
Semi-carbonized nanostructures of carbohydrate for highly efficient photocatalysts†
Yuzhi Han‡
a,
Hengchao Zhang‡b,
Yanmei Yanga,
Chuanxi Lia,
Hui Huang*a,
Yang Liu*a and
Zhenhui Kang*a
aJiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Institute of Functional Nano & Soft Materials (FUNSOM), Soochow University, 199 Ren'ai Road, Suzhou, 215123, Jiangsu, PR China. E-mail: zhkang@suda.edu.cn; yangl@suda.edu.cn; hhuang0618@suda.edu.cn
bPatent Examination Cooperation Jiangsu Center of The Patent Office, SIPO, Suzhou, China
First published on 30th June 2015
Abstract
We demonstrate that the semi-carbonized nanostructures of carbohydrate (cellulose and glucose as precursors) show high performance as photocatalysts directly for selective oxidation of cis-cyclooctene with air as an oxidant. Three dimensionally macroporous carbon spheres (3DMPCS) prepared from cellulose carbonized at 500 °C yielded the highest conversion of cis-cyclooctene (32.6%) and a high selectivity of 2-hydroxycyclooctanone (90.4%) after 24 h reaction, while cellulose carbonized at lower heat treatment temperatures (HTTs) (0–400 °C) or higher (600–900 °C) gave lower cis-cyclooctene conversion. The 3DMPCS synthesized from glucose carbonized at 600 °C shows higher performance in selective oxidation of cis-cyclooctene (12.2% conversion of cis-cyclooctene and 75.4% selectivity of 2-hydroxycyclooctanone) than glucose carbonized at other HTTs (200–500 °C and 700–900 °C) as catalysts. The results indicate that the semi-carbonized nanostructures of carbohydrates are highly efficient photocatalysts.
Introduction
Carbon materials have been used for a long time in heterogeneous catalysis, which can satisfy most of the desirable properties required for a suitable catalyst support.1,2 A considerable volume of literature has been devoted to carbon materials as catalyst supports due to the fact that carbon materials are low cost, available in huge amounts, easily accessible, have a high surface area, have a diverse porous structure, and exhibit resistance to acidic or basic environments.3–8 Only a few researchers reported that carbon materials can be used as catalysts directly. In fact, the carbon materials can be considered as efficient catalysts directly in many reactions, such as, oxidative dehydrogenation (ODH), the hydroxylation of aromatic hydrocarbons, esterification and phenol degradation,2,9–14 due to they possess a large specific surface area (most with porous structure), high resistant to abrasion and acidic/basic environments, good dimensional and thermal stability and high purity to avoid self-poisoning.10 For example, in 2001, the catalytic properties of carbon nanofilaments for the ODH of ethylbenzene to styrene were tested.9 In 2006, we demonstrated that multi-walled carbon nanotubes can be directly used as catalyst for the hydroxylation of aromatic hydrocarbons at low temperature (50–70 °C) with high selectivity without the assistance of any solvent or additive.10 In 2008, as a breakthrough progress, Su et al. reported carbon nanotubes with modified surface as a high performance catalyst for the ODH of the much less active butane and displayed an enhanced selectivity to C4 alkenes.11 Recently, carbon quantum dots, a new class of carbon nanomaterials with sizes below 10 nm, were also demonstrated to be efficient photocatalysts for selective oxidation, light-driven acid-catalysis and hydrogen bond catalysis.15–19 However, it is still indistinct how we can rationally design the process of preparation to obtain the carbonaceous materials with superior performance and broaden the sources of precursors. Solving these challenges will further facilitate and strengthen the capability for rational design of a variety of carbon materials and extended practical applications. The three dimensionally macroporous carbon spheres may be an ideal candidate for their large specific surface area with pore structure, good stability and high purity.Here, we demonstrate the semi-carbonized nanostructures of carbohydrate (cellulose and glucose as precursors) show high performance as photocatalysts directly for selective oxidation of cis-cyclooctene. Three dimensionally macroporous carbon spheres (3DMPCS) prepared from cellulose carbonized at 500 °C (C-3DMPCS-500) yielded the highest conversion of cis-cyclooctene (32.6%) and a high selectivity of 2-hydroxycyclooctanone (90.4%) after 24 h reaction, while cellulose carbonized at lower heat treatment temperatures (HTTs) (0–400 °C) or higher (600–900 °C) gave lower cis-cyclooctene conversion. For 3DMPCS prepared from glucose, similar conclusion was obtained. Glucose carbonized at 600 °C (G-3DMPCS-600) shows higher performance in selective oxidation of cis-cyclooctene (12.2% conversion of cis-cyclooctene and 75.4% selectivity of 2-hydroxycyclooctanone) than glucose carbonized at others HTTs (200–500 °C and 700–900 °C) as catalysts.
Experimental section
Materials
Tetraethyl orthosilicate, aqueous ammonia (28 wt%), hexane, silicone oil, cellulose, glucose, sulfuric acid (H2SO4), tert-butyl hydroperoxide (TBHP), hydrofluoric acid (HF) and ethanol were purchased from the Shanghai Chemical Factory (Shanghai, China) and used as received without further purification. The cis-cyclooctene was obtained from Aldrich. Deionized water (H2O) was used throughout all experiments.Synthesis for C-3DMPCS and G-3DMPCS
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The mixture was placed in drying oven to dry, and then retreated once in the solution of cellulose or glucose to ensure that there is enough cellulose or glucose to form nanostructure. The carbonization process was carried out by heating the mixture at different temperatures (100–900 °C) under nitrogen atmosphere to form the carbon–silica composite. Highly ordered three dimensionally macroporous structures were formed by removing SiO2 bead with HF. The template-free samples were filtered, washed three times with deionized water and ethanol, and finally dried at 60 °C to obtain solid-state sample. The names of samples were simplified and were shown in Table 1.
:1. The mixture was placed in drying oven to dry, and then retreated once in the solution of cellulose or glucose to ensure that there is enough cellulose or glucose to form nanostructure. The carbonization process was carried out by heating the mixture at different temperatures (100–900 °C) under nitrogen atmosphere to form the carbon–silica composite. Highly ordered three dimensionally macroporous structures were formed by removing SiO2 bead with HF. The template-free samples were filtered, washed three times with deionized water and ethanol, and finally dried at 60 °C to obtain solid-state sample. The names of samples were simplified and were shown in Table 1.
| Structure | Precursors | HTTs/°C | Sample name |
|---|---|---|---|
| Three dimensionally macroporous carbon spheres (3DMPCS) | Cellulose | 0 | C-3DMPCS-0 |
| 100 | C-3DMPCS-100 | ||
| 200 | C-3DMPCS-200 | ||
| 300 | C-3DMPCS-300 | ||
| 400 | C-3DMPCS-400 | ||
| 500 | C-3DMPCS-500 | ||
| 600 | C-3DMPCS-600 | ||
| 700 | C-3DMPCS-700 | ||
| 800 | C-3DMPCS-800 | ||
| 900 | C-3DMPCS-900 | ||
| Glucose | 200 | G-3DMPCS-200 | |
| 300 | G-3DMPCS-300 | ||
| 400 | G-3DMPCS-400 | ||
| 500 | G-3DMPCS-500 | ||
| 600 | G-3DMPCS-600 | ||
| 700 | G-3DMPCS-700 | ||
| 800 | G-3DMPCS-800 | ||
| 900 | G-3DMPCS-900 |
Catalytic selective oxidation of cis-cyclooctene
The catalytic selective oxidation of cis-cyclooctene was carried out in a 50 mL quartz three-neck flask with water condenser and continuous magnetic stirring. The as-synthesized catalysts (0.1 g), cis-cyclooctene (10 mL) and TBHP (0.12 g) were put into the flask to form solution, which was stirred with or without illumination (a 150 W Xe arc lamp with a CUT filter to cut off light of wavelength < 420 nm) for 24 h at 80 °C to evaluate the catalytic performance. The oxidation products were analyzed by gas chromatography (GC) and gas chromatography-mass spectroscopy (GC-MS).Characterization
Scanning electron microscopy (SEM) images were taken on a FEI-quanta 200 scanning electron microscope with acceleration voltage of 20 kV. The transmission electron microscopy (TEM) images were obtained with a FEI/Philips Tecnai G2 F20 transmission electron microscope operated at 200 kV with energy dispersive X-ray (EDX) analyses. The normal TEM samples were prepared by dropping the solution onto a copper grid covered with carbon film and dried in air. Raman spectra were collected on an HR 800 Raman spectroscope (J Y, France) equipped with a synapse CCD detector and a confocal Olympus microscope. The spectrograph uses 600 g mm−1 gratings and a 633 nm He–Ne laser. The photoluminescence (PL) spectra were carried out on a Horiba JobinYvon (FluoroMax 4) Luminescence Spectrometer. The samples were excited at 350 nm and the fluorescence spectra were recorded. Firstly, the fluorescence intensity τo was measured without the quencher, and then the fluorescence intensity τ was measured at different quencher concentrations at a fixed sample concentration. The fluorescence lifetimes were measured by a FluoroLog-211-TCSPC. The GC measurements were performed in a Varian 3400 GC column with a cross-linked 5% PhMe silicone column (25 m × 0.2 mm × 0.33 μm) and a FID detector under the following conditions: carrier gas (N2) at 140 K; temperature program 60 °C, 1 min, 15 °C min−1, 180 °C, 15 min; split ratio, 10:1; injector, 300 °C; detector, 300 °C.
Results and discussion
Structure and characterization
The colloidal silica beads were composed of 250 nm silica, and subsequent immersed in the aqueous solution of cellulose or glucose. After removing the silica with HF, the 3DMPCS can be achieved. An illustration of the present synthesis strategy for 3DMPCS is shown in Fig. 1. The Brunauer–Emmett–Teller (BET) surface area of colloidal silica beads was measured to be 15.62 m2 g−1 due to the tightly stacked silica colloidal nanoparticles. The N2 adsorption–desorption isotherms for colloidal silica beads in Fig. S1a† exhibit a type-II isotherm with a type H3 hysteresis loop in P/P0 range of 0.8–1.0, which indicate facile pore connectivity.21 The corresponding pore size distribution exhibits the characteristic of macroporous shown in Fig. S1b.†22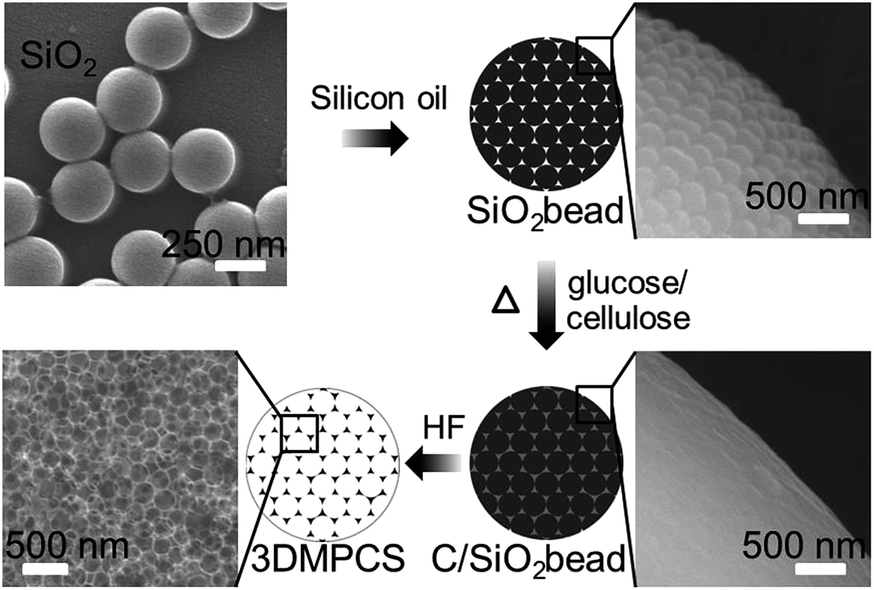 | ||
| Fig. 1 Schematic diagram of the synthesis strategy for 3DMPCS using cellulose or glucose as precursors. | ||
Cellulose was firstly used as precursor to produce a series of thermally treated samples under different temperatures. Fig. 2a shows a typical SEM image of C-3DMPCS-500 with diameter about 25 μm. The silica bead was coated with the cellulose, which surface is smoother than the surface of the obtained colloidal silica beads shown in Fig. 1. The enlarged SEM image of C-3DMPCS-500 is shown in Fig. 2b, from which we can see that the carbon frame was constituted by the accumulation of a large number of uniform and rough spherical pores (diameter about 250 nm). Many overlapping ordered pore structures (Fig. 2c) further demonstrated that the semi-carbonized cellulose spheres are three dimensionally interconnected.
 | ||
| Fig. 2 (a and d) SEM images of C-3DMPCS-500 and G-3DMPCS-600; (b and e) enlarged SEM images of C-3DMPCS-500 and G-3DMPCS-600; and (c and f) TEM images of C-3DMPCS-500 and G-3DMPCS-600. | ||
In the followed experiments, glucose was used as precursor to obtain G-3DMPCS at different HTTs (100–900 °C) with colloidal silica beads as templates. The typical SEM image of the obtained G-3DMPCS-600 with diameter approximately 25 μm is shown in Fig. 2d. Fig. 2e reveals the enlarged SEM image of the G-3DMPCS-600, which shows the carbon framework of G-3DMPCS is similar to the C-3DMPCS. They both have the spherical and close packed pores interconnected to each other with diameter about 250 nm. Fig. 2f displays a TEM image of G-3DMPCS-600, in which the macroporous structure is ordered overlapping. As shown in Fig. 2c and f, the pore size is about 250 nm in diameter and the wall thickness is about 25 nm, which indicates, during the preparation of semi-carbonized C-3DMPCS and G-3DMPCS, the carbon produced from cellulose and glucose is well dispersed into the voids between the silica nanoparticles.
Raman spectroscopy is a widely used tool which was not only able to evaluate the microstructure of carbon materials, the distribution and state of sp2 bonded carbon, but also help to understand the evolution of conductive carbon clusters as a function of HTTs. Raman spectra of C-3DMPCS are shown in Fig. 3a in the region from 1000 to 2000 cm−1, in which there are two relatively broad Raman bands in all curves at about 1356 and 1596 cm−1 corresponding to the D and G bands, respectively. As HTTs increases, the crystalline sp2 carbon atoms grow resulting in two Raman bands become sharper and increase in intensity.23–25 As shown in Fig. 3a, at higher HTTs, the Raman spectra of C-3DMPCS show a discernable band at about 1165 cm−1, which attributed to the appearance of sp3-carbon in carbonaceous materials at the same wavenumber.26 The ratio of the integrated areas of the D-band and G-band (ID/IG) are often used to provide information of the carbon microstructure,27–32 in which the D-band was used to measure the surface area of carbon atoms at the edge of graphitic planes and the G-band was used to measure the average volume of the ordered graphite crystals.33 Thus, the average crystallite thickness (La, depicted in Fig. 3b) of graphitic materials can be measured through the analysis of the ID/IG ratio.34 For heat treating C-3DMPCS, the ID/IG ratios as a function of HTTs is summarized in Fig. 3c. With HTTs increase from 100 to 800 °C, the ID/IG ratios increase due to depolymerisation, which resulted in the formation of isolated sp2 carbon atoms and the increase in overall conductivity.35 For increasing HTTs at 900 °C, the ID/IG ratio decreases (Fig. 3c), indicating the conversion of disordered to ordered sp2 carbon crystallites result in the increase of the La.35 Thus, the highly conductive clusters which were composed of crystalline sp2 carbon are gradually formed, leading to increases in electrical and thermal conductivity.36 Fig. S2† shows the Raman spectra (intensity versus Raman shift) and ID/IG ratios as a function of HTTs for glucose as precursor, from which we also can obtain the similar results.
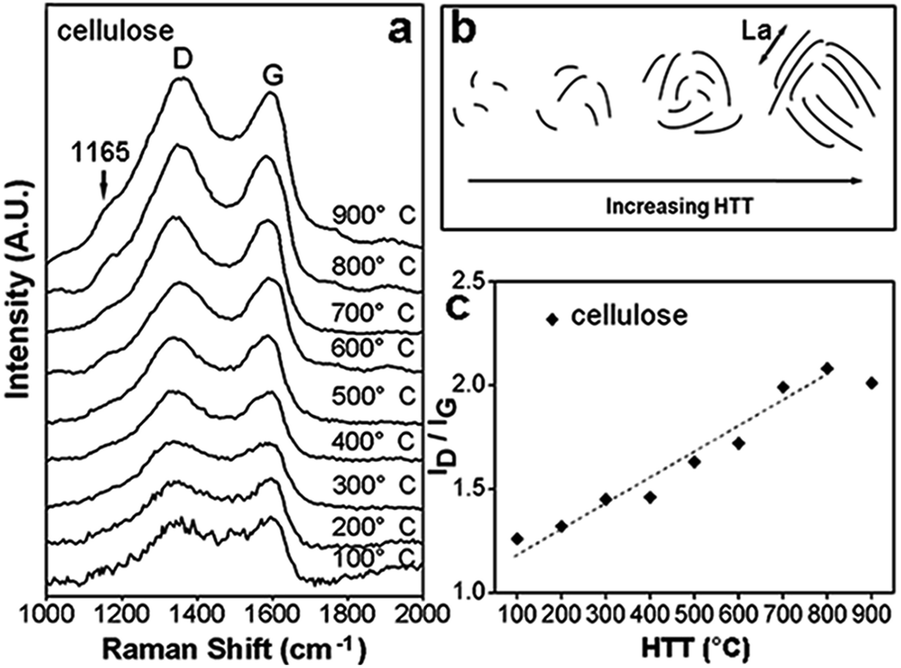 | ||
| Fig. 3 (a) Raman spectra (intensity versus Raman shift) of C-3DMPCS with cellulose as precursor under different HTTs; (b) depiction of the two-dimensional growth of carbon clusters; and (c) ID/IG ratios as a function of HTTs. | ||
Photocatalytic properties for the selective oxidation of cis-cyclooctene
The catalytic activities of this series of carbon materials prepared from cellulose (C-3DMPCS) and glucose (G-3DMPCS) directly as catalysts were investigated by photo-enhanced selective oxidation of cis-cyclooctene. In the catalytic experiments, the different HTTs C-3DMPCS were added in flask with water condenser and continuous magnetic stirring containing cis-cyclooctene under visible light (a 150 W Xenon lamp) irradiation, with TBHP as radical initiator and air as oxidant at 80 °C. The conversion of cis-cyclooctene and selectivity of oxidation products were recorded by GC-MS, and the results are shown in Fig. 4 and Table S1.† Fig. 4a shows the conversion of cis-cyclooctene with C-3DMPCS-0, C-3DMPCS-200, C-3DMPCS-500, C-3DMPCS-600 and C-3DMPCS-800 as catalysts, respectively. As shown in Fig. 4a, the highest conversion of cis-cyclooctene yields of 32.6% using C-3DMPCS-500 (red trace) as catalyst after 24 h reaction. While C-3DMPCS-200, C-3DMPCS-600 and C-3DMPCS-800 as catalysts, the conversions reach 10.1%, 15.9%, and 4.8%, respectively, which are lower than that of C-3DMPCS-500 as catalyst. More important, as shown in the black line, pure cellulose (C-3DMPCS-0) also exhibited the catalytic ability for the cyclooctene oxidation (17.8%) due to the oxygen-containing functional groups on cellulose. The others carbonized temperatures catalysts (100, 300, 400, 700, and 900 °C) also showed lower conversion than that of C-3DMPCS-500 shown in Table S1.† In addition, after 500 °C, the more carbonized temperature, the lower catalytic ability. Fig. 4b shows the selectivity of 2-hydroxycyclooctanone using C-3DMPCS-0, C-3DMPCS-200, C-3DMPCS-500, C-3DMPCS-600 and C-3DMPCS-800 as catalysts, respectively. After 24 h, the selectivity of 2-hydroxycyclooctanone reached 50.3%, 40.1%, 90.4%, 61.1% and 44.2%, respectively, from which we can see that the highest selectivity of 2-hydroxycyclooctanone up to 90.4% was achieved when C-3DMPCS-500 as catalyst. Detailed comparison of the catalytic experimental results (Fig. 4a and b) shows, under the same experimental conditions, the conversion of cis-cyclooctene and selectivity of 2-hydroxycyclooctanone catalyzed by C-3DMPCS-500 are higher than that of other temperatures C-3DMPCS as catalysts, indicating the semi-carbonized C-3DMPCS possesses excellent photocatalytic performance.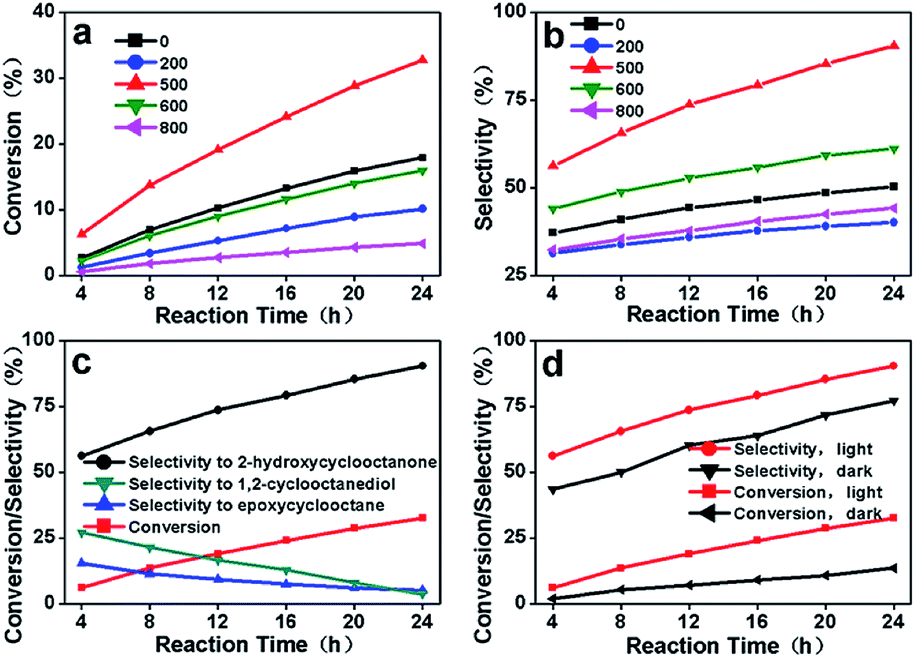 | ||
| Fig. 4 Reaction profiles of the selective oxidation of cis-cyclooctene: (a) the conversion of cis-cyclooctene and reaction time with C-3DMPCS-0, C-3DMPCS-200, C-3DMPCS-500, C-3DMPCS-600 and C-3DMPCS-800 as catalysts; (b) the selectivity of 2-hydroxycyclooctanone and reaction time with C-3DMPCS-0, C-3DMPCS-200, C-3DMPCS-500, C-3DMPCS-600 and C-3DMPCS-800 as catalysts; (c) the relationship between the conversion of cis-cyclooctene/selectivity to different products and reaction time with C-3DMPCS-500 as catalyst; and (d) the relationship between the conversion of cis-cyclooctene/selectivity of 2-hydroxycyclooctanone and reaction time with or without visible light irradiation by C-3DMPCS-500 as catalyst. | ||
The relationship between the conversion of cis-cyclooctene/selectivity to different products and reaction time with C-3DMPCS-500 as catalyst is shown in Fig. 4c, from which we can also see that a high conversion efficiency of 32.6% and a high selectivity of 2-hydroxycyclooctanone up to 90.4% were achieved simultaneously after 24 h reaction. Moreover, the selectivities to epoxycyclooctane and 1,2-cyclooctanediol fell to 5.3% and 3.8% from 15.5% and 27.1% with increasing reaction time. These results indicate that the epoxycyclooctane and 1,2-cyclooctanediol are formed initially as primary products, and 2-hydroxycyclooctanone is formed as a consequence of sequential oxidation, suggesting that the catalytic selectivity of C-3DMPCS can be tunable by careful control of experimental conditions, especially when C-3DMPCS-500 as catalyst. In the following experiments, the photo-responsive catalytic property of C-3DMPCS-500 for the selective oxidation of cis-cyclooctene was explored. Fig. 4d shows the relationship between the conversion of cis-cyclooctene/selectivity of 2-hydroxycyclooctanone and reaction time with C-3DMPCS-500 as catalysts with or without (in dark) visible light irradiation. It can be clearly observed that, under lighting conditions, the conversion of cis-cyclooctene and selectivity of 2-hydroxycyclooctanone are both higher than that in dark environment, which proves that light irradiation is necessary to obtain the high conversion efficiency and selectivity for selective oxidation of cis-cyclooctene catalyzed by C-3DMPCS composites.
In the followed experiments, different HTTs G-3DMPCS directly as catalysts were also tested for photo enhanced cis-cyclooctene selective oxidation. The photocatalytic behaviors of glucose with different carbonized degrees were shown in Fig. S3 and Table S1.† Using G-3DMPCS-300, G-3DMPCS-500, G-3DMPCS-600, G-3DMPCS-700 and G-3DMPCS-900 as catalysts, the conversions of cis-cyclooctene and the selectivities of 2-hydroxycyclooctanone are shown in Fig. S3a and b,† respectively. As shown in Fig. S3a,† after 24 h reaction, the highest conversion of cis-cyclooctene reached 12.2% catalyzed by G-3DMPCS-600. When G-3DMPCS-300, G-3DMPCS-500, G-3DMPCS-700 and G-3DMPCS-900 respectively as catalysts, the conversions yield of 4.8%, 10.1%, 8.2%, and 4.1%, respectively. The selectivity of 2-hydroxycyclooctanone reached 46.8%, 56.4%, 75.4%, 60.5%, and 41.9%, respectively (Fig. S3b†), and the highest selectivity of 2-hydroxycyclooctanone (up to 75.4%) was achieved when G-3DMPCS-600 as catalyst. Analogously, these catalytic results proved that the conversion of cis-cyclooctene and selectivity of 2-hydroxycyclooctanone catalyzed by G-3DMPCS-600 are higher than that achieved using other G-3DMPCS, suggesting that semi-carbonized G-3DMPCS has higher catalytic activity in selective oxidation of cis-cyclooctene.
The detailed catalytic data are shown in Table S1.† In general, higher conversions of cis-cyclooctene yielded with visible light irradiation rather than that of in dark, indicating the visible light can improve the catalytic activity. When cellulose was used as precursor, the highest (32.6%) and lowest (4.6%) conversions under visible light can be obtained with C-3DMPCS-500 and C-3DMPCS-900 as catalysts, respectively. In the three oxidation products included 2-hydroxycyclooctanone, epoxycyclooctane and 1,2-cyclooctanediol, the main product is 2-hydroxycyclooctanone, and the selectivity up to 90.4% when C-3DMPCS-500 was used. When glucose was used as precursor, the highest (12.2%) and lowest (2.3%) conversions in visible light were obtained with G-3DMPCS-600 and G-3DMPCS-200 as catalysts, respectively. These results suggest the semi-carbonized 3DMPCS (C-3DMPCS-500 and G-3DMPCS-600) is suitable for directly as catalyst, which possesses the higher catalytic activity than others forms.
To further compare the influence of HTTs on conversion, the conversion and selectivity of 2-hydroxycyclooctanone for selective oxidation of cis-cyclooctene as functions of HTT were investigated shown in Fig. 5 using different C-3DMPCS and G-3DMPCS as catalysts. As shown in Fig. 5a, the highest conversions of cis-cyclooctene for C-3DMPCS and G-3DMPCS can achieve with the pyrolytic temperature ≈500 °C and ≈600 °C, respectively. Particularly, with HTT increased from 0 to 200 °C, the conversion decreased when using C-3DMPCS as catalyst, which can be ascribed to the removal of large amount of oxygen-containing functional groups. Fig. 5b provided the plots of the selectivity of 2-hydroxycyclooctanone as a function of HTT. Similarly, with HTTs between 200 and 500 °C, C-3DMPCS showed increasing selectivity of 2-hydroxycyclooctanone. For HTTs above 500 °C, the selectivity decrease constantly. For G-3DMPCS, the critical value is 600 °C. Therefore, we can summarize, from 100 to 900 °C, there is an optimal condition between catalytic activity and carbonized carbohydrate heated to obtain the highly efficient catalyst. Based on our experiments, the semi-carbonized carbohydrate is the promising candidate for high performance catalyst in the selective oxidation of cis-cyclooctene. Such an optimization process will be of great value in the design and synthesis of carbon-based materials and extended practical applications.
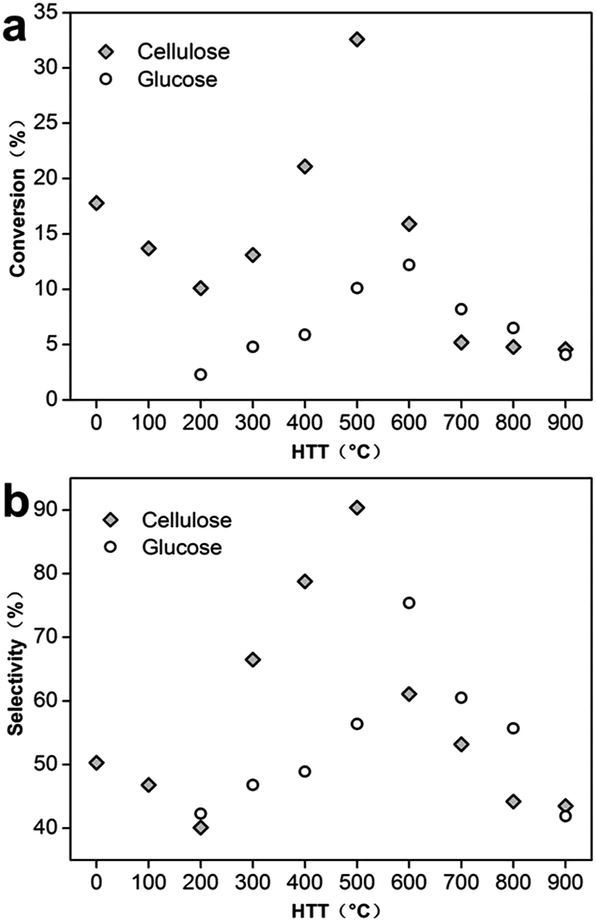 | ||
| Fig. 5 Plots of conversion (a) and selectivity of 2-hydroxycyclooctanone (b) as functions of HTT for the selective oxidation of cis-cyclooctene using different C-3DMPCS and G-3DMPCS as catalysts, respectively. | ||
In addition to photocatalytic activity, the stability of photocatalysts is another important issue for their practical applications. The repeated catalytic experiments were performed by recycling the semi-carbonized carbon material catalysts five times. Fig. 6 shows the relationship between the conversion of cis-cyclooctene and recycling times with C-3DMPCS and G-3DMPCS as catalysts over five catalytic cycles. It was found that the conversions were almost the same in all five runs. When C-3DMPCS-500 was used as catalyst, the conversions for the first and fifth cycle were 32.6% and 30.2%, respectively. For G-3DMPCS-600, the conversions for the first and fifth cycle were 12.2% and 11.1%, respectively, indicating that the semi-carbonized carbohydrate has excellent stability. All the catalytic results above indicate that the semi-carbonized carbohydrates are the promising candidates for high performance catalysts, and our simple process for preparation of semi-carbonized carbohydrate will have promising applications in the future.
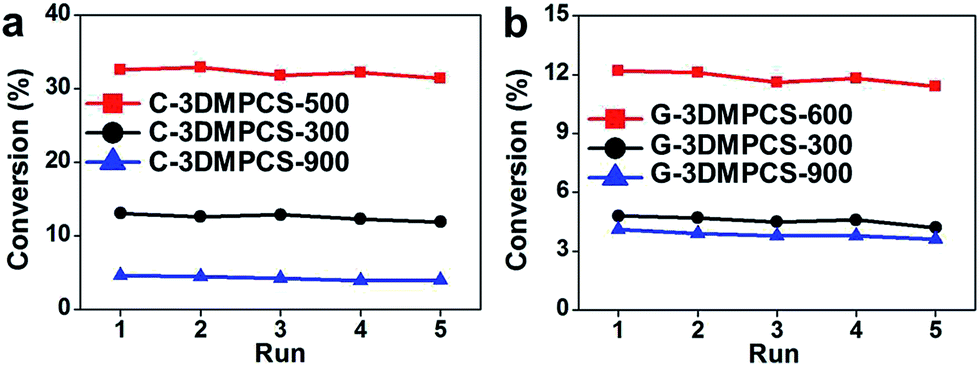 | ||
| Fig. 6 Relationship between the conversion of cis-cyclooctene and recycling times with (a) C-3DMPCS-500, C-3DMPCS-300 and C-3DMPCS-900; (b) G-3DMPCS-600, G-3DMPCS-300 and G-3DMPCS-900 as catalysts over five catalytic cycles. | ||
To further compare the catalytic performance of the obtained catalysts (C-3DMPCS-500 and G-3DMPCS-600) in photocatalytically selective oxidation of cis-cyclooctene, the active carbon and graphene (both pure carbon materials, direct purchase) as catalysts directly were employed for the comparisons. The same catalytic conditions were carried out. 0.1 g catalyst, 10 mL cis-cyclooctene and 0.12 g TBHP were put into the flask to form solution, which was stirred with illumination (a 150 W Xe arc lamp with a CUT filter to cut off light of wavelength < 420 nm) for 24 h at 80 °C. As shown in Table S2,† after 24 h, the conversion of cis-cyclooctene and the selectivity of 2-hydroxycyclooctanone with active carbon as catalyst are respectively 2.9% and 54.8%. While for graphene as catalyst, the conversion of cis-cyclooctene and the selectivity of 2-hydroxycyclooctanone are 5.3% and 62.6%, respectively. The above results are all lower than those of C-3DMPCS-500 (32.6%, 90.4%) and G-3DMPCS-600 (12.2%, 75.4%), which may attribute to the macroporous structures and high electron transfer abilities of 3DMPCS, indicating the semi-carbonized 3DMPCS is high efficient photocatalyst.
Mechanism
Why did the semi-carbonized carbohydrate show excellent catalytic performance directly used as catalyst? The detailed chemical mechanism concerned with many factors, such as the components of catalysts, particle size of catalysts, electron transfer abilities.37–39To measure the electron transfer abilities of the heat treatment samples, the time-correlated single-photon counting (TCSPC) experiments were performed to verify the photoinduced electron accepting properties of C-3DMPCS samples. The charge transfer of carbonized C-3DMPCS were tested in the presence or absence of electron donor N,N-diethylaniline (DEA, 0.88 V vs. NHE)40 and electron acceptor 2,4-dinitrotoluene (−0.9 V vs. NHE).41 C-3DMPCS-300, C-3DMPCS-500 and C-3DMPCS-900 were tested. As shown in Fig. 7, the luminescence of carbonized C-3DMPCS was quenched by DEA and 2,4-dinitrotoluene, indicating the C-3DMPCS is excellent electron donors and electron acceptors. The Stern–Volmer plots for the quenching of luminescence by DEA were shown for the carbonized C-3DMPCS-300, C-3DMPCS-500 and C-3DMPCS-900 in Fig. 7b. The quenching constants (KSV = τF kq) from linear regression were calculated to be 17.1 (500 °C), 14.3 (300 °C), and 11.4 M−1 (900 °C), respectively. The quenching constant for 2,4-dinitrotoluene was 14.4 M−1 (500 °C, Fig. 7d). Obviously, the constant for DEA (17.1 M−1) is larger than that of 2,4-dinitrotoluene (14.4 M−1), indicating that the as-synthesized carbonized C-3DMPCS-500 are highly efficient electron acceptors. Moreover, the electrons accept ability of C-3DMPCS-500 is stronger than others two samples (C-3DMPCS-300 and C-3DMPCS-900), considering the difference value of the constant for DEA and 2,4-dinitrotoluene to these three series of samples, which result in excellent catalytic performance of semi-carbonized carbohydrate.
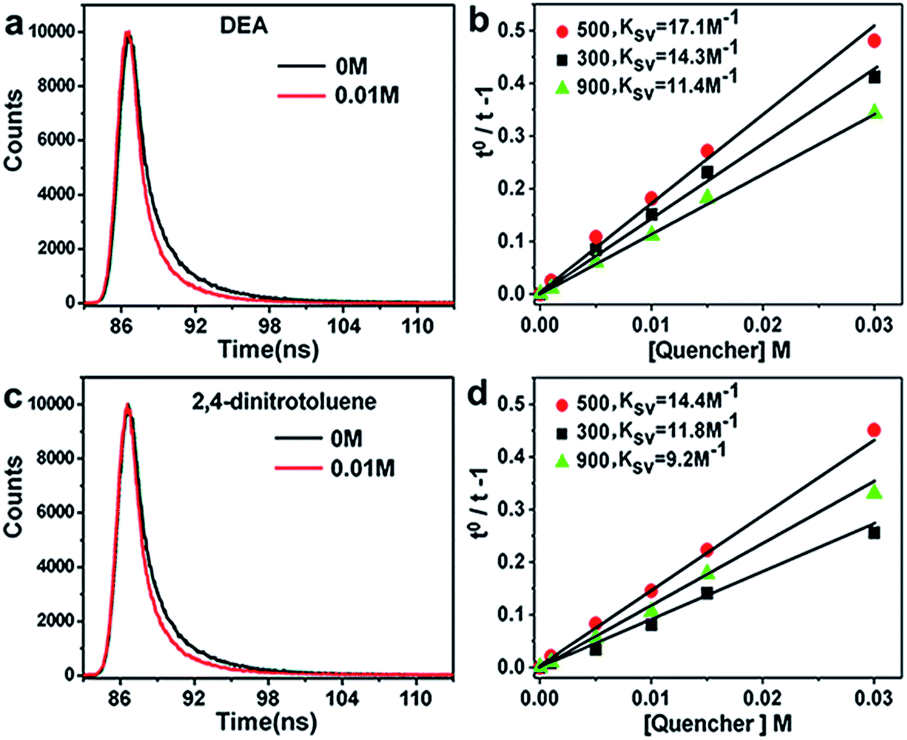 | ||
| Fig. 7 Luminescence decays (485 nm excitation, monitored with 550 nm narrow band pass filter) of the carbonized cellulose with (a) DEA and (c) 2,4-dinitrotoluene. Stern–Volmer plots for the quenching of luminescence quantum yields (485 nm excitation) of the C-3DMPCS-300, C-3DMPCS-500 and C-3DMPCS-900 by (b) DEA and (d) 2,4-dinitrotoluene. | ||
As reported by previous works,35,42–44 in carbonized carbon materials from cellulose, the overall conductivity was determined by the connectivity path of carbon conducting nano-clusters among the insulating matrix and the porosity within the matrix, the conductive carbon nano-clusters form and grow as HTTs increased. The electrical conductivity is shown to increase with increasing HTTs. For our system, the increase in conductivity has been observed for materials in previous studies.45,46 With the increase of the HTTs (100–900 °C), an increasing conductivity is caused by geometrical effects involving the connectivity of some conductive phases47,48 and may also be attributed to the changing intrinsic conductivity of the conductive carbon clusters. The catalytic efficiency of the semi-carbonized materials ultimately was improved.
Taking all these determinants into account, the possible reasons for the high catalytic performance of semi-carbonized carbohydrate are as follows: the porous structure among the carbon spheres is analogous to a kind of microreactor, which can accelerate the diffusion of the reaction reagents and further enhance the conversion efficiency and selectivity.49,50 In addition, during the reaction, the TBHP acts as a radical initiator, while the oxygen in air acts as the oxidant. The cyclooctene can diffuse evenly to both the surface as well as inside the porous spheres, which act as the microreactors. The electrons accept ability of semi-carbonized carbohydrate is stronger than the other samples. Under the combined effect of these factors, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in cyclooctene are directly oxidized by a peroxide species generated from TBHP and oxygen on the numerous active sites of the carbon spheres, indicating the high catalytic efficiency of semi-carbonized carbohydrate when used as photocatalysts.
C bonds in cyclooctene are directly oxidized by a peroxide species generated from TBHP and oxygen on the numerous active sites of the carbon spheres, indicating the high catalytic efficiency of semi-carbonized carbohydrate when used as photocatalysts.
Conclusions
We designed and fabricated a series of 3DMPCS using cellulose and glucose as precursors with HTTs in the range of 100–900 °C, as photocatalyst, which show high performance for selective oxidation of cis-cyclooctene. Three dimensionally macroporous carbon spheres prepared from cellulose carbonized at 500 °C (C-3DMPCS-500) yielded the highest conversion of cis-cyclooctene (32.6%) and a high selectivity of 2-hydroxycyclooctanone (90.4%) after 24 h reaction, while cellulose carbonized at lower HTTs (0–400 °C) or higher HTTs (600–900 °C) gave lower cis-cyclooctene conversion. For 3DMPCS prepared from glucose, similar conclusion was obtained. Glucose carbonized at 600 °C (G-3DMPCS-600) showed higher performance in selective oxidation of cis-cyclooctene (12.2% conversion of cis-cyclooctene and 75.4% selectivity of 2-hydroxycyclooctanone) than glucose carbonized at others HTTs (200–500 and 700–900 °C) as catalysts. These experimental results suggested that the semi-carbonized nanostructures of carbohydrate showed high performance as photocatalysts. The high catalytic activities of semi-carbonized nanostructures from carbohydrate were ascribed to the following factors: (i) the high electron transfer rate of semi-carbonized materials, which have enough conductive carbon nano-clusters, which are responsible for the acceleration of the electrical conductivity. (ii) The semi-carbonized materials show strong electrons accept ability. Furthermore, our simple process for preparation of semi-carbonized nanostructures of carbohydrate will have promising applications in the future. Using this method, we can better take advantage of these valuable carbon materials synthesized from biomass, which is available in high quality and huge amount, and is an environmental friendly renewable resource.Acknowledgements
This work is supported by Collaborative Innovation Center of Suzhou Nano Science and Technology, the National Basic Research Program of China (973 Program) (2012CB825803, 2013CB932702), the National Natural Science Foundation of China (51422207, 51132006, 21471106), the Specialized Research Fund for the Doctoral Program of Higher Education (20123201110018), a Suzhou Planning Project of Science and Technology (ZXG2012028), the Natural Science Foundation of Jiangsu Province of China (BK20140310), China Postdoctoral Science Foundation (2014M560445), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- F. Rodríguez-Reinoso, Porosity in Carbons: Characterization and Applications, ed. J. W. de Patrick, Edward Arnold, London, 1995, p. 253 Search PubMed.
- L. R. Radovic and F. Rodríguez-Reinoso, Chemistrv and Physics of Carbon, ed. P. A. Thrower, Marcel Dekker, New York, 1997, vol. 25, p. 243 Search PubMed.
- F. Rodríguez-Reinoso, Carbon, 1998, 36, 159–175 CrossRef.
- V. Lordi, N. Yao and J. Wei, Chem. Mater., 2001, 13, 733–737 CrossRef CAS.
- R. Z. Yang, X. P. Qiu, H. R. Zhang, J. Q. Li, W. T. Zhu, Z. X. Wang, X. J. Huang and L. Q. Chen, Carbon, 2005, 43, 11–16 CrossRef CAS PubMed.
- L. R. Radovic and C. Sudhakar, Introduction to Carbon Technologies, ed. H. Marsh, E. A. Heintz and F. Rodriguez-Reinoso, Secretariado de Publicaciones, University of Alicante, Alicante, Spain, 1997, p. 107 Search PubMed.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed.
- W. Z. Li, C. H. Liang, W. J. Zhou, J. S. Qiu, Z. H. Zhou, G. Q. Sun and Q. Xin, J. Phys. Chem. B, 2003, 107, 6292–6299 CrossRef CAS.
- G. Mestl, N. I. Maksimova, N. Keller, V. V. Roddatis and R. Schlögl, Angew. Chem., Int. Ed., 2001, 40, 2066–2068 CrossRef CAS.
- Z. H. Kang, E. B. Wang, B. D. Mao, Z. M. Su, L. Gao, L. Niu, H. Y. Shan and L. Xu, Appl. Catal., A, 2006, 299, 212–217 CrossRef CAS PubMed.
- J. Zhang, X. Liu, R. Blume, A. H. Zhang, R. Schlögl and D. S. Su, Science, 2008, 322, 73–77 CrossRef CAS PubMed.
- F. Gao, X. Y. Guo, J. Yin, D. Zhao, M. G. Li and L. Wang, RSC adv., 2011, 1, 1301–1309 RSC.
- H. Huang, J. M. Zhang, Y. X. Zhang, S. Y. Lian and Y. Liu, Solid State Sci., 2013, 24, 115–119 CrossRef CAS PubMed.
- H. Q. Sun, G. L. Zhou, Y. X. Wang, A. Suvorova and S. B. Wang, ACS Appl. Mater. Interfaces, 2014, 6(19), 16745–16754 CAS.
- Z. H. Kang, E. B. Wang, L. Gao, S. Y. Lian, M. Jiang, C. W. Hu and L. Xu, J. Am. Chem. Soc., 2003, 125, 13652–13653 CrossRef CAS PubMed.
- H. T. Li, R. H. Liu, S. Y. Lian, Y. Liu, H. Huang and Z. H. Kang, Nanoscale, 2013, 5, 3289–3297 RSC.
- H. T. Li, X. D. He, Z. H. Kang, H. Huang, Y. Liu, J. L. Liu, S. Y. Lian, C. H. A. Tsang, X. B. Yang and S. T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- Z. Ma, H. Ming, H. Huang, Y. Liu and Z. H. Kang, New J. Chem., 2012, 36, 861–864 RSC.
- Y. Z. Han, H. Huang, H. C. Zhang, Y. Liu, X. Han, R. H. Liu, H. T. Li and Z. H. Kang, ACS Catal., 2014, 4, 781–787 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- S. Kang, J. S. Yu, M. Kruk and M. Jaroniec, Chem. Commun., 2002, 1670–1671 RSC.
- H. C. Zhang, H. Huang, Y. Liu, X. Han, Z. Ma, L. L. Zhang, H. T. Li and Z. H. Kang, J. Mater. Chem., 2012, 22, 20182–20185 RSC.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401–187403 CrossRef CAS.
- T. Gruber, T. W. Zerda and M. Gerspacher, Carbon, 1994, 32, 1377–1382 CrossRef CAS.
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martínez-Alonso and J. M. D. Tascón, J. Mater. Chem., 1998, 8, 2875–2879 RSC.
- K. Ishimaru, T. Hata, P. Bronsveld, T. Nishizawa and Y. Imamura, J. Wood Sci., 2007, 53, 442–448 CrossRef CAS.
- A. Sadezkya, H. Muckenhuberb, H. Grotheb, R. Niessnera and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef PubMed.
- K. Nakamura, M. Fujitsuka and M. Kitajima, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 12260–12263 CrossRef CAS.
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martínez-Alonso and J. M. D. Tascón, Carbon, 1994, 32, 1523–1532 CrossRef CAS.
- L. Nikiel and P. W. Jagodzinski, Carbon, 1993, 31, 1313–7131 CrossRef CAS.
- S. J. Henley, J. D. Carey and S. R. P. Silva, Appl. Phys. Lett., 2004, 85, 6236–6238 CrossRef CAS PubMed.
- S. K. Sze, N. Siddique, J. J. Sloan and R. Escribano, Atmos. Environ., 2001, 35, 561–568 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS PubMed.
- Y. R. Rhim, D. J. Zhang, D. H. Fairbrother, K. A. Wepasnick, K. J. Livi, R. J. Bodnar and D. C. Nagle, Carbon, 2010, 48, 1012–1024 CrossRef CAS PubMed.
- N. Shopova, V. Minkova and K. Markova, J. Therm. Anal., 1997, 48, 309–320 CrossRef CAS.
- C. X. Zhang, J. Hu, X. K. Wang, X. D. Zhang, H. Toyoda, M. Nagatsu and Y. D. Meng, Carbon, 2012, 50, 3731–3738 CrossRef CAS PubMed.
- H. S. Liu, C. J. Song, L. Zhang, J. J. Zhang, H. J. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95–110 CrossRef CAS PubMed.
- J. S. Huang, Y. Liu and T. Y. You, Anal. Methods, 2010, 2, 202–211 RSC.
- X. Wang, L. Cao, F. S. Lu, M. J. Meziani, H. T. Li, G. Qi, B. Zhou, B. A. Harruff, F. Kermarrec and Y. P. Sun, Chem. Commun., 2009, 25, 3774–3776 RSC.
- J. M. Rehm, G. L. Mclendon and P. M. Fauchet, J. Am. Chem. Soc., 1996, 118, 4490–4491 CrossRef CAS.
- A. Celzard, J. F. Marêché, F. Payot, D. Bégin and G. Furdin, Carbon, 2000, 38, 1207–1215 CrossRef CAS.
- A. Celzard, O. Treusch, J. F. Marêché and G. Wegener, J. Mater. Sci., 2005, 40, 63–70 CrossRef CAS.
- K. M. Jäger and D. H. McQueen, Polymer, 2001, 42, 9575–9581 CrossRef.
- A. Celzard, E. Mcrae, G. Furdin and J. F. Marêché, J. Phys.: Condens. Matter, 1997, 9, 2225–2237 CrossRef CAS.
- A. Celzard, E. McRae, C. Deleuze, M. Dufort, G. Furdin and J. F. Marêché, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 6209–6214 CrossRef CAS.
- D. S. McLachlan, M. Blaszkiewicz and R. E. Newnham, J. Am. Ceram. Soc., 1990, 73, 2187–2203 CrossRef CAS PubMed.
- D. S. McLachlan and G. Sauti, J. Nanomater., 2007, 1, 1–9 CrossRef.
- X. J. Bo and L. P. Guo, Phys. Chem. Chem. Phys., 2013, 15, 2459–2465 RSC.
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detail information of samples. See DOI: 10.1039/c5ra11408j |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |