
Assessment of biochar as feedstock in a direct carbon solid oxide fuel cell
Michalis Konsolakis*a,
Nikolaos Kaklidisb,
George E. Marnellosbcd,
Dimitra Zaharakie and
Kostas Komnitsase
aSchool of Production Engineering and Management, Technical University of Crete, GR-73100 Chania, Crete, Greece. E-mail: mkonsol@science.tuc.gr; Tel: +30 28210 37682
bDepartment of Mechanical Engineering, University of Western Macedonia, GR-50100 Kozani, Greece
cChemical Process & Energy Resources Institute, Centre for Research & Technology Hellas, Thermi, Thessaloniki, GR-57001, Greece
dDepartment of Environmental Engineering, University of Western Macedonia, GR-50100 Kozani, Greece
eSchool of Mineral Resources Engineering, Technical University of Crete, GR-73100 Chania, Crete, Greece
First published on 21st August 2015
Abstract
The feasibility of employing biochar as a fuel in a direct carbon fuel cell (DCFC) or a hybrid carbon fuel cell (HCFC) is investigated in the present study, by utilizing bare biochar or biochar/carbonate mixture as feedstock, respectively. Three different types of biochars, i.e., pistachio shells (PI), pecan shells (PE) and sawdust (SD) are used as feedstock in a solid oxide fuel cell (SOFC) of a type: Biochar|Co–CeO2/YSZ/Ag|Air. All samples were characterized by means of chemical composition (ultimate/proximate analysis), mercury porosimetry, N2 adsorption–desorption isotherms (BET method), thermogravimetric analysis (TGA), Fourier Transform Infrared Spectroscopy (FTIR), X-ray Diffraction (XRD) and Scanning Electron Microscopy (SEM), to obtain a close correlation between cell performance and biochar characteristics. The electrochemical measurements reveal that the optimum performance, in terms of maximum power density (Pmax), is obtained for the PI biochar, which demonstrated a power output of 15.5 mW cm−2 at 800 °C, compared to 14 and 10 mW cm−2 for PE and SD biochars, respectively. The obtained cell performance results are interpreted on the basis of biochar physicochemical characteristics and AC impedance spectroscopy studies. The superior performance of PI biochar is attributed to a synergistic effect of several physicochemical characteristics, involving the porosity, the acidity, the volatile matter, the carbon and hydrogen content as well as the population of oxygenated surface functionalities.
Introduction
The increasing depletion of fossil energy sources in conjunction with the growing awareness on the greenhouse effect of associated CO2 emissions in the earth's climate is of very high concern nowadays. In this regard, the development of novel technologies to efficiently convert the chemical energy of carbon-based materials into electricity through an environmentally friendly way is of paramount importance for a sustainable future.1Biomass is a renewable energy resource that is abundantly widespread and can be employed at any arbitrary time to produce energy.2,3 Biomass utilization in the energy sector is associated with significantly lower CO2 emissions in relation to fossil fuels exploitation, given that its formation is part of the carbon cycle in nature.4–7 Biomass can be converted to bio-oil and biochar through well-established processes, such as pyrolysis.8–10 Bio-oil can be further converted to transportation fuels, whereas biochar can be burned to co-generate heat and power.7 However, the employment of biomass as fuel in thermal power plants is still economically infeasible due to the thermodynamically limited conversion efficiency and high cost of logistics (e.g. transportation, storage etc.).4–6
Direct Carbon Fuel cells (DCFCs) are amongst the most promising energy conversion alternatives, since they represent the only technology that can effectively exploit the chemical energy stored in solid carbonaceous materials.11–16 In contrast to conventional coal-fired plants, in DCFCs the chemical energy of carbonaceous feedstock can be directly converted to electricity through an efficient electrochemical route, bypassing the thermodynamic limitations of conventional heat cycles (e.g. Rankine, Brayton etc.).17–19 Moreover, DCFCs have several advantages, compared to conventional power plants and gas-fed solid oxide fuel cells (SOFCs), such as: (i) very high, nearly 100%, theoretical efficiency, due to the very low entropy change of carbon oxidation (ΔS0 = 2.9 J K−1 mol−1),20 (ii) abundance and low cost of raw materials (coke, biomass, municipal solid wastes, etc.), (iii) lower emissions per unit of produced energy compared to coal-fired plants.21
Three different types of DCFCs can be discriminated in relation to the electrolyte employed: molten carbonates, molten hydroxides and solid oxides.19 Among them, SOFCs is an already commercialized technology, offering the well-known advantages of oxygen anion (O2−) conducting ceramic SOFCs, such as thermal stability and fuel flexibility. However, in the case of carbon-fed SOFCs, the limited interaction between the solid fuel and the solid electrolyte/electrode interface hinders the direct electro-oxidation of carbon and hence the achieved DCFC performance. Lately, in order to overcome the restrictions imposed by the currently employed SOFC configurations, a hybrid carbon fuel cell (HCFC) concept has been proposed. It combines the solid oxide and molten carbonate fuel cell technologies, by utilizing: (i) a dense solid electrolyte (e.g. YSZ) to separate the anode and cathode compartments and (ii) a molten carbonate mixture in the anode chamber to facilitate the diffusion of solid feedstock. The enhanced electrochemical performance of carbon-fed HCFCs compared to DCFCs has been clearly demonstrated. This improvement has been mainly ascribed to the easier diffusion of solid carbon into the active electrochemical zone (AEZ) where the electro-oxidation reactions are taking place.11,14,21
The reaction scheme involved in carbon-fed SOFCs is very complex, compared to gas fueled SOFCs, involving both direct and multi-step electro-oxidation reactions.19 More specifically, the following reactions should be taken into account in the case of DCFCs:
| C + 2O2− → CO2 + 4e− | (1) |
| C + O2− → CO + 2e− | (2) |
| CO + O2− → CO2 + 2e− | (3) |
Reactions (1) and (2) correspond to the direct interaction of carbon particles with the oxygen anions (O2−)-transported from the air exposed cathode though the electrolyte membrane-towards CO2 and/or CO formation. Carbon dioxide can be also derived from carbon through a sequential two-step charge transfer process, combining reactions (2) and (3). Reactions (1) and (2) are notably hindered by the limited interactions between the solid carbon and the solid electrolyte/electrode interface. In addition, the CO2 formed by reaction (1) and (3) or directly employed as gasifying agent, can result to the formation of CO through the reverse Boudouard reaction:
| C + CO2 → 2CO | (4) |
Reaction (4) is strongly favored at temperatures higher than ∼700 °C and although non-electrochemical it has a key role in the DCFC performance, since its gaseous product, CO, notably contributes to power generation via reaction (3).12,14
In the presence of carbonates (HCFC concept), the following reactions should be further accounted for:
| C + 2CO32− → 3CO2 + 4e− | (5) |
| C + CO32− → CO + CO2 + 2e− | (6) |
| 2C + CO32− → 3CO + 2e− | (7) |
To date, various carbonaceous materials have been examined as fuels in DCFCs to assess their efficiency as energy carriers. It has been found that their physical and chemical properties notably affect their electrochemical reactivity and the lifetime of the DCFCs.18,22–24 However, in most DCFC studies the most commonly used fuel is carbon black.16 Hence, the employment of other types of readily available and cheaper carbon sources would be highly desirable. In this regard, charcoals derived from biomass or organic wastes, could be used as alternative fuels in DCFCs, since they are renewable in nature, inexpensive, easy to store, available worldwide and highly conductive.
The feasibility of employing various types of biochar materials, derived from apple, energetic willow, sunflower husks and pine, in a DCFC with a molten hydroxide electrolyte, was explored by Kacprzak et al.25,26 and power densities in the range of 18.3 to 22.4 mW cm−2 have been obtained. Munnings et al.27 assessed the viability of using both high grade (coconut char) and low grade (agricultural waste derived biochar) biomass derived fuels in a DCFC. The biochar sample demonstrated the highest OCV (1.07 V) but the lowest overall peak power density. This performance was ascribed to the in situ formed CO either from the decomposition of oxygen-containing ash components (such as Fe2O3) or from the reverse Boudouard reaction. Elleuch et al.,28,29 tested a renewable fuel derived from Tunisian almond shell biomass via carbonization in a DCFC system based on a bi-layer structure. The achieved peak power density was 30% higher than that obtained by a commercial activated carbon (AC) employing the same cell configuration and operating conditions. The achieved performance was attributed to the high concentration of oxygen-containing groups that generate CO.
Based on the above aspects the present study aims at assessing the impact of using biochar as feedstock on DCFC or HCFC performance. Three different types of biochars, i.e., pistachio shells (PI), pecan shells (PE) and sawdust (SD), were assessed as fuels under DCFC or HCFC operation. The obtained results are interpreted in terms of biochar characteristics and their impact on the achieved cell performance.
Experimental
Raw materials – pyrolysis
In the present study three raw materials were subjected to slow pyrolysis for the production of biochar. The raw materials were: (i) pistachio (Pistacia vera L.) shells (PIr) obtained from pistachio trees cultivated in Aegina island, Greece, (ii) pecan (Carya illinoinensis) shells (PEr) obtained from pecan trees cultivated in the region of Chania, Crete, Greece and (iii) sawdust (SDr) from a carpentry workshop located in Akrotiri, Chania. These raw materials were selected since they are produced in large quantities in Greece and many other countries and their valorization is considered beneficial in terms of environmental, economic and social terms. The raw materials were oven-dried (ON-O2, MEDLINE) for 24 hours prior to use in order to remove moisture. Then, pyrolysis was carried out in a modified laboratory furnace Ν-8L SELECTA at 350 °C for 60 min. The heating rate was 10 °C min−1. Nitrogen was fed in the oven for 60 min at a rate of 100 cm3 min−1 to remove air. The obtained biochars are hereinafter denoted as PI, PE and SD.Pyrolysis yield (yP), pH, volatile matter (VM) and ash content of all biochars were determined. The char fraction, consisting of partially or fully ashed inorganic material and any unconverted organic solids and carbonaceous residues, produced during the thermal decomposition of the organic components, was calculated as the difference between 100% and %VM. The fixed carbon (FC) content including the elemental carbon of the original sample and the carbonaceous residue formed after heating, was calculated as the % difference between char and ash content.
The elemental C, H, S and N analysis, as well as the analytical techniques, namely X-ray diffraction (XRD) analysis, thermogravimetric (TG) analysis, Fourier Transform Infrared Spectroscopy (FTIR) analysis, and Scanning Electron Microscopy (SEM) analysis used for all raw materials and biochars are described in detail in Komnitsas et al.30,31 Porosity was measured by mercury intrusion porosimetry using a Micromeritics AutoPore 9400 porosimeter. Surface area was determined by N2 adsorption–desorption isotherms in a NOVA Surface Area Analyzer (Quantachrome instruments).
Fuel cell fabrication
Fuel cell experiments were performed in an oxygen-anion (O2−) conducting cell (Fig. 1) comprising of an 8 mol% yttria-stabilized zirconia (YSZ) tube (15 cm long, 16 mm inside diameter, 1.2 mm thickness) as electrolyte, closed flat at the bottom end. The open end of the YSZ tube was clamped to a stainless-steel gas-tight cap, which had provisions for inlet and outlet gas lines as well as a hollow cooling ring, where water is flowing to keep the temperature low enough to protect the o-rings employed for cell gas tightness. The cathode electrode (Ag) deposited on the outside bottom wall of the YSZ tube, was prepared from silver paste (05X metallo-organic Ag resinate), calcined in static air at 850 °C for 2 h. The anodic (working) electrode was prepared from 20 wt% Co/CeO2 powder (synthesized via wet impregnation from the corresponding nitrate salts) mixed with ethylene glycol.23 This viscous suspension was deposited by painting on the inside bottom of the YSZ tube to form the electrode with a superficial surface area of 1.7 cm2 and a thickness ranging from 5–20 μm. The calcination procedure involved heating in air to 250 °C for 1 h and 850 °C for 2 h, and then cooling in a reducing atmosphere (10% v/v H2 in He). The heating and cooling rate was always retained equal to 4 °C min−1. Two Ag wires anchored on the electrodes surface were employed to establish the necessary electrical contacts. | ||
| Fig. 1 Schematic illustration of the direct biochar fuel cell reactor. Fuel mixture: 800 mg biochar (DCFC mode) or 800 mg biochar + 200 mg carbonates (HCFC mode). | ||
Fuel cell testing
Pure CO2 (99.99% purity, Air Liquide) was employed as purging gas. The inlet gas flow was controlled by a mass-flow meter (Tylan FM 360), and introduced into the reactor cell at a rate of 30 cm3 min−1 (STP conditions). In each test the cell was loaded either with bare biochar (800 mg) or a biochar/carbonate mixture (800 mg biochar/200 mg carbonates). In the latter case, the biochar sample was initially diluted in 250 mL of n-hexane and agitated in an ultrasonic device for 15 min before the addition of the carbonate mixture. Then, the resulting solution was stirred on a heating plate at 70 °C for 4 h until all the n-hexane evaporated. The experiments were performed at 700–800 °C under atmospheric pressure. The cell voltage and electrical current were monitored with digital multimeters (Uni-T UT 55), and the external resistive load was controlled by a resistance box (Time Electronics 1065). The electrochemical impedance spectra were obtained under open circuit conditions in the frequency range between 0.1 Hz and 1 MHz with an amplitude of 30 mV RMS, using the Versa Stat 4 electrochemical workstation by Princeton Applied Research and the corresponding software (Versa Studio) for data processing.Results and discussion
Materials characterization
Table 1 shows the characterization of raw pistachio shells (PIr), pecan shells (PEr), sawdust (SDr) as well as of the biochars produced after pyrolysis at 350 °C for 60 min (PI, PE and SD, respectively). The pyrolysis yield varies between 30.6 and 44.5% for the produced biochars.| PIr | PI | PEr | PE | SDr | SD | |
|---|---|---|---|---|---|---|
| yP, % | — | 31.6 | — | 44.5 | — | 30.6 |
| pH | 4.2 | 5.1 | 4.8 | 4.9 | 3.7 | 4.5 |
| VM, % | 86 | 48.1 | 71.3 | 40.9 | 89.6 | 42.9 |
| Char, % | 14 | 51.9 | 28.7 | 59.1 | 10.4 | 57.1 |
| FC, % | 12.4 | 50.2 | 27 | 57.3 | 8.8 | 55.4 |
| Ash, % | 1.6 | 1.7 | 1.7 | 1.8 | 1.6 | 1.7 |
| % C | 45.9 | 70.0 | 47.4 | 65.6 | 46.7 | 63.2 |
| % H | 6.04 | 3.3 | 5.4 | 3.0 | 5.8 | 2.1 |
| % N | 0.42 | 0.23 | 0.63 | 0.60 | 0.51 | 0.32 |
| Porosity, % | 15.2 | 27.9 | 12.8 | 24.2 | 10.4 | 21.9 |
| Surface area, m2 g−1 | — | 6.4 | — | 3.2 | — | 2.6 |
The pH of biochars PI, PE and SD, is 5.1, 4.9 and 4.5, respectively. These values are higher compared to that of the respective raw materials, indicating an increase in basicity by the pyrolysis process. The volatile matter (VM) content of the biochars is substantially decreased compared to the raw materials, while the char and fixed carbon (FC) content increase accordingly. The VM is typically high for low-temperature biochars and may be correlated with the levels of biodegradable carbon.32 The VM content of biochar PI is substantially higher (48.1%), compared to PE and SD biochars (40.9% and 42.9%, respectively). The ash content, which expresses the inorganic matter content, is low and quite similar in all samples and varies between 1.6 and 1.8%. The % C content in all biochars increases compared to the raw materials, while the hydrogen and nitrogen content decreases accordingly. No sulphur was identified in the samples. Biochar PI has the higher C content (70%), followed by PE (65.6%) and SD (63.2%). The same trend is noted for the hydrogen, H, content: PI (3.3%) > PE (3%) > SD (2.1%). In contrast, PI sample possesses the lowest FC content (50.2%) as compared to SD (55.4%) and PE (57.3%) biochars. All these parameters are expected to have a significant impact on DCFC characteristics, as discussed in the sequence.
The BET surface area of the biochars pyrolysed at 350 °C is quite low, following, however, the sequence: PI (6.4 m2 g−1) > PE (3.2 m2 g−1) > SD (2.6 m2 g−1). The same order is obtained in relation to porosity values: PI (27.9%)> PE (24.2%) > SD (21.9%). An improvement on the textural characteristics can be achieved by employing higher pyrolysis temperatures;30 for instance, after pyrolysis at 550 °C, surface area values as high as 351, 299 and 85 m2 g−1 can be obtained for PI, PE and SD, respectively. These results are in accordance with SEM analysis data, shown below.
Fig. 2 shows the XRD patterns of raw materials as well as of their corresponding biochars. Cellulose, which is one of the important structural components of the primary cell wall of green plants, is detected in all raw materials.
 | ||
| Fig. 2 XRD patterns of raw pistachio shells (PIr), pecan shells (PEr), sawdust (SDr) and the produced biochars (PI, PE and SD, respectively). | ||
However, after pyrolysis the intensity of the peaks is lower and they become broader indicating a less ordered structure. The peaks of the residual inorganic phases, such as kalicinite, calcite and whewellite, are more easily detected in biochars due to the decomposition of organic phases at 350 °C. Halite present in PI is due to the residual salt that was not fully removed from the raw pistachio shells. The XRD patterns of the biochars are characterized by the more or less elevated background between 2-theta of 15 to 30°, due to the presence of organic matter.33 More specifically, the typical peak of the carbonaceous structure at (002) can be observed at ca. 25°. However, it is evident that this peak is broader and weaker in PI and PE samples, as compared to SD sample, probably implying a less ordered structure. Given that the carbon structure in biochars can affect their reactivity and conductivity, a corresponding alteration in the DCFC performance could be expected (see below).
The mass change of raw materials and biochars, as a function of temperature, is evaluated by means of thermogravimetric analysis under nitrogen flow. Fig. 3 and 4 depict the TG (remaining weight) and DTG (weight loss rate) curves, respectively, versus temperature, for PIr, PEr and SDr raw materials as well as for the corresponding biochars. According to Fig. 3, the total calculated remaining weight for SDr, PIr and PEr is almost 10, 16 and 27% respectively, while for the produced biochars is 54.5, 57 and 54% (SD, PI and PE, respectively). DTG curves in Fig. 4 show that the major weight loss for all raw materials was initiated at around 250 °C as is also confirmed by the respective TG curves (Fig. 3).
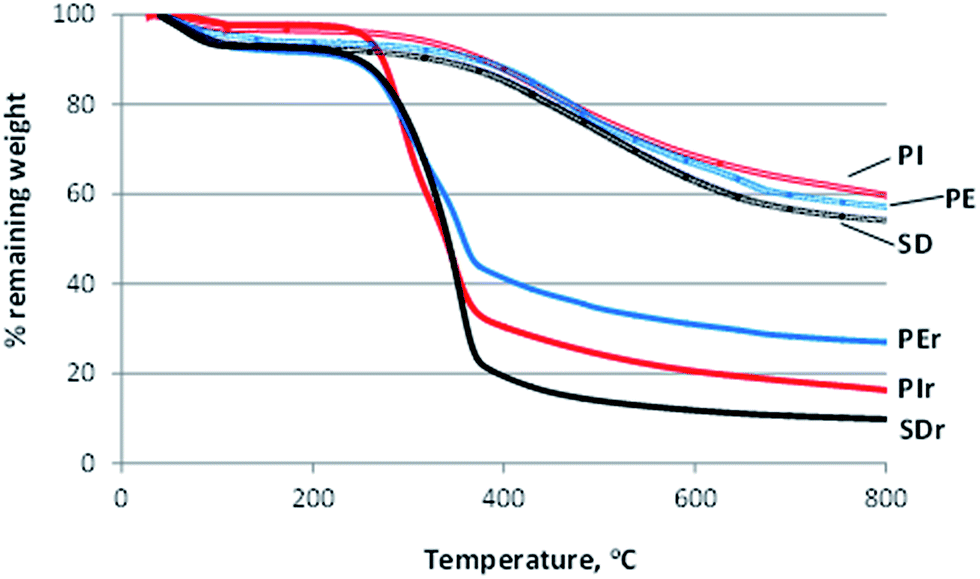 | ||
| Fig. 3 TG curves versus temperature for pistachio shells (PIr), pecan shells (PEr), sawdust (SDr) and the produced biochars (PI, PE and SD, respectively). | ||
 | ||
| Fig. 4 DTG curves versus temperature for pistachio shells (PIr), pecan shells (PEr), sawdust (SDr) and the produced biochars (PI, PE and SD, respectively). | ||
Two distinct peaks are clearly shown in the DTG curve of PIr and PEr, which are typical for pyrolysis of lignocellulose materials. The first peak at around 300 °C represents the decomposition of hemicellulose which takes place between 150 and 350 °C. The second peak at around 350 °C is assigned to the decomposition of cellulose which usually takes place in a relatively narrow temperature range of 275–350 °C. The presence of cellulose in PIr and PEr is also confirmed by the XRD analysis, as discussed earlier.
As it is also shown in Fig. 4, the thermal decomposition of biochars was initiated at around 300 °C and the major loss of weight is shown at the temperature range 300–400 °C. The gradual decomposition of lignin over a wider temperature range (usually between 275 and 500 °C) is represented by the flat section of the DTG curves of biochars.34,35
Fig. 5 shows the FTIR spectra of raw pistachio shells (PIr), pecan shells (PEr), sawdust (SDr) and the produced biochars (PI, PE and SD, respectively), while the corresponding band assignments are shown in Table 2. All spectra are characteristic of a generic oxygenated hydrocarbon due to its cellulose content, while a loss of chemical diversity is shown for biochars compared to the respective raw materials, which results in major bands attenuation.
 | ||
| Fig. 5 FTIR spectra of raw pistachio shells (PIr), pecan shells (PEr), sawdust (SDr) and the produced biochars (PI, PE and SD, respectively). | ||
| Band (cm−1) | Assignment | References |
|---|---|---|
| 3400 | Hydroxyl group (–OH) stretching | 36 |
| 2950–2850 | Aliphatic C–H deforming vibration | 37 and 38 |
| 2398 | Atmospheric carbon dioxide | 39 |
| 1740 | ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibration in carbonyl group or presence of carboxylic bonds O) vibration in carbonyl group or presence of carboxylic bonds |
40 |
| 1600 | Aromatic CO ring or CC stretching |
41 and 42 |
| 1500–1400 | C6 ring modes | 43 |
| 1310 | δ(CH) vibration in alkanes and alkyl groups |
44 |
| 1260 | CC stretching |
33 |
| 1050, 1020 | O–H deformation vibrations, b-glycosidic bonds in cellulose and hemicellulose | 45 |
| 900–600 | C–H wagging vibrations | 46 |
| 796 | C–H stretching vibrations | 37 |
| 554 | –OH out of plane bending modes | 38 |
The broad peaks, shown mainly in raw materials at around 3430 cm−1, indicate the presence of hydroxyl group (–OH) stretching. The peaks between 2950 and 2850 cm−1, also present in raw materials, are ascribed to aliphatic C–H deforming vibration. The small band at 2398 cm−1 is due to the presence of atmospheric carbon dioxide. The bands around 1740 cm−1 for all raw materials, which are slightly shifted for all biochars to 1700 cm−1, are assigned to ν(CO) vibration in carbonyl group or the presence of carboxylic bonds. The bands at around 1600 cm−1 are due to the presence of aromatic CO ring stretching (likely –COOH) or CC stretching of aromatic groups in lignin implying the presence of residual lignin after decomposition.
The characteristic peaks which appear at 1500–1400 cm−1 in raw materials and almost disappear in all biochars are attributed to C6 ring modes. The band at 1260 cm−1, which is mainly noticed in PIr and almost disappears after pyrolysis (PI biochar), is attributed to CC stretching.
The intense bands occurring at 1050 cm−1 for PIr and PEr and 1020 cm−1 for SDr are characteristic of O–H deformation vibrations or b-glycosidic bonds in cellulose and hemicellulose. These peaks disappear in all biochars indicating the decomposition of cellulose and hemicellulose. The presence of aromatic and heteroaromatic compounds in the region 900–600 cm−1 is confirmed by C–H wagging vibrations.
PE biochar shows also three small peaks: at 1310 cm−1 due to δ(CH) vibration in alkanes and alkyl groups, at 796 cm−1 which is attributed to aromatic and heteroaromatic compounds confirmed by C–H stretching vibrations and also at 554 cm−1 due to –OH out of plane bending modes. It is also of worth noticing that the band at ca. 1740 cm−1, which is emerged after pyrolysis, varies notably in intensity, following the order: PI > PE > SD. The latter is in accordance with the C content in biochars (Table 1), revealing the higher concentration of carbonyl and/or carboxylic groups in PI sample, followed by PE and SD samples. Moreover, the same trend seems to be followed in relation to –OH groups (at ca. 3400 cm−1). These findings are of major importance, since the population of oxygenated surface functionalities (e.g., carbonyl, carboxyl and hydroxyl groups) exerts a pronounced effect on DCFC performance.14
In Fig. 6, the SEM images of raw materials and their corresponding biochars are shown. The matrix of PIr is heterogeneous containing particles with varying size (27–228 μm). In PI biochar some agglomeration takes place and a porous structure is revealed while the pore diameter varies between 10 and 32 μm. The matrix of PEr is quite homogeneous and after pyrolysis a microporous structure is formed due to the release of volatiles, while the pore diameter ranges between 8 and 28 μm. The fiber like structure of SDr is slightly altered after pyrolysis at 350 °C and pores with 9–16 μm diameter are emerged as a result of the release of high-molecular-weight volatiles.
 | ||
| Fig. 6 SEM images of pistachio shells (PIr), pecan shells (PEr), sawdust (SDr) and the produced biochars (PI, PE and SD, respectively). | ||
DCFC performance
Fig. 7 shows the impact of biochar type on DCFC characteristics, in terms of cell voltage, current density and power density at 700, 750 and 800 °C under CO2 flow. The best performance is obtained by the PI sample which demonstrates a maximum power density of 3.7, 8.0 and 12.8 mW cm−2 at 700, 750 and 800 °C, respectively. Slightly lower values are recorded for PE sample, while much lower power densities are obtained with the SD sample, i.e., 1.4, 5.4 and 9.2 mW cm−2 at 700, 750 and 800 °C, respectively. Thus, the following order, in terms of achieved maximum power density, is recorded at all temperatures examined: PI > PE > SD. | ||
| Fig. 7 Effect of biochar type on DCFC performance at 700 (a), 750 (b) and 800 °C (c). Feedstock: 800 mg biochar; CO2 flow = 30 cm3 min−1. | ||
A similar DCFC performance has been obtained by employing Cu/CeO2 instead of Co/CeO2 as anode and pine charcoal as feedstock, i.e., 7 and 12 mW cm−2 at 750 and 800 °C, respectively. Contrary, lower values were achieved for conventional carbon fuels, i.e., 3.4 and 4.6 mW cm2 with the anthracite and bituminous coal, respectively, implying the potential of bio-based fuels as feedstock in DCFCs.22 In a similar manner, Kulkarni et al.47 attained a maximum power density of 40 mW cm−2 at 800 °C, by employing 2 wt% Ni-infiltrated Yttria doped Ceria (20 mol% YDC) as anodic electrode and activated coconut charcoal as fuel.
Concerning the developed Open Circuit Voltage (OCV), a clear increase in the absolute OCV values is observed with increasing temperature for all samples (Table 3). For instance, OCV absolute values of 887, 912 and 949 mV are recorded at 700, 750 and 800 °C respectively, for PI. PE sample shows slightly higher absolute open circuit voltages compared to SD and PI biochars, although being lower than the theoretical OCV (ca. 1.0 volt). However, as temperature increases the OCV values are converged for all samples (Table 3).
| Biochar | DCFC operation | HCFC operation | ||
|---|---|---|---|---|
| OCV (mV) | Pmax (mW cm−2) | OCV (mV) | Pmax (mW cm−2) | |
| 700 °C | ||||
| SD | 885 | 1.4 | 1010 | 1.9 |
| PE | 903 | 3.2 | 1007 | 3.9 |
| PI | 887 | 3.7 | 1018 | 4.5 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| 750 °C | ||||
| SD | 914 | 5.4 | 1028 | 6.0 |
| PE | 918 | 7.9 | 1023 | 9.3 |
| PI | 912 | 8.0 | 1034 | 10.5 |
|
||||
| 800 °C | ||||
| SD | 948 | 9.2 | 1052 | 9.7 |
| PE | 948 | 11.4 | 1052 | 14.1 |
| PI | 949 | 12.8 | 1051 | 15.5 |
The corresponding open circuit AC impedance spectra at 700, 750 and 800 °C, obtained at conditions identical to those employed in Fig. 7, are presented in Fig. 8. They are comprised of a small high frequency (HF) arc, overlapped with a large low frequency (LF) arc. In all cases, the size of both arcs, which indicates the electrode resistance, is notably decreased with the cell temperature. More specifically, both the ohmic (intercept of the high frequency arc with the real axis) and electrode resistances are substantially decreased in the order of PI < PE < SD as well as upon increasing cell temperature, in perfect agreement with the observed DCFC performance (Fig. 7). Specifically, the ohmic resistance decreased from 16.6 to 9.5 Ω cm2 upon increasing the cell temperature from 700 to 800 °C for SD feedstock. A further decrease of the ohmic resistance to 6.2 and 4.4 Ω cm2 at 800 °C was obtained when PE and PI were used as feedstock, respectively. A similar value to PI feedstock, equal to 3.98 Ω cm2 at 800 °C, has been reported for activated coconut charcoal when 40 mol% CO2/N2 and 2 wt% Ni/YDC were employed as carrier gas and anode material, respectively.47
 | ||
| Fig. 8 Effect of biochar type on AC impedance spectra under DCFC operation at 700 (a), 750 (b) and 800 °C (c). Feedstock: 800 mg biochar; CO2 flow = 30 cm3 min−1. | ||
The observed changes in ohmic resistance upon utilizing different feedstock at the same temperature can be assigned to their different reactivity towards the reverse Boudouard reaction. The latter results in a different amount of in situ formed CO, which in turn leads to a different reduction degree of Co/CeO2 anode composite, as verified by 4-probe conductivity measurements.23 Similarly, the electrode resistance is notably influenced by the fuel type and the operation temperature. As the cell temperature decreases from 800 to 700 °C the electrode resistances are increasing by ca. 3.5–4.0 times. Specifically, for the PI feedstock, the electrode resistance is 8.8, 3.8 and 2.2 Ω cm2 at 700, 750 and 800 °C, respectively. Moreover, the electrode resistance is strongly depended on fuel type, especially at low cell operation temperatures. The electrode resistances for PI, PE and SD feedstock at 700 °C are equal to 5.9, 8.8 and 10.5 Ω cm2 respectively, closely correlated with the achieved DCFC performance. A similar behavior is also observed at 800 °C, with, however, smaller differences between the samples.
Based on the pseudo-capacitance values of both arcs, which are falling in the order of 10−3–10−5 F cm−2 for the HF arc and 10−1–10−3 F cm−2 for the LF arc, it can be safely argued that the HF arc is attributed to charge transfer processes involving the carbon and CO electro-oxidation reactions, while the LF arc corresponds to mass transfer limitations.
Given that the cathode and electrolyte materials as well as the anodic and cathodic atmospheres are identical in all the examined cases, the corresponding alterations in the AC impedance spectra can be attributed to the biochar characteristics. Moreover, since the electrode resistance depicted by the size of the two arcs in electrochemical impedance spectra (EIS) reflects the electrode kinetics and mass transfer limitations, the observed DCFC behavior (Fig. 7) can be attributed to the feedstock induced modifications in DCFC characteristics.
In particular, the reactivity of the different biochars towards O2− anions at the three phase boundary (reactions (1) and (2)) as well as to CO2 via the reverse Boudouard reaction (4), are reflected in the AC impedance spectra and the achieved power output. Importantly, the increased reactivity of biochars towards CO2 results in a higher amount of in situ formed CO, which due to its faster electro-oxidation kinetics and better diffusion characteristics compared to solid biochar, results in lower activation and concentration overpotentials values and thus to better electrochemical performance. In the light of the above findings, the biochar reactivity seems to be favored by the increased carbon, hydrogen and volatile matter content as well as from the high porosity and acidity, while it is hindered by the increased fixed carbon and char contents (Table 1). In addition, the crystal disorder and the relative population of surface carbonyl/hydroxyl groups are also favoring the reactivity of the different biochars.
The corresponding results obtained in the hybrid carbon fuel cell (HCFC), where biochar is co-fed with an eutectic mixture of 62% mol Li2CO3-38% mol K2CO3 electrolyte at a weight ratio of 4:1, are depicted in Fig. 9. The same trend, in relation to the impact of biochar type on cell performance, is obtained under the hybrid mode of operation: PI > PE > SD. However, it should be noted that an increase in the maximum power density is achieved by utilizing molten carbonates eutectic mixtures as biochar additive at the anode compartment. In all cases, and independent on biochar type and temperature, an enhancement in power output of about 20–30% is recorded, compared to the non-hybrid DCFCs (Table 3).
 | ||
| Fig. 9 Effect of biochar type on HCFC performance at 700 (a), 750 (b) and 800 °C (c). Feedstock: 800 mg biochar + 200 mg carbonates; CO2 flow = 30 cm3 min−1. | ||
The absolute OCV values are in general increased with the operation temperature and when carbonates are infused into the biochar feedstock (Table 3). In contrast, no significant variations on OCV values are obtained upon altering the biochar type at the same temperature, especially at 800 °C, as also observed in the absence of carbonates mixture. The deviation observed from the theoretical OCV values (ca. 1.0 volt) under both DCFC and HCFC operation modes can be ascribed to the complex scheme of chemical and charge transfer reactions occurring in the anode compartment. In a similar manner, it has been well documented in the literature that the developed OCV can be significantly altered by the fuel's physicochemical characteristics, carrier gas type and operating conditions as well as by the presence of carbonates.17,18,22 All these parameters strongly affect the extent of gas phase/surface chemical reactions and charge transfer processes at the anode compartment, determining the anode gas composition and consequently the developed OCV.
The open circuit AC impedance spectra (Fig. 10) are in perfect agreement with the observed cell characteristics. The ohmic resistance is significantly decreased with the addition of carbonates in the feedstock, e.g., from 9.9, 6.2 and 4.4 Ω cm2 to 6.1, 4.3 and 3.4 Ω cm2 at 700, 750 and 800 °C, respectively, for PI biochar. The impact of carbonates in ohmic resistance is more pronounced at lower temperatures and for the less reactive biochars, i.e. PE and SD samples. In addition, a clear improvement on electrode resistance is obtained for all biochar types when the carbonates are admixed to the feedstock and as the cell temperature increases, following the same order as in the case of carbonates-free feedstock experiments, i.e. PI > PE > SD. More specifically, the electrode resistance decreases by carbonates infusion to PI feedstock from 5.9, 3.6 and 1.6 Ω cm2 to 4.3, 3.1 and 1.5 Ω cm2, at 700, 750 and 800 °C, respectively. The corresponding values in the presence of carbonates for PE (1.7 Ω cm2) and SD (2.3 Ω cm2) biochars at 800 °C are higher compared to the PI–carbonates mixture and clearly lower for bare PE (2.2 Ω cm2) and SD (2.9 Ω cm2) feedstock.
 | ||
| Fig. 10 Effect of biochar type on AC impedance spectra under HCFC operation at 700 (a), 750 (b) and 800 °C (c). Feedstock: 800 mg biochar + 200 mg carbonates; CO2 flow = 30 cm3 min−1. | ||
The aforementioned behavior concerning the observed improvements in ohmic and electrode resistances in the co-presence of carbonates mixture can be also attributed to the additional CO formation from reactions (5)–(7) taking place during HCFC operation. This in situ formed CO contributes to the further reduction of Co/CeO2 electrode thus improving its electronic conductivity.24 Moreover, it provides an indirect route to improve cell performance through its faster electrode kinetics and diffusion compared to the direct electro-oxidation and mass transport of solid biochar.22 This explanation is consistent with the AC impedance spectra (Fig. 10).
The first HF arc, attributed to charge transfer processes, has apparently disappeared, whereas the second LF arc has notably decreased, as compared to the absence of carbonates (Fig. 8), implying that in the presence of carbonates the power generation is mainly provided from the fast CO electro-oxidation reaction.
In the light of the aforementioned findings, it can be concluded that the biochar physicochemical characteristics, such as the carbon, hydrogen and volatile matter content, as well as the acidity, the presence of carbonyl/carboxylic groups, the degree of graphitic structure, the porosity and the surface area have a key role in the electrochemical performance. Interestingly, the volatile matter content, the surface area as well as the carbon and hydrogen content follow in general the same trend as the power output, i.e. PI > PE > SD, implying their vital role in cell performance. The latter is more obvious in Fig. 11, which depicts the impact of the above mentioned parameters on the power output at 800 °C. Moreover, the high concentration of carbonyl/hydroxyl groups in PI sample (Fig. 5) in conjunction to its crystal disorder (Fig. 2) can be further accounted for its superior DCFC performance, as compared to PE and SD samples. Furthermore, by improving the textural characteristics of biochars, such as the porosity and BET surface area, an enhanced DCFC performance can be obtained. The latter could be mainly attributed to the better diffusion of reactants/intermediates into the carbon structure, which as a result increases their reactivity. The beneficial effect of the above mentioned parameters has been well documented in literature.14,22
 | ||
| Fig. 11 Correlation of the DCFC power output at 800 °C with biochar physicochemical characteristics. | ||
Similar conclusions, in relation to the impact of carbon physicochemical characteristics in DCFC performance, have been previously obtained by Kaklidis et al.,22 who observed that amongst the different feedstock examined in DCFCs, i.e. anthracite coal, bituminous coal and pine charcoal, the optimum performance was demonstrated by charcoal which presented higher VM and carbon contents while the concentration of carbonyl/hydroxyl groups and the crystal disorder followed exactly the same trend as the observed DCFC performance.
The present findings clearly demonstrated the key role of CO, which is in situ formed via the reverse Boudouard reaction (reaction (4)) or through the interaction of carbon with carbonates (reactions (6) and (7)), on the power output. The different reactivity of biochar fuels towards CO2 (Fig. 7) resulted in a different amount of produced CO, which in turn affects the overall DCFC performance. In addition, when carbonates are infused into the carbon fuel, the surplus formed CO results to a higher cell performance (Fig. 9). This pronounced behavior can be attributed to the influence of CO on anode reducibility23 as well as to its faster electro-oxidation kinetics compared to solid biochar.22–24 The key role of CO on DCFC characteristics has been already demonstrated in our relevant previous studies.22–24 By combining fuel cell measurements with gas phase analysis (GC measurements) an almost linear correlation between the power output and CO formation rate was revealed.22 Moreover, AC impedance spectroscopy studies in conjunction with gas phase analysis clearly confirmed the beneficial effect of in situ produced CO on ohmic and electrode resistances, and consequently on DCFC performance.22–24
Finally, it should be admitted that the power output achieved in the present study under both DCFC and HCFC mode of operation is relatively low. This inferior performance is primarily due to the ohmic losses, derived from the high thickness (1.2 mm) of YSZ electrolyte. In addition, better interfacial resistances and improved cell performance would be achieved by optimizing the electrodes adherence on the electrolyte surface and the associated current collector configuration. Work is in progress toward this direction.
Conclusions
In the present work, three different types of biochars, namely pistachio shells (PI), pecan shells (PE) and sawdust (SD) are employed as feedstock in a solid oxide fuel cell (SOFC) of the type Biochar|Co–CeO2/YSZ/Ag|Air. The hybrid mode of operation was also explored by carbonates infusion to biochar feedstock. The optimum performance, in terms of maximum power density (Pmax), was obtained under HCFC mode of operation and for the PI biochar, which demonstrated a power output of 15.5 mW cm−2 at 800 °C, compared to 14 and 10 mW for PE and SD samples, respectively. On the basis of an extensive characterization study and AC impedance spectroscopy, a direct correlation between biochar physicochemical characteristics and power output is revealed. The carbon, hydrogen and volatile matter content, the porosity and surface area, the acidity, the presence of carbonyl/carboxylic groups as well as the carbon disorder exert a pronounced impact on the electrochemical performance of biochar-fed DCFC. The present results demonstrated the feasibility of employing worldwide available and low cost biomass derived products, such as biochars, as feedstock in direct carbon fuel cells.References
- P. Carneiro and P. Ferreira, Renewable Energy, 2012, 44, 17–22 CrossRef PubMed.
- J. V. Dam, A. P. C. Faaij, I. Lewandowski and G. Fischer, Biomass Bioenergy, 2007, 31(6), 345–366 CrossRef PubMed.
- N. Kautto and P. Peck, Renewable Energy, 2012, 46, 23–30 CrossRef PubMed.
- L. S. Esteban and J. E. Carrasco, Biomass Bioenergy, 2011, 35, S21–S30 CrossRef PubMed.
- J. Heinimö and M. Junginger, Biomass Bioenergy, 2009, 33(9), 1310–1320 CrossRef PubMed.
- J. G. Rogers and J. G. Brammer, Biomass Bioenergy, 2009, 33(10), 1367–1375 CrossRef PubMed.
- J. Yu, Y. Zhao and Y. Li, J. Power Sources, 2014, 270, 312–317 CrossRef CAS PubMed.
- A. V. Bridgwater and G. V. C. Peacocke, Renewable Sustainable Energy Rev., 2000, 4(1), 1–73 CrossRef CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18(2), 590–598 CrossRef CAS.
- D. Mohan, C. U. Pittman Jr and P. H. Steele, Energy Fuels, 2006, 20(3), 848–889 CrossRef CAS.
- D. X. Cao, Y. Sun and G. L. Wang, J. Power Sources, 2007, 167(2), 250–257 CrossRef CAS PubMed.
- N. J. Cherepy, R. Krueger, K. J. Fiet, A. F. Jankowski and J. F. Cooper, J. Electrochem. Soc., 2005, 152, A80–A87 CrossRef CAS PubMed.
- A. C. Chien and S. S. C. Chuang, J. Power Sources, 2011, 196(10), 4719–4723 CrossRef CAS PubMed.
- T. M. Gür, Chem. Rev., 2013, 113(8), 6179–6206 CrossRef PubMed.
- Y. Nabae, K. D. Pointon and J. T. S. Irvine, J. Electrochem. Soc., 2009, 156(6), B716–B720 CrossRef CAS PubMed.
- A. C. Rady, S. Giddey, S. P. S. Badwal, B. P. Ladewing and S. Bhattacharya, Energy Fuels, 2012, 26(3), 1471–1488 CrossRef CAS.
- C. Jiang and J. T. S. Irvine, J. Power Sources, 2011, 196(17), 7318–7322 CrossRef CAS PubMed.
- Y. Nabae, K. D. Pointon and J. T. S. Irvine, Energy Environ. Sci., 2008, 1, 148–155 CAS.
- S. Zecevic, E. M. Patton and P. Parhami, Chem. Eng. Commun., 2005, 192(10–12), 1655–1670 CrossRef CAS PubMed.
- Recent Trends in Fuel Cell Science and Technology, ed. S. Basu, Anamaya Publishers, New Delhi, India, 2007 Search PubMed.
- S. Giddey, S. P. S. Badwal, A. Kulkarni and C. Munnings, Prog. Energy Combust. Sci., 2012, 38(3), 360–399 CrossRef CAS PubMed.
- N. Kaklidis, V. Kyriakou, I. Garagounis, A. Arenillas, J. A. Menéndezd, G. E. Marnellos and M. Konsolakis, RCS Adv., 2014, 4, 18792–18800 CAS.
- N. Kaklidis, I. Garagounis, V. Kyriakou, V. Besikiotis, A. Arenillas, J. A. Menéndez, G. E. Marnellos and M. Konsolakis, Int. J. Hydrogen Energy, 2015 DOI:10.1016/j.ijhydene.2015.02.007.
- M. Konsolakis, G. E. Marnellos, A. Al-Musa, N. Kaklidis, I. Garagounis and V. Kyriakou, Chin. J. Catal., 2015, 36, 509–516 CrossRef CAS.
- A. Kacprzak, R. Kobylecki and Z. Bis, J. Power Sources, 2013, 239, 409–414 CrossRef CAS PubMed.
- A. Kacprzak, R. Kobylecki, R. Włodarczyk and Z. Bis, J. Power Sources, 2014, 255, 179–186 CrossRef CAS PubMed.
- C. Munnings, A. Kulkarni, S. Giddey and S. P. S. Badwal, Int. J. Hydrogen Energy, 2014, 39(23), 12377–12385 CrossRef CAS PubMed.
- A. Elleuch, A. Boussetta, J. Yu, K. Halouani and Y. Li, Int. J. Hydrogen Energy, 2013, 38(36), 16590–16604 CrossRef CAS PubMed.
- A. Elleuch, A. Boussetta, K. Halouani and Y. Li, Int. J. Hydrogen Energy, 2013, 38(36), 16605–16614 CrossRef CAS PubMed.
- K. Komnitsas, D. Zaharaki, G. Bartzas, G. Kaliakatsou and A. Kritikaki, Desalin. Water Treat., 2014, 1–10, DOI:10.1080/19443994.2014.981227.
- K. Komnitsas, D. Zaharaki, I. Pyliotis, D. Vamvuka and G. Bartzas, Waste Biomass Valorization, 2015 DOI:10.1007/s12649-015-9364-5.
- D. A. Laird, R. C. Brown, J. E. Amonette and J. Lehmann, Biofuels, Bioprod. Biorefin., 2009, 3(5), 547–562 CrossRef CAS PubMed.
- X. Cao and W. Harris, Bioresour. Technol., 2010, 101(14), 5222–5228 CrossRef CAS PubMed.
- D. Vamvuka and S. Sfakiotakis, Renewable Energy, 2011, 36(9), 2433–2439 CrossRef CAS PubMed.
- C. Ververis, K. Georghiou, N. Christodoulakis, P. Santas and R. Santas, Ind. Crops Prod., 2004, 19(3), 245–254 CrossRef CAS PubMed.
- Z. Liu, A. Quek, S. K. Hoekman and R. Balasubramanian, Fuel, 2013, 103, 943–949 CrossRef CAS PubMed.
- D. Angin and S. Şensöz, Int. J. Phytorem., 2014, 16(7–8), 684–693 CrossRef PubMed.
- W. A. W. A. K. Ghani, A. Mohd, G. Da Silva, R. T. Bachmann, Y. H. Taufiq-Yap, U. Rashid and A. H. Al-Muhtaseb, Ind. Crops Prod., 2013, 44, 18–24 CrossRef PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons Ltd, England, 3rd edn, 2001 Search PubMed.
- R. K. Sharma, J. B. Wooten, V. L. Baliga, X. Lin, W. Geoffrey Chan and M. R. Hajaligol, Fuel, 2004, 83(11–12), 1469–1482 CrossRef CAS PubMed.
- B. Chen, Z. Chen and S. Lv, Bioresour. Technol., 2011, 102(2), 716–723 CrossRef CAS PubMed.
- M. Gray, M. G. Johnson, M. I. Dragila and M. Kleber, Biomass Bioenergy, 2014, 61, 196–205 CrossRef CAS PubMed.
- X. Cao, L. Zhong, X. Peng, S. Sun, S. Li, S. Liu and R. Sun, Bioresour. Technol., 2014, 155, 21–27 CrossRef CAS PubMed.
- A. Caglar and A. Demirbas, Energy Convers. Manage., 2000, 41(16), 1749–1756 CrossRef CAS.
- T. Mimmo, P. Panzacchi, M. Baratieria, C. A. Davies and G. Tonon, Biomass Bioenergy, 2014, 62, 149–157 CrossRef CAS PubMed.
- M. K. Hossain, V. Strezov, K. Y. Chan, A. Ziolkowski and P. F. Nelson, J. Environ. Manage., 2011, 92(1), 223–228 CrossRef CAS PubMed.
- A. Kulkarni, S. Giddey and S. P. S. Badwal, J. Solid State Electrochem., 2015, 19, 325–335 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |