
Solid phase synthesis of functionalized indazoles using triazenes – scope and limitations†
Ana Maria Garciaab,
Nicole Jungac,
Carmen Gilb,
Martin Niegerd and
Stefan Bräse*ac
aInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology, Campus North, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
bCentro de Investigaciones Biológicas (CSIC), Ramiro de Maeztu 9, 28040 Madrid, Spain
cInstitute of Organic Chemistry, Karlsruhe Institute of Technology, Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu
dLaboratory of Inorganic Chemistry, Department of Chemistry, University of Helsinki, P.O Box 55 (A. I. Virtasen aukio 1), 00014 Helsinki, Finland
First published on 16th July 2015
Abstract
Indazoles are important heterocycles as they are a substantial part in many drugs. In this study, we present a modular synthesis of highly substituted indazoles via a strategy on solid supports. The heterocyclic nitrogen atoms originate from diazonium salts, being cleaved from triazene containing resins. The scope and limitations of this process are explored, considering especially the competitive occurrence of triazines and the cleavage of hydrolyzed and traceless side products.
Introduction
Benzoannelated nitrogen heterocycles are known to be interesting scaffolds to find compounds which possess high biological activity and are considered as a source of many privileged compound classes in medicinal chemistry.1–7 In particular, some indazole-containing compounds have a substantial impact when used as therapeutic agents (Fig. 1). Well-known examples are Bendazac (A)8 which is an anti-inflammatory agent used as an anti-cataract drug and Benzydamine (B),9 a serotonin 5-HT3 receptor antagonist used to treat and prevent nausea and vomiting induced by cancer chemotherapy. Apart from this, famous indazole derivatives include DNA-intercalating agents such as the benzothiopyranoindazole CI-958,10 activators of the nitric oxide receptor,11 immunosuppressors such as Bindarit (C),12 calmodulin antagonists such as DY-9760 (D),13 and indazoles with anticancer activity such as Lonidamine (E) (Fig. 1).14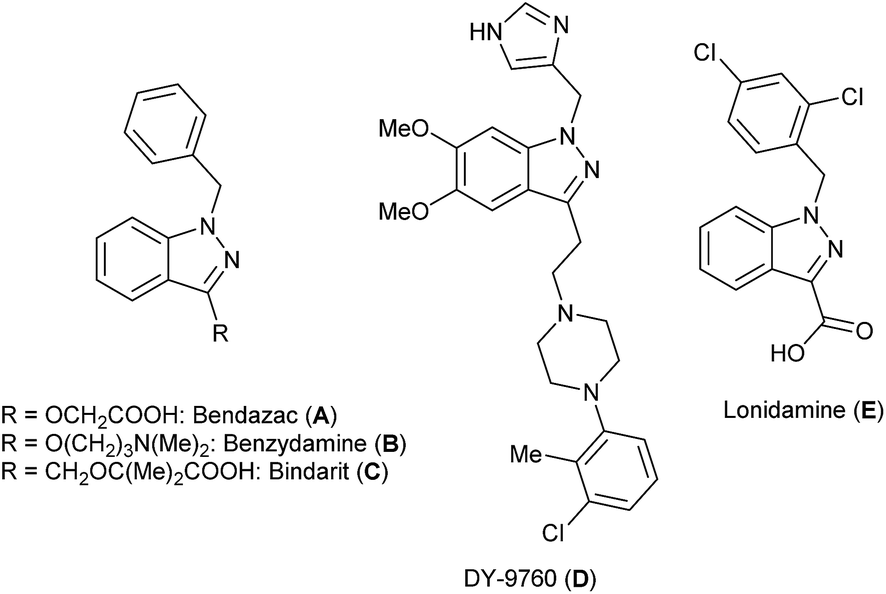 | ||
| Fig. 1 Molecules containing an indazole moiety with proven biological activity. | ||
Moreover, indazoles are interesting molecules because they may act as bioisosteres15 of relevant heterocycles such as indoles16 and benzimidazoles.17 Unlike the latter ones, the indazole heterocycle is one of the least exploited ones from a synthetic point of view, especially regarding solid phase procedures.18 While the solid phase organic synthesis (SPOS) of small-sized molecules has emerged as an important tool for the generation of heteroaromatic scaffolds in drug discovery,19 there are only a few reported examples for the synthesis of indazoles in solid phases.20–22 Therefore, in order to benefit from the possibility of rapid syntheses without tedious and time-consuming purification steps, we intended to develop a straightforward method to gain diverse indazoles via solid phase chemistry. While according to the first synthesis of indazoles on solid supports,20 the indazole unit was formed by a Lewis acid-catalyzed cyclization and the cleavage of the indazole from the solid phase in a second step, we decided to follow a route that would allow the formation of the indazole and cleavage off the resin in only one step.
We envisaged the triazene functionality to act as a suitable linker system to cleave indazoles as we observed the formation of a single 3-acylaminoindazole during the reaction of a 3,3-diisopropyltriazene derivative with an acyl chloride and the subsequent acid-mediated cleavage of the triazene. This result was obtained in our group as a side-reaction in the solution phase synthesis of 3-acylbenzotriazines in 2009.23 In the former procedure, the triazenes, which have been shown to be remarkably versatile starting materials for the liquid and solid phase synthesis of numerous nitrogen-based heterocycles,6,24 had been used as a protected diazonium salt.25,26
Results and discussion
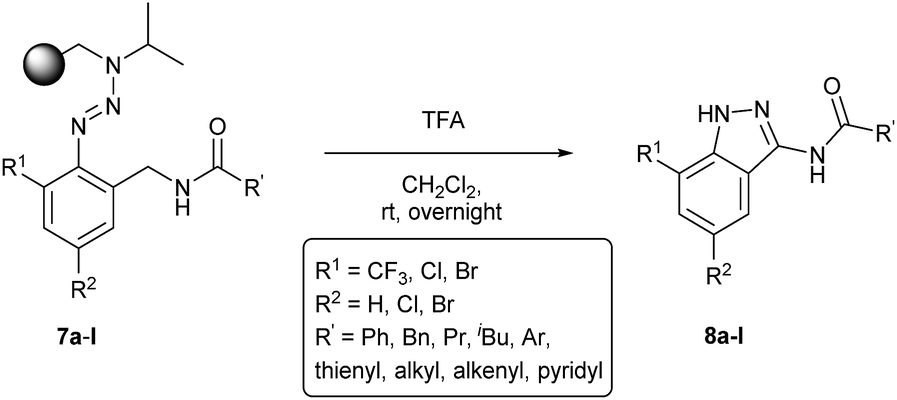
Encouraged by these results in conventional solution-phase reactions, we envisaged the development of an alternative route for the synthesis of indazole heterocycles on solid supports. This manuscript summarizes a novel established procedure for the solid phase synthesis of indazole heterocycles and enlightens the influence of the substituents in different positions on the molecule on the formation of the heterocyclic core. The immobilization of building blocks on a polymeric support using the triazene T1 linker requires the syntheses of diazonium salts which were prepared by the diazotization of o-aminobenzonitriles 1a–1h with isoamylnitrite. Their subsequent coupling with isopropylaminomethylpolystyrene 2 yielded triazene resins 3a–3h according to described procedures.23 While almost all literature-known reactions in solid phases using triazene linkers have been performed with a benzylaminomethylpolystyrene backbone,6 we switched to the given isopropyl-derivative (2) in order to reflect the conditions of a successful reaction in solution as well as possible. Subsequently, the nitrile groups of the immobilized triazenes 3a–3h were reduced to the corresponding amines by using a solution of lithium aluminium hydride in tetrahydrofuran (Scheme 1).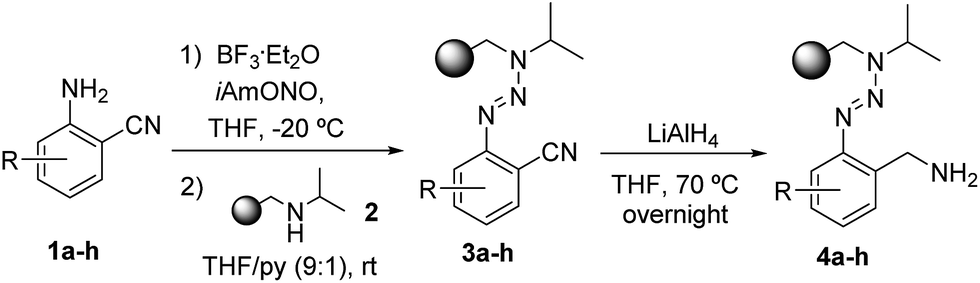 | ||
| Scheme 1 Synthesis of the triazene resins 4a–4h. | ||
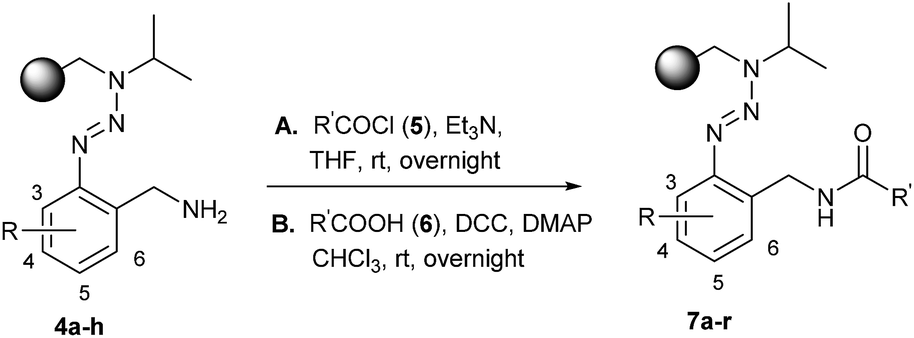
Resins 4 were then modified via the formation of an amide bond which was carried out to give resins 7 (Table 1) via two different ways: (A) an acylation strategy of amine 4 with the corresponding acyl chloride 5 in the presence of triethylamine or (B) the coupling between amine 4 and carboxylic acid 6 by using the coupling reagents DCC and DMAP. The latter procedure allows a wide variety of substituents on the introduced building block due to the large amount of carboxylic acids that are commercially available.
|
||||
|---|---|---|---|---|
| Resin 4 | R | Method | R′ | Resin 7 |
| 4a | 3-CF3 | A | Phenyl | 7a |
| 4a | 3-CF3 | A | 2,6-F2C6H3 | 7b |
| 4a | 3-CF3 | A | 4-MeOC6H4 | 7c |
| 4a | 3-CF3 | A | Isobutyl | 7d |
| 4a | 3-CF3 | A | n-Propyl | 7e |
| 4a | 3-CF3 | A | 2-Thiophenyl | 7f |
| 4a | 3-CF3 | A | Isobutenyl | 7g |
| 4a | 3-CF3 | A | Benzyl | 7h |
| 4a | 3-CF3 | B | 4-Pyridyl | 7i |
| 4b | 3-Cl, 5-Cl | A | Phenyl | 7j |
| 4c | 3-Br, 5-Br | A | Phenyl | 7k |
| 4d | 3-Cl | A | Phenyl | 7l |
| 4a | 3-CF3 | A | Methyl | 7m |
| 4e | H | A | Phenyl | 7n |
| 4f | 4-Cl | A | Phenyl | 7o |
| 4g | 5 F | A | Phenyl | 7p |
| 4h | 5-Cl | A | Phenyl | 7q |
| 4a | 3-CF3 | B | 4-IC6H4 | 7r |
The cleavage of resins 7 was performed with trifluoroacetic acid in anhydrous dichloromethane in order to obtain the desired indazoles (8) (Table 2). According to the conventional synthesis in solution,23 resin 4a was chosen as the starting material for the first experiments, along with 2-amine-3-trifluoromethylbenzonitrile, coupled with several acyl chlorides/carboxylic acids. Independent of the nature of the second building block (carrying R′), we could obtain the target indazoles (8a–8i) in moderate yields (29–34%) in solid phases when R′ was not methyl. Several substituents in the R′ position have been introduced to the immobilized compounds 7 including aromatic, aliphatic, thienyl, alkenyl and pyridyl residues without being able to find crucial differences concerning the success of the given synthetic route. Moreover, by changing the original 3-trifluoromethylbenzonitrile building block (1a) to 3-chlorine and 3-bromine-substituted benzonitriles (1b–1d), we could also obtain the corresponding indazoles (8j–8l) in moderate yields.
|
||||
|---|---|---|---|---|
| Indazole | R1/R2 | R′ | Structure | Yield [%] |
| 8a | R1 = CF3, R2 = H | Phenyl | 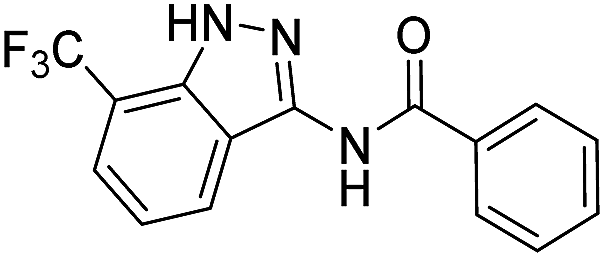 |
31 |
| 8b | R1 = CF3, R2 = H | 2,6-F2C6H3 | 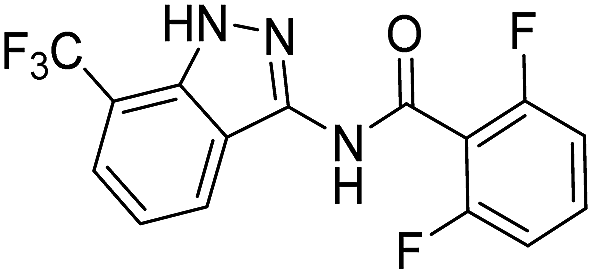 |
30 |
| 8c | R1 = CF3, R2 = H | 4-MeOC6H4 | 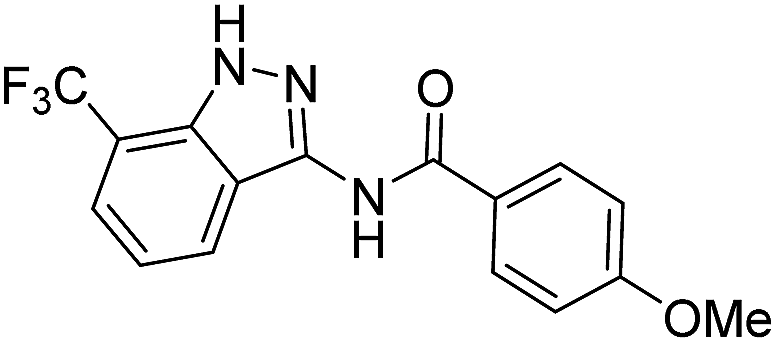 |
30 |
| 8d | R1 = CF3, R2 = H | Isobutyl | 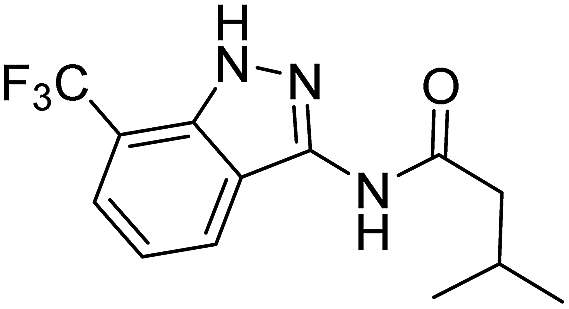 |
30 |
| 8e | R1 = CF3, R2 = H | n-Propyl | 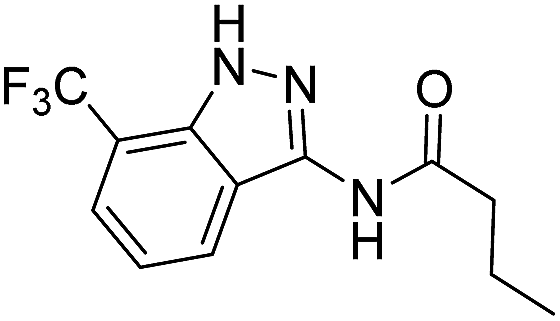 |
34 |
| 8f | R1 = CF3, R2 = H | 2-Thiophenyl | 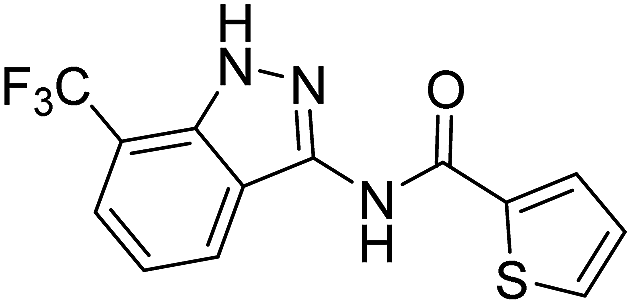 |
30 |
| 8g | R1 = CF3, R2 = H | Isobutenyl | 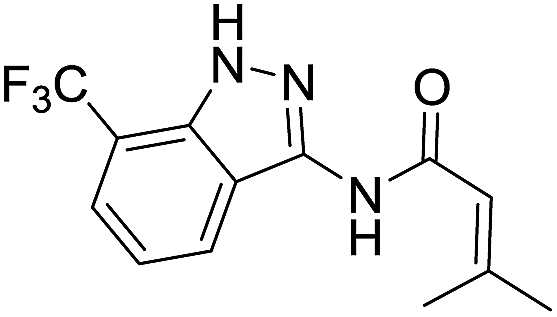 |
31 |
| 8h | R1 = CF3, R2 = H | Benzyl | 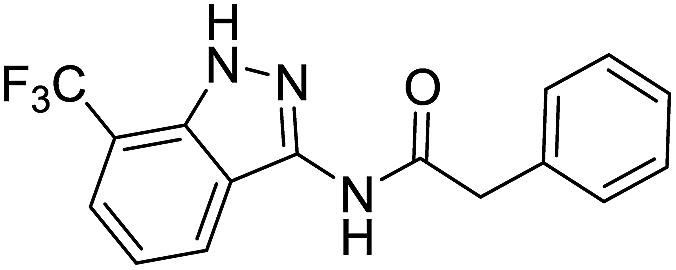 |
34 |
| 8i | R1 = CF3, R2 = H | 4-Pyridyl | 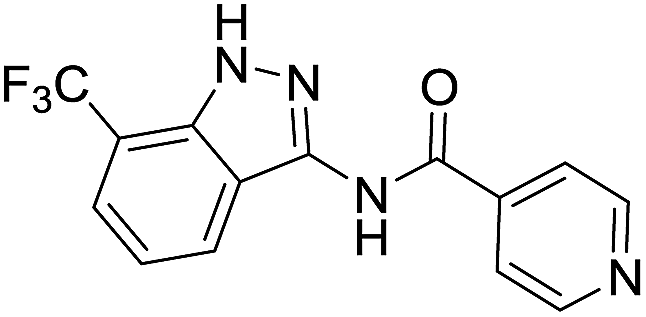 |
29 |
| 8j | R1 = R2 = Br | Ph | 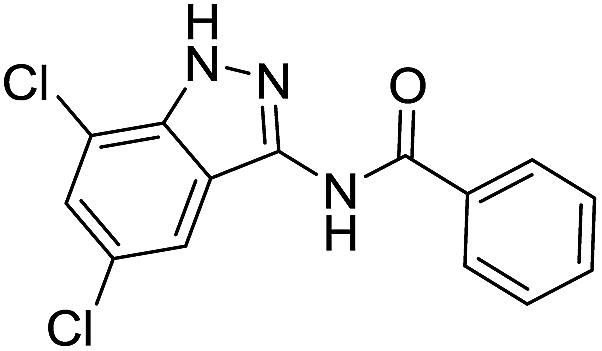 |
29 |
| 8k | R1 = R2 = Br | Ph | 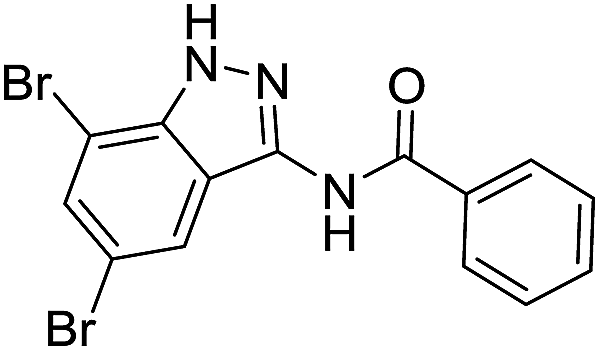 |
26 |
| 8l | R1 = Cl, R2 = H | Ph | 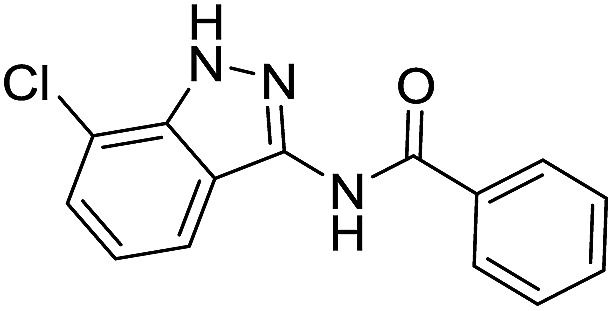 |
30 |
While the reactions of resins 7 including R′ residues larger than methyl gave the desired indazoles, which could be confirmed by crystallizing compound 8e and investigating it via X-ray crystallography (Fig. 2), the reaction of resin 4a with acetyl chloride followed by the cleavage of the resulting resin 7m did not give the expected indazole but the related benzotriazine 9, which was isolated through solution phase approaches (Scheme 2). In accordance with the former results,23 we assume that if 3-isopropyl-(2-(trifluoromethyl)phenyl)triaz-1-enes are used as possible sources in the triazine vs. indazole formation, precursors bearing a methyl R′ residue would favor the formation of benzotriazines (9) while precursors bearing an R′ residue larger than methyl would favor the formation of indazoles (8).
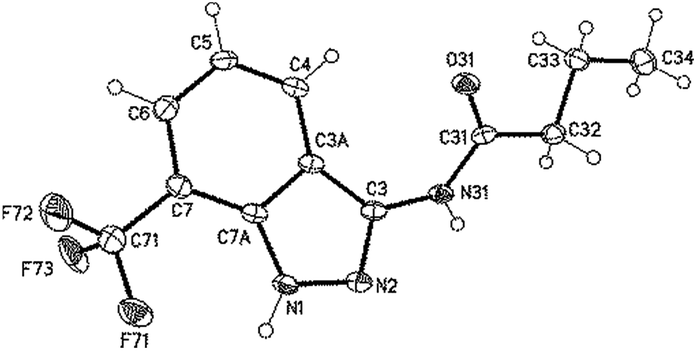 | ||
| Fig. 2 Crystal structure of 8e (minor part of the disordered F-atoms omitted for clarity, displacement parameters are drawn at the 50% probability level). | ||
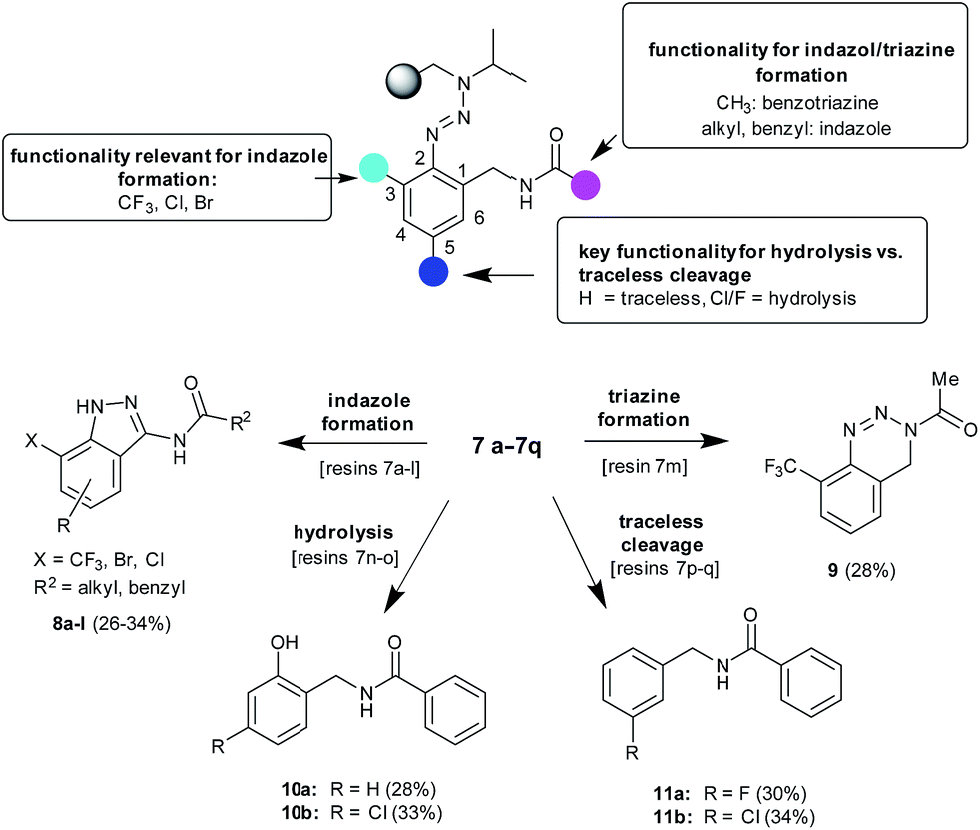 | ||
| Scheme 2 Products obtained via the cleavage of resins 7a–7q and dependencies regarding three points of diversity. | ||
Further experiments have been conducted to examine the scope of the indazole formation on solid supports. Besides the successfully used resins 4a–4d, carrying electron withdrawing substituents in the ortho-position to the triazene linkage, other amine building blocks were selected (1e–1h) giving resins 4e–4h that were used for the presented synthetic procedure. It has been shown that the substitution in the ortho-position as given in resins 7a–7l is mandatory for the cleavage of indazoles from solid supports. Resins where the ortho-position remains unsubstituted (7n–7q) do not yield indazoles via the herein presented procedure. The examination of the cleavage products of the latter resins showed that, depending on the substitution pattern of the triazene-containing aromatic moiety, either the hydrolysis or the traceless cleavage product was formed (10a–10b or 11a–11b respectively). We assume that with an unsubstituted ortho-position, the para-position (to the triazene-linkage) would have an especially strong influence on the formation of compounds of the type of 10 or 11 due to possible diazonium salt stabilization.27 If an atom with a lone electron pair is present in the 5-position, e.g. a chlorine, it can stabilize the positive charge of the diazonium moiety, whereas if the chlorine atom is in the 4-position, this conjugation is not possible and traces of water present in trifluoroacetic acid could attack the salt to yield the hydrolyzed product (Scheme 2). It is important to remark that these four different products (traceless 11, hydrolyzed 10, indazole 8, and the triazine derivative 9) are obtained exclusively without any traces of the others, which means cyclization occurs in a different way depending on the substitution of the initial benzonitrile. In other words, the stability of the resulting diazonium salt after cleavage, which strongly depends on the nature of the substituents at the ortho- and para-positions, has a strong influence on the cyclization step.
The herein presented procedure was evaluated towards its potential to synthesize complex indazole heterocycles on solid supports and the newly established method was used for the introduction of additional arene functionalities by a cross-coupling reaction. The polymer-bound aryl iodide 7r was coupled with 2-methylboronic acid via a Suzuki reaction to form triazene resin 12 in the presence of Pd(PPh3)4 as a catalyst (Scheme 3). After the cleavage, indazole 13 was isolated in a moderate yield of 36% in the solid phase.
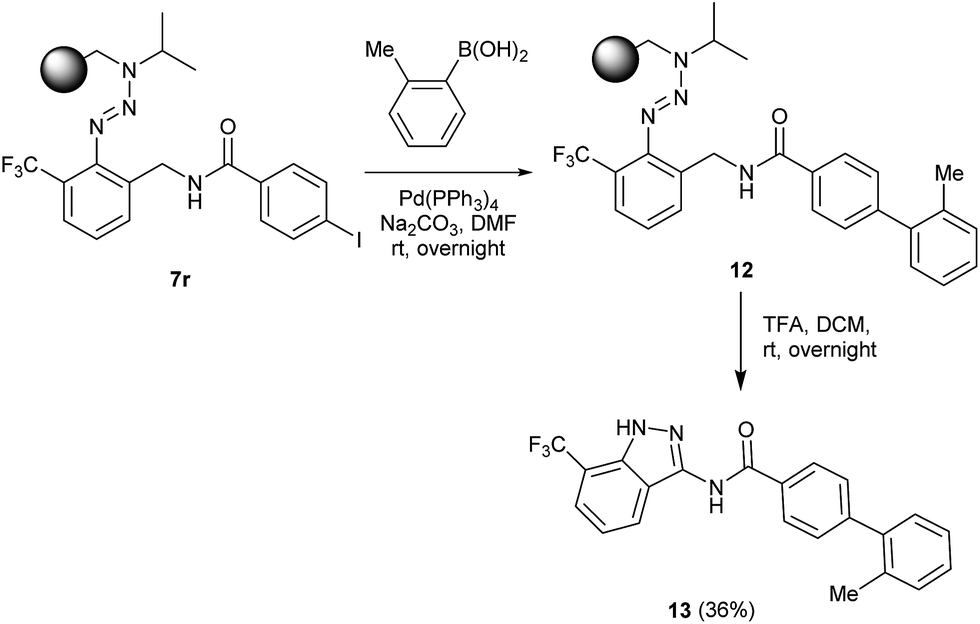 | ||
| Scheme 3 Cross-coupling reaction on solid supports and subsequent cleavage to give indazole 13. | ||
Conclusions
In this study, we present a modular synthesis of highly substituted indazoles on solid supports. The scope and limitations of this process are explored and side products are identified. The modularity was demonstrated by a synthetic procedure consisting of five steps using three different building blocks, one of which could be successfully introduced via an on-bead modification by Suzuki coupling. Only moderate yields were shown for the cleaved target compounds but the purity of the crude material allowed fast purification via flash chromatography. The solid supported reaction furnished enough material for biological evaluations via a straightforward protocol using only commercially available compounds. The herein developed methodology enables the extension of the protocol to increase the diversity of synthesized indazoles in future applications, offering a novel access to libraries of nitrogen-rich heterocycles with fluorine substitution patterns.Single crystal structure determination of 8e
The single crystal X-ray diffraction study of 8e was carried out on a Bruker-Nonius KappaCCD diffractometer at 123 K using MoKα radiation (λ = 0.71073 Å). Direct methods (for 8e, SHELXL-97)28 were used for structure solution and refinement was carried out using SHELXL-2014 (ref. 28) (full-matrix least-squares on F2). Hydrogen atoms were localized by difference Fourier synthesis map and refined using a riding model [H (N) free]. A semi-empirical absorption correction was applied. The trifluoromethyl group is disordered.Compound 8e
C12H12F3N3O, Mr = 271.25 g mol−1, colourless crystals, size 0.30 × 0.06 × 0.04 mm, triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 4.8788(4) Å, b = 10.1605(6) Å, c = 12.1307(9) Å, α = 79.331(5)°, β = 81.744(7)°, γ = 86.251(5)°, V = 584.35(7) Å3, Z = 2, Dcalcd = 1.542 Mg m−3, F(000) = 280, μ = 0.134 mm−1, T = 123 K, 6971 measured reflections (2θmax = 55°), 2681 independent reflections [Rint = 0.032], 206 parameters, 83 restraints, R1 [for 2010 I > 2σ(I)] = 0.042, wR2 (for all data) = 0.104, S = 1.04, largest diff. peak and hole = 0.320 e Å−3/−0.233 e Å−3.
(no. 2), a = 4.8788(4) Å, b = 10.1605(6) Å, c = 12.1307(9) Å, α = 79.331(5)°, β = 81.744(7)°, γ = 86.251(5)°, V = 584.35(7) Å3, Z = 2, Dcalcd = 1.542 Mg m−3, F(000) = 280, μ = 0.134 mm−1, T = 123 K, 6971 measured reflections (2θmax = 55°), 2681 independent reflections [Rint = 0.032], 206 parameters, 83 restraints, R1 [for 2010 I > 2σ(I)] = 0.042, wR2 (for all data) = 0.104, S = 1.04, largest diff. peak and hole = 0.320 e Å−3/−0.233 e Å−3.
Acknowledgements
We acknowledge the DAAD and the JAE predoctoral program from CSIC for their financial support (A.M.G.). The work was further supported by the Helmholtz programme Biointerfaces in Technology and Medicine (N.J. and S.B.), the Compound’s Platform (ComPlat) and by MINECO (SAF2012-33600) (C.G.).Notes and references
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed.
- K. Knepper, S. Vanderheiden and S. Bräse, Beilstein J. Org. Chem., 2012, 8, 1191–1199 CrossRef CAS PubMed.
- C. Gil and S. Bräse, J. Comb. Chem., 2009, 11, 175–197 CrossRef CAS PubMed.
- R. E. Ziegert, J. Toräng, K. Knepper and S. Bräse, J. Comb. Chem., 2005, 7, 147–169 CrossRef CAS PubMed.
- K. Knepper, R. E. Ziegert and S. Bräse, Tetrahedron, 2004, 60, 8591–8603 CrossRef CAS PubMed.
- S. Bräse, Acc. Chem. Res., 2004, 37, 805–816 CrossRef PubMed.
- S. Bräse, C. Gil and K. Knepper, Bioorg. Med. Chem., 2002, 10, 2415–2437 CrossRef.
- A. Guglielmotti, A. Capezzone de Joannon, N. Cazzolla, M. Marchetti, L. Soldo, G. Cavallo and M. Pinza, Pharmacol. Res., 1995, 32, 369–373 CrossRef CAS.
- N. H. Fanaki and M. A. el-Nakeeb, J. Chemother., 1992, 4, 347–352 CAS.
- E. C. Dees, L. R. Whitfield, W. R. Grove, S. Rummel, L. B. Grochow and R. C. Donehower, Clin. Cancer Res., 2000, 6, 3885–3894 CAS.
- D. L. Selwood, D. G. Brummell, J. Budworth, G. E. Burtin, R. O. Campbell, S. S. Chana, I. G. Charles, P. A. Fernandez, R. C. Glen, M. C. Goggin, A. J. Hobbs, M. R. Kling, Q. Liu, D. J. Madge, S. Meillerais, K. L. Powell, K. Reynolds, G. D. Spacey, J. N. Stables, M. A. Tatlock, K. A. Wheeler, G. Wishart and C. K. Woo, J. Med. Chem., 2001, 44, 78–93 CrossRef CAS PubMed.
- E. Mora, A. Guglielmotti, G. Biondi and P. Sassone-Corsi, Cell Cycle, 2012, 11, 159–169 CrossRef CAS PubMed.
- S. Tachibana, Y. Fujimaki, M. Tachibana, M. Tanaka, T. Kurata, O. Okazaki and K. Sudo, Arzneimittelforsch., 2005, 55, 135–144 CAS.
- M. de Lena, V. Lorusso, A. Latorre, G. Fanizza, G. Gargano, L. Caporusso, M. Guida, A. Catino, E. Crucitta, D. Sambiasi and A. Mazzei, Eur. J. Cancer, 2001, 37, 364–368 CrossRef CAS.
- P. Fludzinski, D. A. Evrard, W. E. Bloomquist, W. B. Lacefield, W. Pfeifer, N. D. Jones, J. B. Deeter and M. L. Cohen, J. Med. Chem., 1987, 30, 1535–1537 CrossRef CAS.
- F. R. de Sa Alves, E. J. Barreiro and C. A. Fraga, Mini-Rev. Med. Chem., 2009, 9, 782–793 CrossRef.
- M. Boiani and M. Gonzalez, Mini-Rev. Med. Chem., 2005, 5, 409–424 CrossRef CAS.
- R. Attaur, S. Malik, S. S. Hasan, M. I. Choudhary, C.-Z. Ni and J. Clardy, Tetrahedron Lett., 1995, 36, 1993–1996 CrossRef.
- F. Balkenhohl, C. von dem Bussche-Hünnefeld, A. Lansky and C. Zechel, Angew. Chem., Int. Ed., 1996, 35, 2288–2337 CrossRef CAS PubMed.
- B. Yan and H. Gstach, Tetrahedron Lett., 1996, 37, 8325–8328 CrossRef CAS.
- S. Krupkova, G. A. Slough and V. Krchnak, J. Org. Chem., 2010, 75, 4562–4566 CrossRef CAS PubMed.
- K. Kisseljova, P. Smyslova and V. Krchnak, ACS Comb. Sci., 2014, 16, 573–577 CrossRef CAS PubMed.
- R. Reingruber, S. Vanderheiden, T. Muller, M. Nieger, M. Es-Sayed and S. Bräse, Tetrahedron Lett., 2009, 50, 3439–3442 CrossRef CAS PubMed.
- K. C. Nicolaou, H. J. Mitchell, N. F. Jain, N. Winssinger, R. Hughes and T. Bando, Angew. Chem., Int. Ed., 1999, 38, 240–244 CrossRef CAS.
- S. Bräse, D. Enders, J. Köbberling and F. Avemaria, Angew. Chem., Int. Ed., 1998, 37, 3413–3415 CrossRef.
- S. Bräse, J. Köbberling, D. Enders, R. Lazny, M. Wang and S. Brandtner, Tetrahedron Lett., 1999, 40, 2105–2108 CrossRef.
- S. Dahmen and S. Bräse, Angew. Chem., Int. Ed., 2000, 39, 3681–3683 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data in cif-format (8e) and synthetic procedures and characterization for synthesized compounds (8a–8l, 9–13). CCDC 1038684. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra09705c |
| This journal is © The Royal Society of Chemistry 2015 |