DOI:
10.1039/C5RA11212E
(Paper)
RSC Adv., 2015,
5, 65321-65327
Optimization of an analytical method for the spectrophotometric determination of copper in tea and water samples after ultrasonic assisted cloud point extraction using a benzothiazole fluorescein derivative complexing agent†
Received
12th June 2015
, Accepted 23rd July 2015
First published on 24th July 2015
Abstract
In this work, 2′,7′-di(2-benzothiazolyl)-fluorescein (BTF) was synthesized by using 2-(2′,4′-dihydroxyphenyl) benzothiazole (BT) and phthalic anhydride. BTF is colorful and its absorption maximum occurs at 559 nm. Interestingly, the introduction of Cu2+ affords a dramatic decrease of the absorbance at 559 nm, which is due to the spirolactonization of BTF induced by Cu2+. BTF shows high selectivity toward Cu2+ over other common metal ions in real samples, such as K+, Mg2+, Ca2+, Na+, Fe3+, Cd2+, Al3+, Co2+, Cr3+, Mn2+, Ni2+, La3+ and Zn2+. On this basis, a cloud point extraction (CPE) was developed for copper determination by a UV-vis method. The BTF–Cu2+ complex was explored for pre-concentration, and dissolved in a micellar phase composed by octylphenoxypolyethoxyethanol (Triton X-114). Under optimal experimental conditions, the decrease of the absorbance value (ΔA) at 559 nm is linear with Cu2+ concentration in the range from 0.03 to 2 μmol L−1 (1.90–127 μg L−1) with a limit of detection of 7.48 nmol L−1 (3σ). The method has been successfully applied for the determination of copper in tea and water samples.
1. Introduction
Copper (Cu) is a micronutrient that serves as an essential factor to all organisms, and plays important roles in many physiological functions, including antioxidant effects, and hemoglobin synthesis.1,2 However, excessive intake or long-term exposure of copper would lead to the damage to liver and kidneys due to accumulation of the heavy metal. According to the U.S. Environmental Protection Agency (US EPA), the maximum contaminant level goal (MCLG) for copper in drinking water was 1.3 mg L−1. In the past decades, various techniques such as atomic absorption spectrometry,3,4 chemiluminescence,5,6 ICP-OES7,8 and spectrophotometric9 methods have been developed for the detection of copper. Among these, spectrophotometric method received attractive attention due to the common availability of the instrumentation, the simplicity of apparatus, fastness, precision and accuracy of the technique. However, spectrophotometric methods suffer from its insufficient sensitivity due to very low concentration of copper in many environmental and biological samples. Hence, a preconcentration step prior to its spectrophotometric determination is usually necessary in order to reach an appropriate level of sensitivity. Many preconcentration procedures for copper determination have been developed, such as liquid–liquid extraction,10 solid-phase extraction,11 cloud point extraction,12 flotation,13 etc.
Cloud point extraction (CPE) has been emerged as an alternative to conventional liquid–liquid extraction due to its lower toxic reagent use, use of nontoxic surfactants and safe, fast, simple and more economical procedure for analysis of various metal ions in various samples. In CPE, selection of the complexing agent is a crucial step. Azo dyes are the most frequently used as complexing agents due to their low solubility in water and ability to form hydrophobic complexes with a large number of metals. The low solubility of complex-forming ligands and their metallic complexes in water, most frequently were used in CPE. At the same time, more sensitive and selective complexing agents were exploited, which is a new trend for the cloud point extraction process.14 Several chelating extractants, such as 8-hydroxyquinoline (oxine),15 diethyldithiocarbamate (DDTC),16 thiamine,17 4-(2-pyridylazo) resorcinol (PAR)18 and 1,5-diphenylthiocarbazone (dithizone),19 have been used to extract Cu2+ in various samples. Several researchers have applied compounds by synthesis for developing fluorescence method for Cu2+.20–28
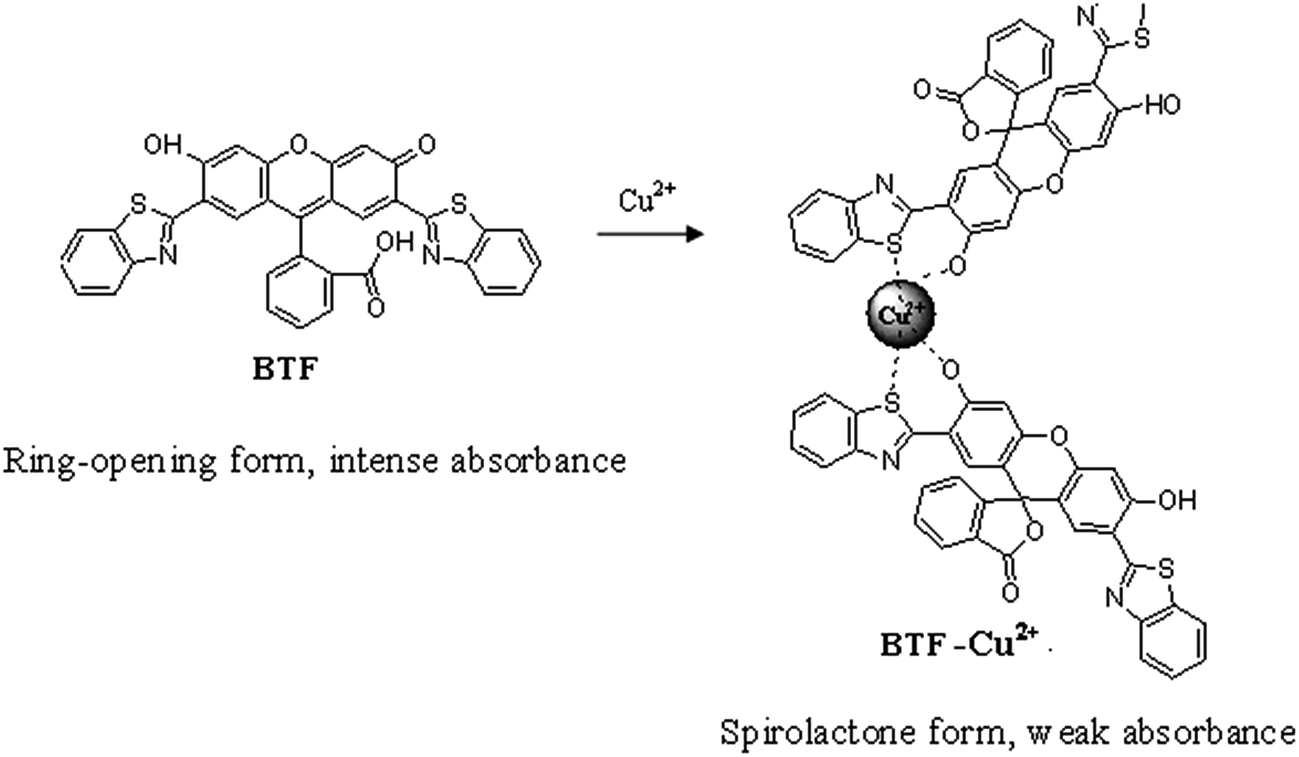
In recent years, spirocyclization of xanthene dyes have become a powerful strategy for developing chemosensing systems.29 These probes have already attracted great interest due to the valuable characteristics of xanthene derivatives, such as high molar extinction coefficient, longer excitation wavelength and a unique conformational transformation from the spirolactone (colorless and nonfluorescent) to an ring-opened structure (colorful and fluorescent) upon reaction with the analyte.30–33 Here, we synthesized 2′,7′-di(2-benzothiazolyl)-fluorescein (BTF) by using 2-(2′,4′-dihydroxyphenyl) benzothiazole (BT) and phthalic anhydride. The BTF displays absorption maximum at 559 nm. Interestingly, it was observed that upon mixing with Cu2+, the absorbance of BTF at 559 nm decreased significantly, which is apparently due to the spirocyclization of BTF induced by Cu2+ (Scheme 1). On this basis, a new spectrophotometric method for Cu2+ assay was proposed, in which BTF was used for preconcentration of Cu2+ based on cloud point extraction (CPE) of BTF–Cu2+ complex using Triton X-114. The decrease of the absorbance value at 559 nm is linear with Cu2+ concentration in the 0.03–2 μmol L−1 range. The proposed method has been applied to the determination of copper in tea and water samples.
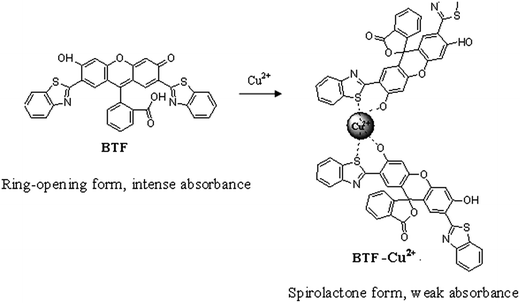 |
| | Scheme 1 The possible binding mode of Cu2+ with BTF. | |
2. Experimental
2.1 Instrumentations
A lambda ultraviolet, visible, near infrared absorption spectrometer with a quartz cell path length of 0.5 cm (model 750S, USA) was used for measurements. A shimadzu 7510 inductively coupled plasma atomic emission spectrometer (ICP-AES) was used for copper concentration determination. The ICP-AES instrumental parameters are shown in Table 1. 1H NMR spectra were recorded on an INOVA-400 spectrometer (Varian Unity). The pH values were measured by a pH-meter model pHS-204 (model 204, Switzerland). A low speed centrifuge model (model TD3, China) was used to facilitate the phase separation. High pressure seal digester (inner cup for the Teflon, Shenyang, China) was used for samples digestion.
Table 1 ICP-AES operation conditions for determination of copper
| Parameter |
|
| R.F. incident power (kW) |
1.2 |
| Plasma argon gas flow rate (L min−1) |
15 |
| Auxiliary argon gas flow rate (L min−1) |
1.2 |
| Observation height (mm) |
15 |
| Nebulizer gas flow rate (L min−1) |
0.7 |
| Analytical line wavelengths (nm) |
327.396 |
2.2 Reagents
A stock solution of BTF (0.1 mmol L−1) was prepared by dissolving 5.98 mg of BTF in 100 mL of dimethylformamide (DMF). The stock solution of Cu2+ (1.0 mmol L−1) was prepared by dissolving 24.97 mg of CuSO4·5H2O in 100 mL water. Working standard solutions were obtained daily by stepwise dilution from standard stock solution with doubly distilled water. The concentration of Cu2+ in the prepared solutions was verified by ICP-AES using GSB07-1257-2000 (Yingze-Naxin Chemical Technology Institute, Beijing, China, 1000 μg mL−1) as standard. Octylphenoxypolyethoxyethanol (Triton X-114) was obtained from Aldrich-Sigma (NJ, USA). 2-Aminothiophenol and 3-diethylaminophenol were obtained from Aladdin (Aladdin Chemical Reagent Co., Ltd, Shanghai, China). 2, 4-Dihydroxybenzaldehyde was obtained from Da Rui Fine Chemicals Co., Ltd (Shanghai, China). 2,4-Sodium metabisulfite (Na2S2O5), mesylate and hydrazine hydrate (80%) were obtained from Kelong Chemical Reagent Factory (Chengdu, Sichuan, China). Other reagents used in the experiments were of analytical grade, the water was doubly distilled water.
A standard reference material (SRM) for trace elements in tea GBW10016, labeled amount of copper: 18.6 ± 0.7 μg g−1, supplied by the ministry of geology and geophysical and geochemical research institute (Beijing, China), was used to assess the accuracy of the proposed method. Green tea was purchased from the local market (Haikou, China).
2.3 Synthesis
2.3.1 Synthesis of BT. BTF was prepared from BT. BT was synthesized according to the procedure reported previously.34 Briefly, to a solution of 2-aminothiophenol (3.12 mmol, 0.33 mL) and 2,4-dihydroxybenzaldehyde (3.16 mmol, 0.446 g) in DMF (10 mL), sodium metabisulfite (Na2S2O5, 0.610 g) was added and the reaction mixture was refluxed for 2 h. After cooling, the resulting solution was added dropwise into water (200 mL). The precipitate was filtered off, washed with water (20 mL × 3) and dried to afford the crude product, which was further purified by recrystallization from methanol to give BT as a light yellow solid (0.38 g, yield 50%). 1H NMR (400 MHz, d6-DMSO): δ 11.68 (s, 1H), 10.19 (s, 1H), 8.07 (d, 1H, J = 7.6 Hz), 7.97–7.90 (m, 2H), 7.49 (t, 1H, J = 7.4 Hz), 7.38 (t, 1H, J = 7.4 Hz), 6.46 (bs, 2H).
2.3.2 Synthesis of BTF. BTF (Scheme 1) was synthesized according to the published procedure35 with some modifications. BT (1 g, 4.00 mmol) was mixed with phthalic anhydride (0.3 g, 2 mmol) in methanesulfonic acid (14 mL). After the mixtures were heated at 150 °C for 24 h, it cooled to room temperature, and then added dropwise to ice-cold water (40 mL) with continuous stirring until the water was all used up. The precipitated product was filtered and washed with water until pH value of filtrate was in range of 5–6. The crude product was recrystallized from ethyl acetate and afforded the desired product (0.1 g, 36.7% yield). 1H NMR (DMSO-d6, 400 MHz): δ 12.07 (s, 1H), 8.08 (s, 3H), 7.84 (m, 5H), 7.66 (s, 1H), 7.43 (m, 4H), 7.01 (s, 1H), 6.50 (d, 2H, J = 17.2 Hz); 13C NMR (DMSO-d6, 100 MHz): δ 168.86, 166.96, 166.22, 164.76, 162.19, 161.71, 158.29, 158.00, 152.58, 152.50, 151.46, 151.11, 134.83, 133.30, 131.67, 131.52, 129.90, 128.66, 127.77, 126.28, 126.21, 124.76, 124.43, 124.31, 122.12, 121.80, 121.35, 117.07, 110.99, 110.11, 108.49, 103.76, 102.75; MS (ESI): calculated for MH+(C34H17N2O5S2) 597.0584; found 597.0600.
2.4 Procedures for sample preparation
Before digestion, the green tea was dried in an oven at 60 °C for 2 h. After cooling, the dried tea samples were ground to powders using an agate mortar. Then powder of tea was passed through 40 mesh sieve and put in dryer to dry again at 60 °C for 1 h.
Three samples (SRM or green tea powders) were weighed exactly (0.3 g) and transferred to Teflon reactors. After adding 10 mL of HNO3 (65%), the reactors were sealed and maintained at 160 °C for 4 h and then cooled down to room temperature. In order to remove the remaining acid, the reactor was placed on an electric hot plate to concentrate its final volume to 1.0 mL. After cooling to the room temperature, each sample was diluted to 10 mL with water. Suitable aliquots were taken and diluted with appropriate multiples for the general procedure. A blank solution subjected to the same procedure was measured in parallel with the samples.
2.5 Procedure for CPE
The CPE procedure was based on two steps. For the first stage, the complex reaction was performed in a 10 mL volumetric centrifuge tube. Typically, to a calibrated centrifuge tube containing 1 mL of phosphate buffer solution (pH 7.4) and 0.5 mL of 0.1 mmol L−1 BTF (the equivalent of 5 μmol L−1), different concentrations of Cu2+ was added and the reaction mixture was diluted to 7.0 mL with water. The reaction solution was kept at room temperature (25 °C) for 6 min. Then, 0.3 mL of 1 mol L−1 NaCl and 0.5 mL of 5% Triton-114 (w/v) were added to the mixture solutions and the solution was diluted to 10 mL with water, which then were kept in an ultrasonic bath at room temperature for 8 min. Separation of the phases was achieved by centrifugation for 10 min at 3000 rpm. Then, the aqueous phase was decanted, and the surfactant-rich phase was diluted to 2 mL with 0.6 mL methanol and a certain amount water to decrease the viscosity, thus facilitating the absorbance signal measurement. A portion of this solution was transferred into a 0.5 cm glass cell and its absorbance was measured at 559 nm. A blank solution was also prepared in the same way without the addition of Cu2+ solution.
3. Results and discussion
3.1 Complexation characteristics of BTF with Cu2+
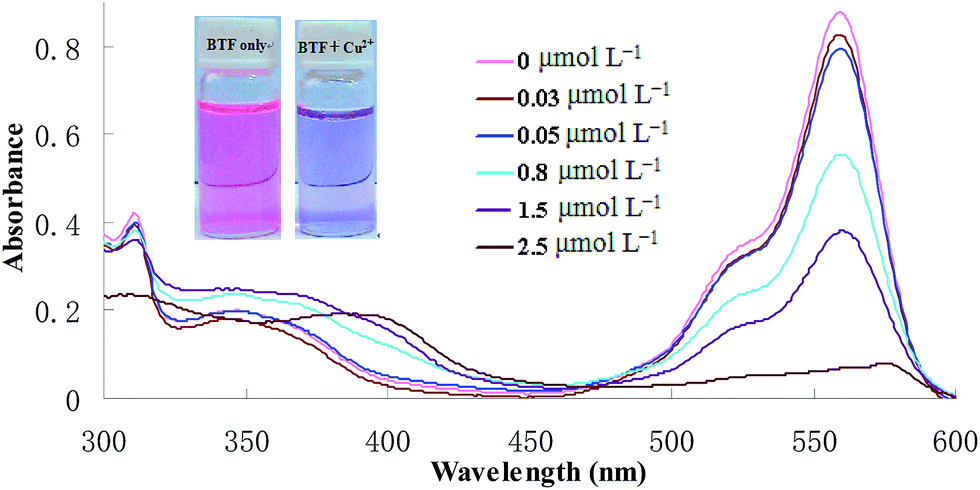
In this section, the binding interaction of BTF with Cu2+ was investigated. As can be seen from Fig. 1, BTF is a colorful, fluorescent compound showing intense absorption at 559 nm. Upon incubation with increasing concentrations of Cu2+, it can be seen that the absorbance at 559 nm was decreased gradually. When 0.5 equiv. of Cu2+ was added to BTF, the absorbance value at 559 nm is decreased significantly. However, the absorbance value at 559 nm is hardly changed when more than 0.5 equiv. of Cu2+ was added to BTF solution. The changes of absorbance at 559 nm of BTF with the addition of increasing concentrations of Cu2+ are shown in ESI as Fig. S1.† Furthermore, the pink color of the solution almost disappeared and a purple color was observed when a large mount (0.5 equiv. or more than 0.5 equiv.) of Cu2+ was added. The above results clearly indicated that spirolactone was formed upon the addition of Cu2+ to the solution of BTF, and a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry is most possible for the binding mode of BTF and Cu2+.
:1 stoichiometry is most possible for the binding mode of BTF and Cu2+.
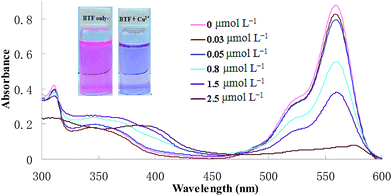 |
| | Fig. 1 Absorption spectra of BTF (5 μmol L−1) after CPE with increasing concentrations of Cu2+ (0.03–2.5 μmol L−1). | |
3.2 Spectral studies
In order to carry out the quantification analysis, the absorption spectra of BTF was measured and compared with BTF–Cu2+ (the structure is shown in Scheme 1). As can be seen from Fig. 1, the absorption maximum of BTF is located at 559 nm. Fig. 1 also depicts the changes of absorbance intensity at 559 nm of BTF with the addition of increasing concentrations of Cu2+. With an increasing amount of Cu2+, the absorbance value at 559 nm decreased accordingly. Furthermore, the decrease of the absorbance value at 559 nm is linear with Cu2+ concentration in the range from 0.03 to 2 μmol L−1; hence the absorbance of the detection system was measured at 559 nm in the following experiment.
3.3 Effect of pH
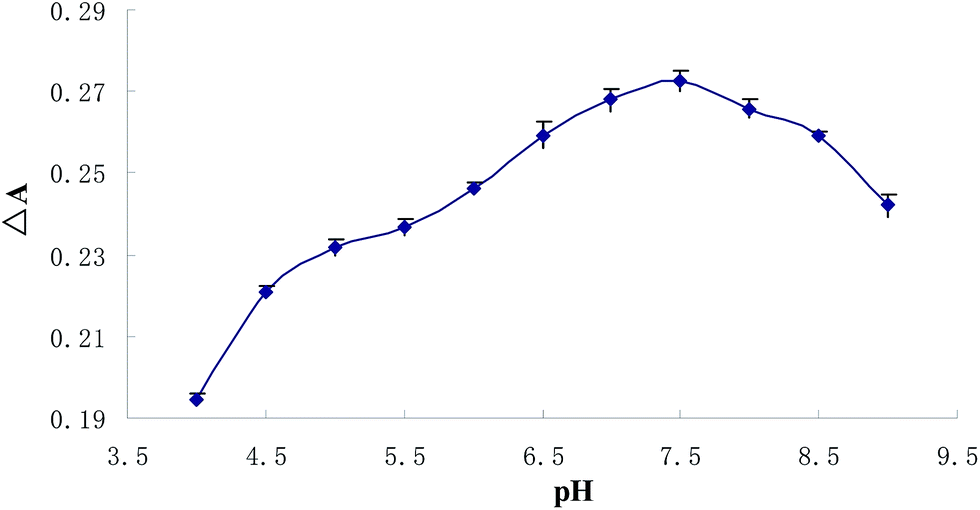
As preconcentration effectiveness of an analyte with a given CPE method may vary depending on the solution pH, finding the proper pH is one of the most crucial issues in the optimization of working conditions. For this reason, a set of Cu2+ solutions (0.5 μmol L−1) were prepared within the pH range of 4–9 and subjected to the CPE procedure. The pH values of the solutions were adjusted by using of appropriate buffer solutions. The influence of the solution pH on analytical signal was presented in Fig. 2, indicates that the quantitative extraction of complex was observed within the pH range of 7–8, therefore for further studies all samples were buffered to pH around 7.4.
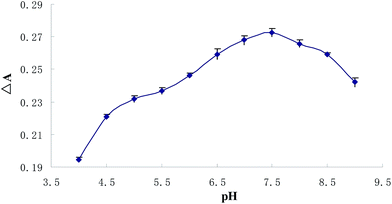 |
| | Fig. 2 pH-dependent absorbance value changes of BTF (5 μmol L−1) upon adding 0.5 μmol L−1 copper after CPE (ΔA: the decrease of the absorbance value at 559 nm between 5 μmol L−1 BTF and 5 μmol L−1 BTF upon adding 0.5 μmol L−1 copper). | |
3.4 Influence of reaction time
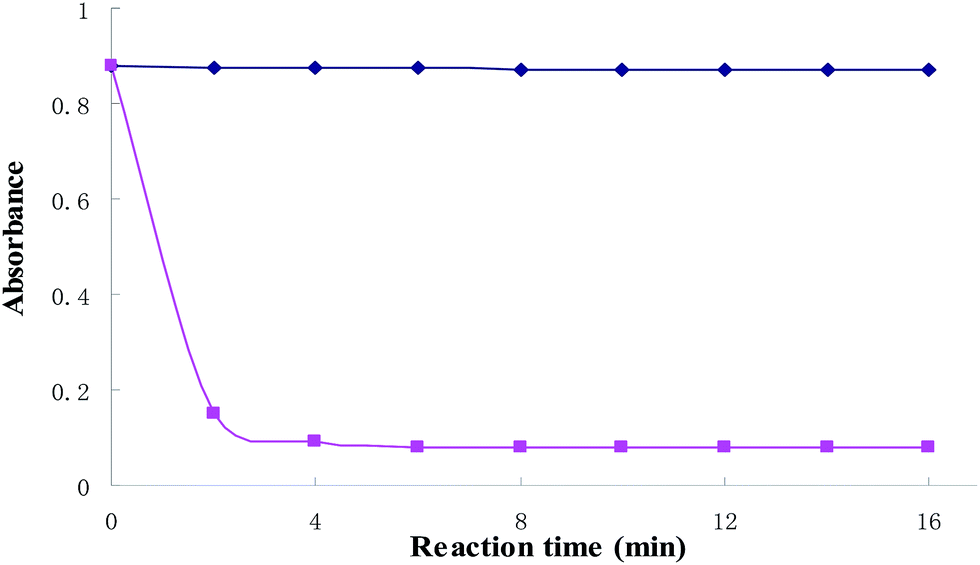
At room temperature, the kinetics of the reaction system was investigated. The results were shown in Fig. 3. The figure shows that the absorbance intensity of the blank solution is hardly changed with time. However, the absorbance is decreased in a short period of time with the Cu2+ added, the absorbance at 559 nm is decreased significantly after 6 min when BTF and 0.5 equiv. of Cu2+ was added, and thereafter remained stable. Thus, 6 min was selected for further studies.
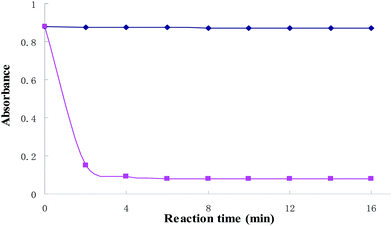 |
| | Fig. 3 Time-dependent absorbance value changes of BTF (5 μmol L−1) upon adding 2 equiv. of Cu2+. | |
3.5 Effect of NaCl concentration
Salt addition can decrease the cloud point temperature of micellar solutions36 and also increase the density of the dilute phase, making the separation of the water and surfactant-rich phase easy,37 the impact of electrolytes on this work was evaluated by adding different salts to the solutions before CPE. It can be observed that the addition of inorganic salts including MgCl2 and KCl in the range of 0–0.07 mol L−1 had no significant effect on the proposed cloud point extraction efficiency. However, an increasing of absorbance intensities was obtained from 0 to 0.03 mol L−1 NaCl, and the decrease in absorbance with increasing NaCl concentrations (0.03–0.07 mol L−1) was observed. It is possible that the molecular architecture of the surfactant and consequently the micelle formation process were changed in the presence of NaCl concentration.38 Hence, 0.03 mol L−1 NaCl was selected for further experiments.
3.6 Effect of Triton X-114 concentration
Due to its advantages such as commercial availability with high purity, low toxicity and cost, and relatively low cloud point temperature, Triton X-114 is one of the non-ionic surfactant extensively used in CPE.39 The effect of Triton X-114 concentration on the CPE efficiency of Cu2+ was studied in the range of 0.04–0.4% (w/v). The results reveal that the amounts of Triton X-114 ≤0.25% have not significant effect on copper extraction. However, a considerable decrease in absorbance is observed with increasing the surfactant amounts higher than 0.25% (w/v). This can be attributed to an increase in volume and viscosity of the micellar phase.40 Hence, a 0.25% (w/v) Triton X-114 was chosen for subsequent experiments.
3.7 Effect of ultrasound extraction time
In this work, ultrasonic bath and NaCl solution were adopted, instead of the conventional heating and incubation. The ultrasound extraction time plays an important role in CPE. It affected the extraction efficiency of the established method remarkably. In the present work, the ultrasound extraction time was investigated in the range from 0 to 12 min. It was found that the decrease of the absorbance value at 559 nm dramatically increased from 0 to 8 min. However, the decrease of the absorbance value at 559 nm remained unchanged when the ultrasound extraction time was beyond 8 min. As a result, the optimal condition of ultrasound extraction time was found to be 8 min.
3.8 Choice of solvent
The transparency of the resultant samples can affect UV-vis determination. So, proper dilution of the enrichment phase is necessary before determination. In this study, the amount and type of dilution solvent were investigated. Some conventional solvents including methanol, ethanol, acetone, and DMF were investigated. The experimental results indicated that 30% methanol in water was the best choice for the determination of copper after the CPE methods. The concentrated samples were diluted to 2 mL with 0.6 mL methanol and certain amount water before UV-vis determination.
3.9 Interferences
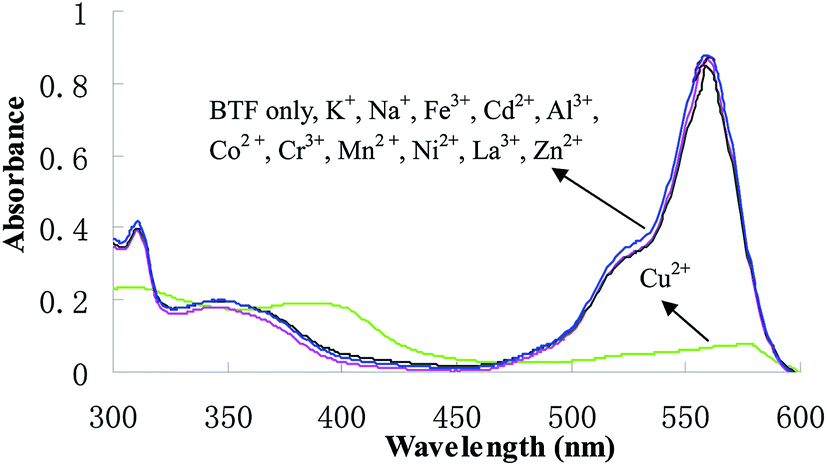
Changes in the absorption spectra of BTF caused by other cations, such as K+, Mg2+, Ca2+, Na+, Fe3+, Cd2+, Al3+, Co2+, Cr3+, Mn2+, Ni2+, La3+and Zn2+ were investigated. After the addition of 1.0 equiv. of each of these cations for 6 min, the solutions were subjected to general CPE procedure, and then the absorption spectra of BTF were measured and shown in Fig. 4. It can be observed that only Cu2+ induced dramatic changes in the absorption spectra of BTF, while other cations did not cause obvious changes under identical conditions. This result implies that the selectivity of BTF to Cu2+ over other cations is remarkably high. Furthermore, the effect of coexisting ions on copper determination were studied by analyzing a standard solution of Cu2+ (0.5 μmol L−1) to which increasing amounts of interfering species were added, using an error ≤5% as the criterion. Among the tested ions, K+, Mg2+ and Na+ did not interfere even at 1000-fold of Cu2+; Ca2+, Fe3+, Cd2+, Al3+, Co2+ and Cr3+ did not interfere at 200-fold of Cu2+; La3+, Mn2+ and Zn2+ did not show interferences at a concentration of 50-fold higher than Cu2+.
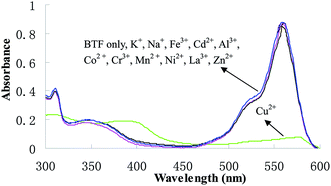 |
| | Fig. 4 Absorption spectra of BTF (5.0 μmol L−1) in the presence of 1 equiv. of various metal ions after CPE. | |
3.10 Figure of merits
The standard solutions of Cu2+ were processed according to the proposed method. The calibration curve is linear over the range of 0.03–2.0 μmol L−1 with a correlation coefficient (R) of 0.9989 (n = 9). The limit of detection was 7.48 nmol L−1 (3σ), which was calculated as the ratio of three times of the blank standard deviation over the slope of the calibration curve. The relative standard deviation was 1.1% for 0.5 μmol L−1 Cu2+ subjected to nine individual performances of CPE.
To confirm the accuracy of the proposed method, the method has been applied to determination of copper in the certified reference tea samples (GBW10016). The obtained results (Table 2) were in good agreement with the certified values using a t-test at 95% confidence limits.41 The RSD value was 1.5% and the relative recoveries of copper were in the range of 98.2% to 101.8%. Evidently, the method is reliable and feasible.
Table 2 The results for the determination of copper in tea of standard reference material after CPE (mean ± SD, n = 3)
| Amount of sample g |
Labeled μg |
Found μg |
Recovery% |
Average recovery% |
RSD% |
| 0.3105 |
5.8 ± 0.2 |
5.8 ± 0.3 |
100.0 |
99.8 |
1.5 |
| 0.3002 |
5.6 ± 0.2 |
5.5 ± 0.2 |
98.2 |
| 0.3064 |
5.7 ± 0.2 |
5.8 ± 0.2 |
101.8 |
A comparison of the proposed method with other methods42–48 for the determination of copper reported in references is given in Table 3. The detection limit of method is superior to those of CPE procedures.43,44,46 The proposed method is more sensitive than other methods, that based on atomic absorption spectrometry47,48 and spectrophotometry.42–44,46 To the best of our knowledge, this system is the first to employ BTF and CPE for the spectrophotometric detection of Cu2+ without masking agents. This fast determination, good selectivity, and inexpensive technique offer practical potential for the detection of copper in real samples.
Table 3 Comparison of the analytical figures of merit for the determination of Cu(II) among different methodsa
| Detection |
Agents |
Linear range (μg L−1) |
RSD (%) |
Detection limit (μg L−1) |
Ref. |
| RSD: relative standard deviation; SPM: spectrophotometry; RS-CPE: rapidly synergistic cloud point extraction; 4-BPDC: 4-bencylpiperidine-ditiocarbamate; BTAS: 4-(2′-benzothiazolylazo)-salicylic acid; DDTC: sodium diethyldithiocarbamate; PDBDM: 4-(phenyl diazenyl)benzene-1,3-diamine; BTF: 2′,7′-di(2-benzothiazolyl)-fluorescein. |
| SPM |
BTAS |
630–5040 |
0.6–0.7 |
7.0 |
42 |
| CPE/SPM |
Dithizone |
15–250 |
2.9–3.4 |
4.6 |
43 |
| CPE/SPM |
Isoleucine |
10–1000 |
2.8 |
5 |
44 |
| SPM |
Bromosulphonazo III |
0–1 |
0.6–4.7 |
0.7 |
45 |
| CPE/SPM |
4-BPDC |
5–200 |
2.8 |
1.6 |
46 |
| CPE/FAAS |
PDBDM |
10–260 |
1.0 |
0.6 |
47 |
| CPE/FAAS |
DDTC |
5–300 |
3.4 |
1.1 |
48 |
| CPE/SPM |
BTF |
1.9–127 |
1.1 |
0.5 |
This work |
3.11 Analytical applications
Considering the biological and environmental significance, copper in the real water samples and green tea were analyzed to demonstrate and the application of the established method. Three replicates determinations for real samples were performed and satisfactory results were obtained. The analytical results are summarized in Table 4. An ICP-AES method was used as a reference method and the results are also shown in Table 4. The obtained relative recoveries of copper calculated for the added standards were always higher than 98.0%. The result suggested the reliability and accuracy of the method for copper detection in real samples.
Table 4 Recovery studies of copper in water and green tea samples (mean ± SD, n = 3)
| Sample |
Added (μmol L−1) |
Found (μmol L−1) |
Recovery% |
| Proposed |
ICP-AES |
| |
0 |
0.464 ± 0.011 |
0.461 ± 0.008 |
— |
| River water |
0.3 |
0.759 ± 0.008 |
0.754 ± 0.013 |
98.3 |
| |
0.6 |
1.077 ± 0.009 |
1.074 ± 0.010 |
101.2 |
| |
0 |
0.071 ± 0.009 |
0.070 ± 0.09 |
— |
| Tap water |
0.3 |
0.379 ± 0.013 |
0.374 ± 0.018 |
102.7 |
| |
0.6 |
0.677 ± 0.012 |
0.680 ± 0.015 |
101.0 |
| |
0 |
0.448 ± 0.007 |
0.450 ± 0.005 |
— |
| Green tea |
0.25 |
0.694 ± 0.004 |
0.697 ± 0.009 |
98.4 |
| |
0.5 |
0.954 ± 0.006 |
0.951 ± 0.008 |
101.2 |
| |
0.75 |
1.190 ± 0.011 |
1.197 ± 0.009 |
98.9 |
4. Conclusion
In summary, an analytical method was developed for the spectrophotometric determination of copper after ultrasonic assisted cloud point extraction (US-CPE) using BTF as a complexing agent. BTF displayed a selective complexation and colorimetric change upon addition of Cu2+. Such that the spectrophotometric method can be utilized for the detection of Cu2+. Furthermore, the method, which is based on the US-CPE of the BTF–Cu2+ complex, allows the determination of copper as low as 7.48 nmol L−1. The proposed method requires inexpensive instrumentation and offers good selectivity and accuracy that can be applied to the determination of copper in a variety of samples. Triton X-114 has been used for preconcentration of copper in samples, and thus high toxic solvent extraction has been avoided. The method offers a simple, rapid and nonpolluting alternative to other preconcentration techniques.
Acknowledgements
This work was supported by the Natural Science Foundation of Hainan Province, China (20154196) and the Education Department Found of Hainan Province (No. Hjkj2013-01).
References
- T. W. Lin and S. D. Huang, Anal. Chem., 2001, 73, 4319–4325 CrossRef CAS.
- M. Govindaraju and H. S. Shekar, Int. J. Res. Pharm. Chem., 2011, 1, 320–328 CAS.
- X. Zou and Y. M. Huang, Anal. Methods, 2013, 5, 6486–6493 RSC.
- D. Citak and M. Tuzen, Food Chem. Toxicol., 2010, 48, 1399–1404 CrossRef CAS PubMed.
- T. Kamidate, A. Ishikawa and H. Watanabe, Bull. Chem. Soc. Jpn., 1992, 65, 1591–1594 CrossRef CAS.
- A. Katayama, T. Kamidate and H. Watanabe, Bull. Chem. Soc. Jpn., 1992, 65, 2501–2504 CrossRef CAS.
- Y. Yamini, M. Faraji, S. Shariati, R. Hassani and M. Ghambaria, Anal. Chim. Acta, 2008, 612, 144–151 CrossRef CAS PubMed.
- M. C. Bruzzoniti, R. M. de Carlo, C. Sarzanin, R. Maina and V. Tumiatti, Talanta, 2012, 99, 703–711 CrossRef CAS PubMed.
- R. Chaisuksant, W. Palkawong-na-ayuthaya and K. Grudpan, Talanta, 2000, 53, 579–585 CrossRef CAS.
- S. Shariati and M. Golshekan, Acta Chim. Slov., 2011, 58, 311–317 CAS.
- R. Saxena and P. L. Meena, RSC Adv., 2014, 4, 20216–20225 RSC.
- H. Shoaee, M. Roshdi, N. Khanlarzadeh and A. Beiraghi, Spectrochim. Acta, Part A, 2012, 98, 70–75 CrossRef CAS PubMed.
- S. E. Ghazy, S. E. Samra and S. M. El-Morsy, Adsorpt. Sci. Technol., 2001, 19, 175–185 CrossRef CAS.
- K. Pytlakowska, V. Kozik and M. Dabioch, Talanta, 2013, 110, 202–228 CrossRef CAS PubMed.
- L. L. Zhao, S. X. Zhong, K. M. Fang, Z. S. Qian and J. R. Chen, J. Hazard. Mater., 2012, 239, 206–212 CrossRef PubMed.
- Y. Gao, P. Wu, W. Li, Y. L. Xuan and X. D. Hou, Talanta, 2010, 81, 586–590 CrossRef CAS PubMed.
- A. B. Tabrizi, J. Hazard. Mater., 2007, 139, 260–264 CrossRef CAS PubMed.
- E. L. Silva, S. Roldan Pdos and M. F. Giné, J. Hazard. Mater., 2009, 171, 1133–1138 CrossRef CAS PubMed.
- N. Sato, M. Mori and H. Itabashi, Talanta, 2013, 117, 376–381 CrossRef CAS PubMed.
- Y. Zheng, X. Cao, J. Orbulescu, V. Konka, F. M. Andreopoulos, S. M. Pham and R. M. Leblanc, Anal. Chem., 2003, 75, 1706–1712 CrossRef CAS.
- H. J. Kim, S. Y. Park, S. Yoon and J. S. Kim, Tetrahedron, 2008, 64, 1294–1300 CrossRef CAS PubMed.
- Y. Q. Weng, F. Yue, Y. R. Zhong and B. H. Ye, Inorg. Chem., 2007, 19, 7749–7755 CrossRef PubMed.
- H. L. Mu, R. Gong, Q. Ma, Y. M. Sun and E. Q. Fu, Tetrahedron Lett., 2007, 31, 5525–5529 CrossRef PubMed.
- Y. Zheng, Q. Huo, P. Kele, F. M. Andreopoulos, S. M. Pham and R. M. Leblanc, Org. Lett., 2001, 21, 3277–3280 CrossRef PubMed.
- Q. Yang, X. S. Zhu, C. G. Yan and J. Sun, Anal. Methods, 2014, 6, 575–580 RSC.
- J. Xie, M. Menand, S. Maisonneuve and R. Métivier, J. Org. Chem., 2007, 72, 5980–5985 CrossRef CAS PubMed.
- Y. J. Mei, P. A. Bentley and W. Wang, Tetrahedron Lett., 2006, 47, 2447–2449 CrossRef CAS PubMed.
- Y. J. Zheng, K. M. Gattás-Asfura, V. Konka and R. M. Leblanc, Chem. Commun., 2002, 2350–2351 RSC.
- X. Q. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed.
- V. Dujols, F. Ford and A. W. Czarnik, J. Am. Chem. Soc., 1997, 119, 7386–7387 CrossRef CAS.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- K. M. Khan, F. Rahim, S. A. Halim, M. Taha, M. Khan, S. Perveen, Z. Haq, M. A. Mesaik and M. Choudhary, Bioorg. Med. Chem., 2011, 19, 4286–4294 CrossRef CAS PubMed.
- V. S. Patil, V. S. Padalkar, A. B. Tathe and N. Sekar, Dyes Pigm., 2013, 98, 507–517 CrossRef CAS PubMed.
- L. M. Coelho and M. A. Z. Arruda, Spectrochim. Acta, Part B, 2005, 60, 743–748 CrossRef PubMed.
- E. Pramauro, Ann. Chim., 1990, 80, 101–109 CAS.
- M. Ghaedi, A. Shokrollahi, F. Ahmadi, H. R. Rajabi and M. Soylak, J. Hazard. Mater., 2008, 150, 533–540 CrossRef CAS PubMed.
- E. K. Paleologos, D. L. Giokas and M. I. Karayannis, Trends Anal. Chem., 2005, 24, 426–436 CrossRef CAS PubMed.
- S. A. M. Fathi and M. R. Yaftian, J. Colloid Interface Sci., 2008, 334, 167–170 CrossRef PubMed.
- Wuhan University, Analytical Chemistry, Higher Education Press, Beijing, China, 5th edn, 2006, pp. 62–64 Search PubMed.
- E. Y. Hashem, M. M. Seleim and A. M. El-Zohry, J. Appl. Spectrosc., 2011, 78, 586–593 CrossRef CAS.
- L. J. Manzoori and G. Karim-Nezhad, Iran. J. Chem. Chem. Eng., 2005, 24, 47–52 Search PubMed.
- P. Liang and J. Yang, J. Food Compos. Anal., 2010, 23, 95–99 CrossRef CAS PubMed.
- L. Yuan, S. H. Huo, X. N. Ren and H. Chen, Chin. Chem. Lett., 2008, 19, 92–94 CrossRef CAS PubMed.
- F. Shemirani, M. R. Jamali and R. R. Kozani, Chem. Anal., 2007, 52, 327–336 CAS.
- A. Shokrollahi, M. Ghaedi, O. Hossaini, N. Khanjari and M. Soylak, J. Hazard. Mater., 2008, 160, 435–440 CrossRef CAS PubMed.
- G. Q. Xiang, S. P. Wen, X. M. Jiang, X. Liu and L. J. He, Iran. J. Chem. Chem. Eng., 2011, 30, 101–107 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11212e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.