M. Z. Zhang, Z. M. Liu, W. Huang and G. F. Yang, J. Agric. Food Chem., 2008, 56, 10767 CrossRef CAS PubMed.
- M. Wang, J. Zhang, J. Liu, C. Xu and H. Ju, J. Lumin., 2002, 99, 79 CrossRef CAS.
- (a) S. Hanessian, G. McNaughton-Smith, H.-G. Lombart and W. D. Lubell, Tetrahedron, 1997, 53, 12789 CrossRef CAS and references cited therein; (b) H. O. Kim, C. Lum and M. S. Lee, Tetrahedron Lett., 1997, 38, 4935 CrossRef CAS.
- (a) T. Jahn, G. M. König, A. D. Wright, G. Wörheide and J. Reitner, Tetrahedron Lett., 1997, 38, 3883 CrossRef CAS; (b) P. M. When and J. Du Bois, J. Am. Chem. Soc., 2002, 124, 12950 CrossRef PubMed; (c) M. Morgen, S. Bretzke, P. Li and D. Menche, Org. Lett., 2010, 12, 4494 CrossRef CAS PubMed; (d) K. Vickery, A. M. Bonin, R. R. Fenton, S. O'Mara, P. J. Russell, L. K. Webster and T. W. Hambley, J. Med. Chem., 1993, 36, 3663 CrossRef CAS; (e) C. M. Bromba, J. W. Mason, M. G. Brant, T. Chan, M. D. Lunke, M. Petric, M. J. Boulanger and J. E. Wulff, Bioorg. Med. Chem. Lett., 2011, 21, 7137 CrossRef CAS PubMed; (f) P. Chand, P. L. Kotian, A. Dehghani, Y. El-Kattan, T.-H. Lin, T. L. Hutchison, Y. S. Babu, S. Bantia, A. J. Elliott and J. A. Montgomery, J. Med. Chem., 2001, 44, 4379 CrossRef CAS PubMed.
- W. Wang, D. D. Simovic, M. Di, L. Fieber and K. S. Rein, Bioorg. Med. Chem. Lett., 2013, 23, 1949 CrossRef CAS PubMed.
- (a) K.-D. Hesse, Justus Liebigs Ann. Chem., 1971, 743, 50 CrossRef CAS PubMed; (b) G. le Fevre and J. Hamelin, Tetrahedron Lett., 1979, 20, 1757 CrossRef; (c) B. Fouchet, M. Joucla and J. Hauelin, Tetrahedron Lett., 1981, 22, 1333 CrossRef CAS; (d) T. Shimizu, Y. Hayashi, Y. Kitora and K. Teramura, Bull. Chem. Soc. Jpn., 1982, 55, 2450 CrossRef CAS; (e) T. Shimizu, Y. Hayashi, M. Miki and K. Teramura, J. Org. Chem., 1987, 52, 2277 CrossRef CAS; (f) L. O. Davis, W. F. M. Daniel and S. L. Tobey, Tetrahedron Lett., 2012, 53, 522 CrossRef CAS PubMed; (g) H. Xie, J. Zhu, Z. Chen, S. Li and Y. Wu, Synthesis, 2011, 2767 CAS; (h) T. Hashimoto, H. Kimura, Y. Kawamata and K. Maruoka, Nat. Chem., 2011, 3, 642 CrossRef CAS PubMed; (i) T. Hashimoto, M. Omote and K. Maruoka, Angew. Chem., Int. Ed., 2011, 50, 3489 CrossRef CAS PubMed.
- (a) S. Kobayashi, R. Hirabayashi, H. Shimizu, H. Ishitani and Y. Yamashita, Tetrahedron Lett., 2003, 44, 3351 CrossRef CAS; (b) É. Frank, Z. Mucsi, I. Zupkó, B. Réthy, G. Falkay, G. Schneider and J. Wölfling, J. Am. Chem. Soc., 2009, 131, 3894 CrossRef PubMed; (c) A. Zamfir, S. Schenker, W. Bauer, T. Clark and S. B. Tsogoeva, Eur. J. Org. Chem., 2011, 3706 CrossRef CAS PubMed.
- (a) R. Grigg, J. Kemp and N. Thompson, Tetrahedron Lett., 1978, 19, 2827 CrossRef; (b) G. le Fevre and J. Hamelin, Tetrahedron Lett., 1979, 20, 1757 CrossRef; (c) B. B. Snider, R. S. E. Conn and S. Sealfon, J. Org. Chem., 1979, 44, 218 CrossRef CAS; (d) R. Grigg, M. Dowling, M. W. Jordan, V. Sridharan and S. Thianpatanagul, Tetrahedron, 1987, 43, 5873 CrossRef CAS; (e) V. V. Khau and M. J. Martinelli, Tetrahedron Lett., 1996, 37, 4323 CrossRef CAS.
- (a) S. Kobayashi, H. Shimizu, Y. Yamashita, H. Ishitani and J. Kobayashi, J. Am. Chem. Soc., 2002, 124, 13678 CrossRef CAS PubMed; (b) Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 11279 CrossRef CAS PubMed.
- (a) S. Shirakawa, P. J. Lombardi and J. L. Leighton, J. Am. Chem. Soc., 2005, 127, 9974 CrossRef CAS PubMed; (b) A. Zamfir and S. B. Tsogoeva, Synthesis, 2011, 1988 CAS.
- (a) Q. Yang, X. Jiang and S. Ma, Chem.–Eur. J., 2007, 13, 9310 CrossRef CAS PubMed; (b) S. Ma, N. Jiao, Z. Zheng, Z. Ma, Z. Lu, L. Ye, Y. Deng and G. Chen, Org. Lett., 2004, 6, 2193 CrossRef CAS PubMed; (c) R. L. LaLonde, Z. J. Wang, M. Mba, A. D. Lackner and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 598 CrossRef CAS PubMed; (d) T. Hashimoto, Y. Maeda, M. Omote, H. Nakatsu and K. Maruoka, J. Am. Chem. Soc., 2010, 132, 4076 CrossRef CAS PubMed; (e) H. Suga, A. Funyu and A. Kakehi, Org. Lett., 2006, 9, 97 CrossRef PubMed; (f) H. Suga, Y. Furihata, A. Sakamoto, K. Itoh, Y. Okumura, T. Tsuchida, A. Kakehi and T. Baba, J. Org. Chem., 2011, 76, 7377 CrossRef CAS PubMed; (g) B. R. Rosen, J. E. Ney and J. P. Wolfe, J. Org. Chem., 2010, 75, 2756 CrossRef CAS PubMed.
- (a) H. Suga, T. Arikawa, K. Itoh, Y. Okumura, A. Kakehi and M. Shiro, Heterocycles, 2010, 81, 1669 CrossRef CAS; (b) S. Muller and B. List, Angew. Chem., Int. Ed., 2009, 48, 9975 CrossRef PubMed; (c) O. Mahe, I. Dez, V. Levacher and J. F. Briere, Angew. Chem., Int. Ed., 2010, 49, 7072 CrossRef CAS PubMed; (d) Z. C. Geng, J. Chen, N. Li, X. F. Huang, Y. Zhang, Y. W. Zhang and X. W. Wang, Beilstein J. Org. Chem., 2012, 8, 1710 CrossRef CAS PubMed; (e) M. Fernández, E. Reyes, J. L. Vicario, D. Badía and L. Carrillo, Adv. Synth. Catal., 2012, 354, 371 CrossRef PubMed; (f) B. S. Kumar, V. Venkataramasubramanian and A. Sudalai, Org. Lett., 2012, 14, 2468 CrossRef CAS PubMed and references cited therein.
- (a) B. List, Tetrahedron, 2002, 58, 5573 CrossRef CAS; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed.
- (a) R. D. Aher, B. S. Kumar and A. Sudalai, J. Org. Chem., 2015, 80, 2024 CrossRef CAS PubMed; (b) A. Bøgevig, K. Juhl, N. Kumaragurubaran, W. Zhuang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2002, 41, 1790 CrossRef; (c) A. J. Oelke, S. Kumarn, D. A. Longbottom and S. V. Ley, Synlett, 2006, 2548 CAS; (d) N. S. Chowdari, D. B. Ramachary and C. F. Barbas, Org. Lett., 2003, 5, 1685 CrossRef CAS PubMed; (e) S. P. Kotkar, V. B. Chavan and A. Sudalai, Org. Lett., 2007, 9, 1001 CrossRef CAS PubMed.
- (a) S. P. Kotkar and A. Sudalai, Tetrahedron Lett., 2006, 47, 6813 CrossRef CAS PubMed; (b) V. Rawat, P. V. Chouthaiwale, V. B. Chavan, G. Suryavanshi and A. Sudalai, Tetrahedron Lett., 2010, 51, 6565 CrossRef CAS PubMed; (c) V. Venkataramasubramanian, I. N. C. Kiran and A. Sudalai, Synlett, 2015, 26, 355 CrossRef CAS.
- E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1353 CrossRef CAS.
- ESI†.
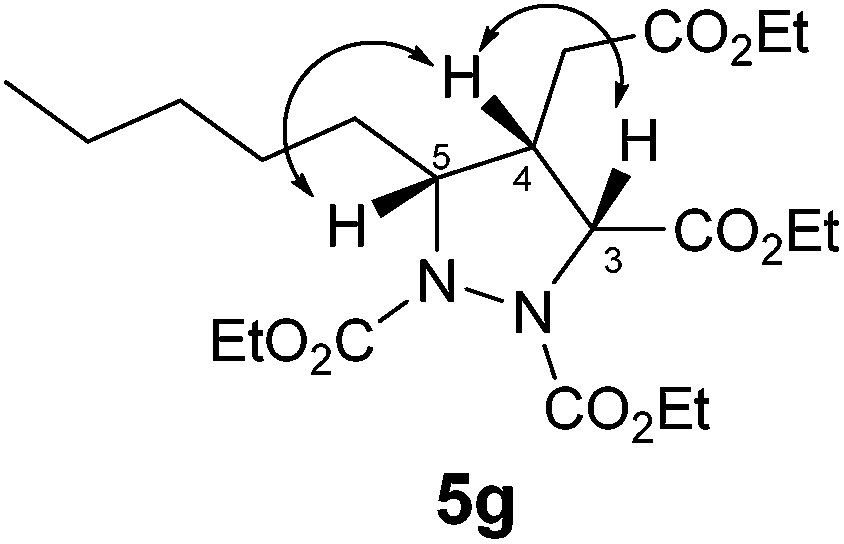
- The relative stereochemistry of 5g was confirmed by COSY and NOESY studies. A significant NOESY correlation was observed between H5–H4 and H4–H3 confirming the syn relationship between H3–H4–H5 (see below).
 .
. - Attempts to deuterate the NH proton of carbamate 6 under various reaction conditions to ascertain the proton exchange phenomena were unsuccessful.
- M. Chérest, H. Felkin and N. Prudent, Tetrahedron Lett., 1968, 9, 2199 CrossRef.
- H. B. Burgi, J. D. Dunitz, J. M. Lehn and G. Wipff, Tetrahedron, 1974, 30, 1563 CrossRef.
- syn, syn diastereoselectivity obtained during Michael addition can be explained using chelation controlled favorable transition state model as shown below:
 .
. - Hydrolysis of ester to carboxylic acid was observed during reductive N–N bond cleavage; for detailed experimental procedure see ESI†.
- Carbamate deprotection in 4, 5 and 7 was unsuccessful with following reaction conditions: (1) R′ = tBu; (a) TFA, CH2Cl2, 25 °C to 70 °C, 1 h to 12 h; (b) methanolic HCl, 25 °C, 12 h. (2) R′ = Bn; (a) 10% Pd, C/H2 (1 atm), MeOH; (b) Raney®-Ni, H2 (60 psi), MeOH. (3) R′ = Et; K2CO3, EtOH, 25 °C to 70 °C.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1041539. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra09583b |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |