
Copper catalyzed direct tert-butyl sulfonylation of alkynes with t-butylsulfinamide leading to (E)-vinyl sulfones†
Zhidong Liua,
Xiaolan Chen*a,
Jianyu Chena,
Lingbo Qu*ab,
Yingya Xiaa,
Haitao Wua,
Huili Maa,
Shaohua Zhua and
Yufen Zhaoc
aCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, 450052, China. E-mail: chenxl@zzu.edu.cn; Fax: +86 371 67767051; Tel: +86 0371 67767051
bChemistry and Chemical Engineering School, Henan University of Technology, Zhengzhou, 450001, China
cDepartment of Chemistry, Xiamen University, Xiamen, 361005, China. E-mail: yfzhao@xmu.edu.cn
First published on 6th August 2015
Abstract
The first copper-catalyzed tert-butyl sulfonylation reaction of alkynes with t-butylsulfinamide for the construction of (E)-vinyl sulfones has been developed. The cleavage of the N–S bond of t-butylsulfinamide, which was catalyzed by CuSO4·5H2O, is the key step for this transformation.
Vinyl sulfones have attracted considerable attention for applications in organic synthesis and medicinal chemistry.1 Very recently, some investigations have also shown that vinyl sulfone derivatives are potent antiparasitic drugs2 and neuroprotective agents.3 To date, various methodologies have been developed to synthesize vinyl sulfones. Traditional methodologies generally involve (1) the Knoevenagel condensation of aldehydes with sulfonylacetic acids (Scheme 1-a),4 (2) wittig reactions,5 and (3) elimination from α- or β-substituted sulfones.6 However, these methods suffer from some limitations such as tedious procedures, poor selectivity or inaccessible raw materials. In recent years, a number of studies targeting the synthesis of vinyl sulfones have been accomplished such as the direct oxidation of the corresponding vinyl sulfides with oxidants (Scheme 1-b);7 the cross-coupling reactions of sulfinate salts with alkynes,8 alkenes9 (Scheme 1-c)9a and their derivative vinyl halides10 and arylboronic acids;11 and the addition of sodium sulfinates or 1,2-bis-(phenylsulfonyl)ethane to alkynes.12 Only recently, the palladium-catalyzed or iodine-catalyzed sulfonylation of alkenes with sulfonyl hydrazides, gold-catalyzed sulfonylation of alkynes with sulfinic acids and copper-catalyzed aerobic decarboxylative sulfonylation of cinnamic acids with sodium sulfinates were developed by Jiang,13 Lei,14 Shi,15 and Guo,16 respectively. In addition, a novel sulfone source, DMSO, has also been discovered for the synthesis of vinyl sulfones. For example, Loh's group described the alternative Cu(I)-catalyzed methyl sulfonylation of alkynes using DMSO as a methyl sulfone source (Scheme 1-d).17 Yuan's group published the iodide-induced sulfonylation of alkenes using DMSO as a methyl sulfone source.18 Our group has also developed the CuSO4·5H2O–H-phosphonate catalyzed synthesis of vinyl alkylsulfones from alkynes and the widely available solvent, DMSO.19 Despite the remarkable improvement in vinyl sulfone synthesis, the discovery of diverse sulfone sources is still highly desired. Herein, we disclose an efficient method for the synthesis of (E)-vinyl sulfones from alkynes and t-butylsulfinamide using a CuSO4-phosphorous acid catalytic system to catalyze the formation of the vinyl–S bond. To the best of our knowledge, this is the first example of the use of t-butylsulfinamide as a sulfur source to prepare vinyl sulfones.
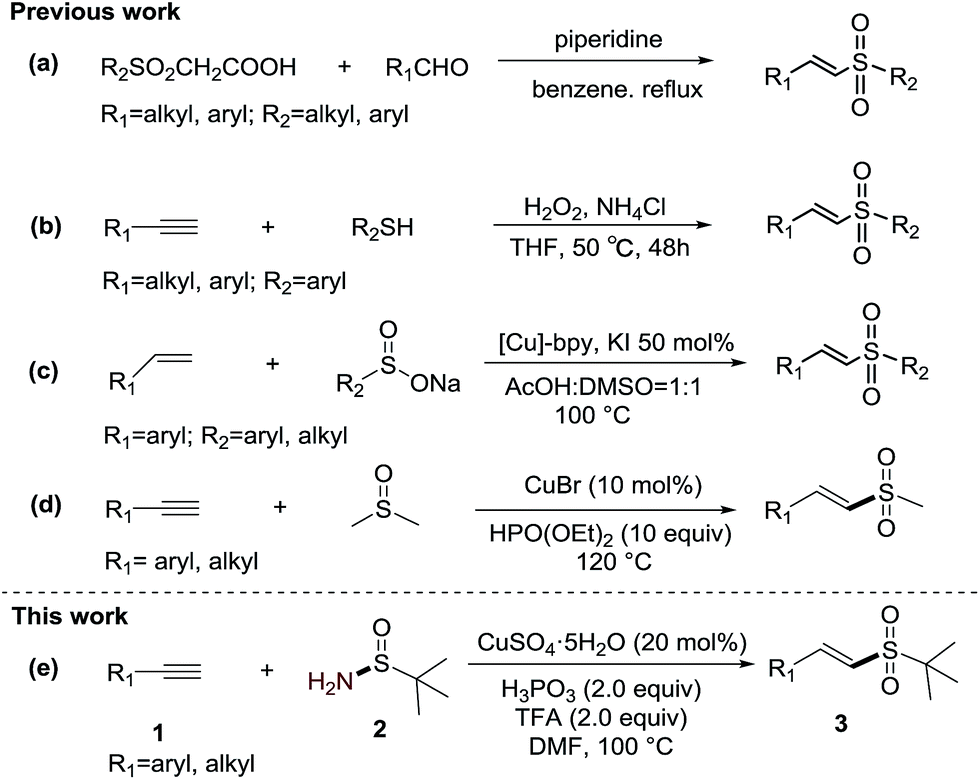 | ||
| Scheme 1 Comparison of previous work with this work. | ||
We initiated this study by establishing optimal experimental conditions using the model reaction of phenylacetylene (1a) with t-butylsulfinamide 2 in the presence of CuSO4·5H2O, phosphorous acid and trifluoroacetic acid (TFA) in DMF under open-air conditions for 12 h at 100 °C, as summarized in Table 1. Initially, the reaction of phenylacetylene (1a) with t-butylsulfinamide (2) was performed in DMF to examine the catalytic activities of several relatively inexpensive transition-metal salts, including Cu, Ag, Pd, Ni and Fe salts (Table 1, entries 1–8). Among the above-mentioned metal salts, copper salts, especially CuSO4·5H2O, turned out to be the most effective catalyst, which afforded a yield of 65%. (Table 1, entry 1). Metal salts, such as AgNO3, Pd(OAc)2, NiCl2·6H2O and FeCl3·6H2O, failed to deliver the product, 3a (Table 1, entries 5–8). The model reaction did not proceed in the absence of a metal, H3PO3 or TFA (Table 1, entries 9–11). The ideal amounts of CuSO4·5H2O, H3PO3 and TFA were also explored (Table 1, entries 1 and 12, 13, 16–19). The amounts shown in entry 12 afforded the highest yield. Besides TFA, other acids, such as HNO3 and H2SO4, were also examined. The result showed that these acids failed to produce the product, 3a (Table 1, entries 14–15). Further screening of solvents showed that DMF was the best choice among the tested solvents. The reaction in toluene and 1,4-dioxane failed to generate the product, 3a (Table 1, entries 21–22), while the reaction conducted in DMSO obtained a yield of 32% 3a and 43% vinyl methyl sulfones (Table 1, entry 20). Therefore, the optimal reaction conditions are 2.0 equiv. H3PO3, 20 mol% CuSO4·5H2O and 2.0 equiv. TFA in DMF at 100 °C for 12 h.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat (mol%) | H3PO3 (equiv.) | Acid (equiv.) | Solvent | T (°C) | Yieldb (%) (E) |
| a Reaction conditions: 1a (0.5 mmol), 2 (0.6 mmol), catalyst (0–30 mol%) H3PO3 (0–3.0 equiv.) and acid (0–3.0 equiv.) at 100 °C for 12 h.b Isolated yields. | ||||||
| 1 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(1) | DMF | 100 | 65 |
| 2 | Cu(OAc)2·H2O(20) | H3PO3(2.0) | TFA(1) | DMF | 100 | 48 |
| 3 | Cul(20) | H3PO3(2.0) | TFA(1) | DMF | 100 | Trace |
| 4 | Cu2O(20) | H3PO3(2.0) | TFA(1) | DMF | 100 | Trace |
| 5 | AgNO3 | H3PO3(2.0) | TFA(1) | DMF | 100 | NR |
| 6 | Pd(OAc)2 | H3PO3(2.0) | TFA(1) | DMF | 100 | NR |
| 7 | NiCl26H2O(25) | H3PO3(2.0) | TFA(1) | DMF | 100 | NR |
| 8 | FeCl3·6H2O | H3PO3(2.0) | TFA(1) | DMF | 100 | NR |
| 9 | — | H3PO3(2.0) | TFA(1) | DMF | 100 | NR |
| 10 | CuSO4·5H2O(20) | — | TFA(1) | DMF | 100 | NR |
| 11 | CuSO4·5H2O(20) | H3PO3(2.0) | — | DMF | 100 | Trace |
| 12 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(2) | DMF | 100 | 81 |
| 13 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(3) | DMF | 100 | 76 |
| 14 | CuSO4·5H2O(20) | H3PO3(2.0) | HNO3(2) | DMF | 100 | Trace |
| 15 | CuSO4·5H2O(20) | H3PO3(2.0) | H2SO4(2) | DMF | 100 | Trace |
| 16 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(2) | DMF | 100 | 52 |
| 17 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(2) | DMF | 100 | 80 |
| 18 | CuSO4·5H2O(20) | H3PO3(1.0) | TFA(2) | DMF | 100 | 60 |
| 19 | CuSO4·5H2O(20) | H3PO3(3.0) | TFA(2) | DMF | 100 | 76 |
| 20 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(2) | DMSO | 100 | 32 |
| 21 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(2) | Toluene | 100 | Trace |
| 22 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(2) | 1,4-Dioxane | 100 | Trace |
| 23 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(2) | DMF | 80 | 35 |
| 24 | CuSO4·5H2O(20) | H3PO3(2.0) | TFA(2) | DMF | 120 | 78 |

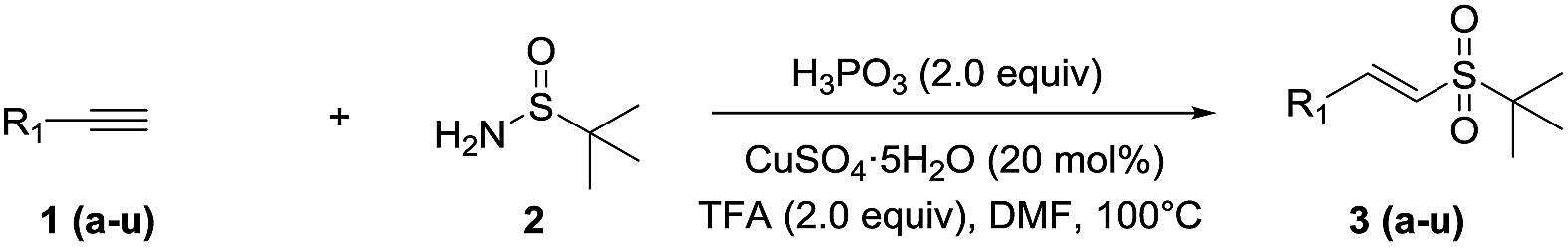
The substrate scope of alkynes and t-butylsulfinamide under the optimized conditions was then explored, as demonstrated in Table 2. Various vinyl sulfones were efficiently obtained via the new reaction. It can be seen that both the electron-donating (OCH3, CH3, C2H5, n-C3H7) and electron-withdrawing groups (F, Cl, Br) of aryl terminal alkynes were suitable for this protocol, and the corresponding vinyl sulfones were obtained in good to excellent yields (3a–o). Common functionalities, which include halogen, methoxy, acetamide and cyano groups, were well tolerated. In addition, the reaction of 1-ethynylcyclohexene with t-butylsulfinamide efficiently afforded the conjugated sulfone, 3p (70%). To our delight, heteroaromatic alkynes also gave the corresponding 3q–s in moderate to good yields, which range from 51% to 69%. 1,3-Diethynylbenzene produced the expected product 3t (53%) in moderate yield. However, the reaction with 1-phenylpropyne did not progress well, and only a trace amount of product was obtained (3u).
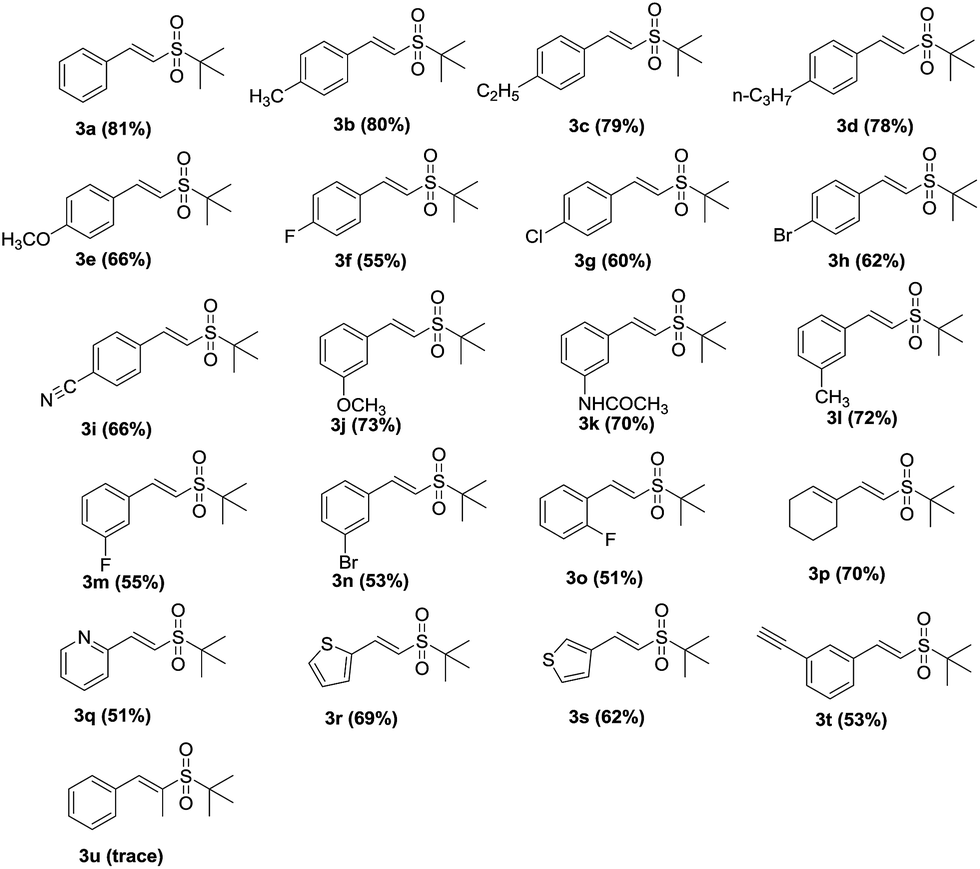
To further probe the reaction mechanism, some experiments were subsequently carried out (Scheme 2). When the reaction was carried out under a nitrogen atmosphere, the corresponding vinyl sulfone, 3a, was not obtained (Scheme 2-a), which indicates that the copper-catalyzed hydrosulfonylation of alkynes requires the presence of oxygen. When TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy), a widely used radical scavenger, was added into the reaction system, the reaction was completely suppressed (Scheme 2-b), which suggests that these reactions undergo a radical mechanism. Moreover, an isotopic labeling experiment was performed. The reaction of phenylacetylene with t-butylsulfinamide in the presence of 10 equiv. H218O afforded 18O-labeled 3a (Scheme 2-c), which clearly demonstrates that the additional oxygen atom of 3a originated from H2O instead of O2 in air.
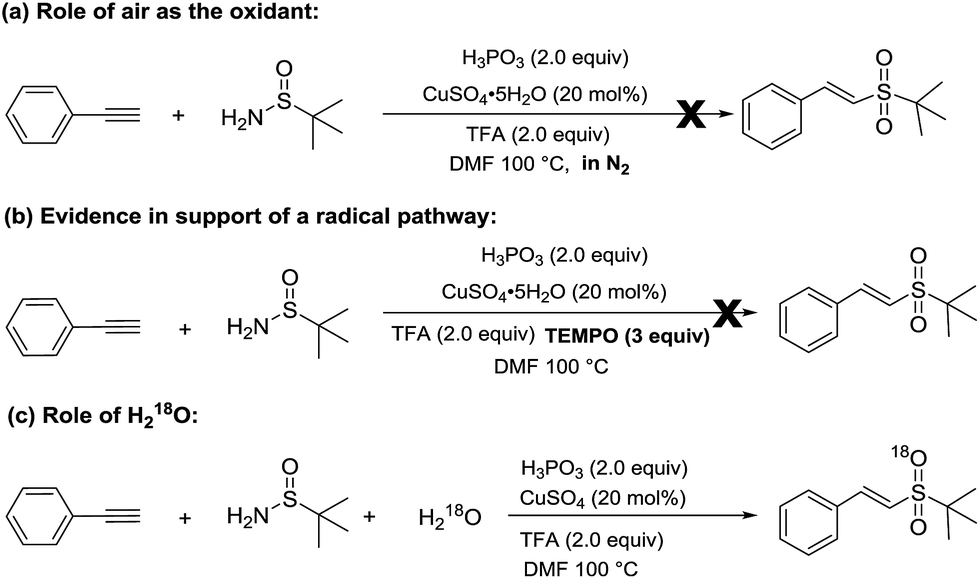 | ||
| Scheme 2 Experiments investigating the reaction mechanism. (a) 1a (1.0 mmol), 2 (1.2 mmol), H3PO3 (2.0 mmol), CuSO4·5H2O (20 mol%), TFA (2.0 mmol) at 100 °C under nitrogen atmosphere for 12 h; (b) 1a (1.0 mmol), 2 (1.2 mmol), H3PO3 (2.0 mmol), CuSO4·5H2O (20 mol%), TFA (2.0 mmol), TEMPO (3 equiv.) at 100 °C for 12 h. (c) 1a (1.0 mmol), 2 (1.2 mmol), H218O (10 equiv.), H3PO3 (2.0 mmol), CuSO4 (20 mol%), TFA (2.0 mmol), at 100 °C for 12 h. | ||
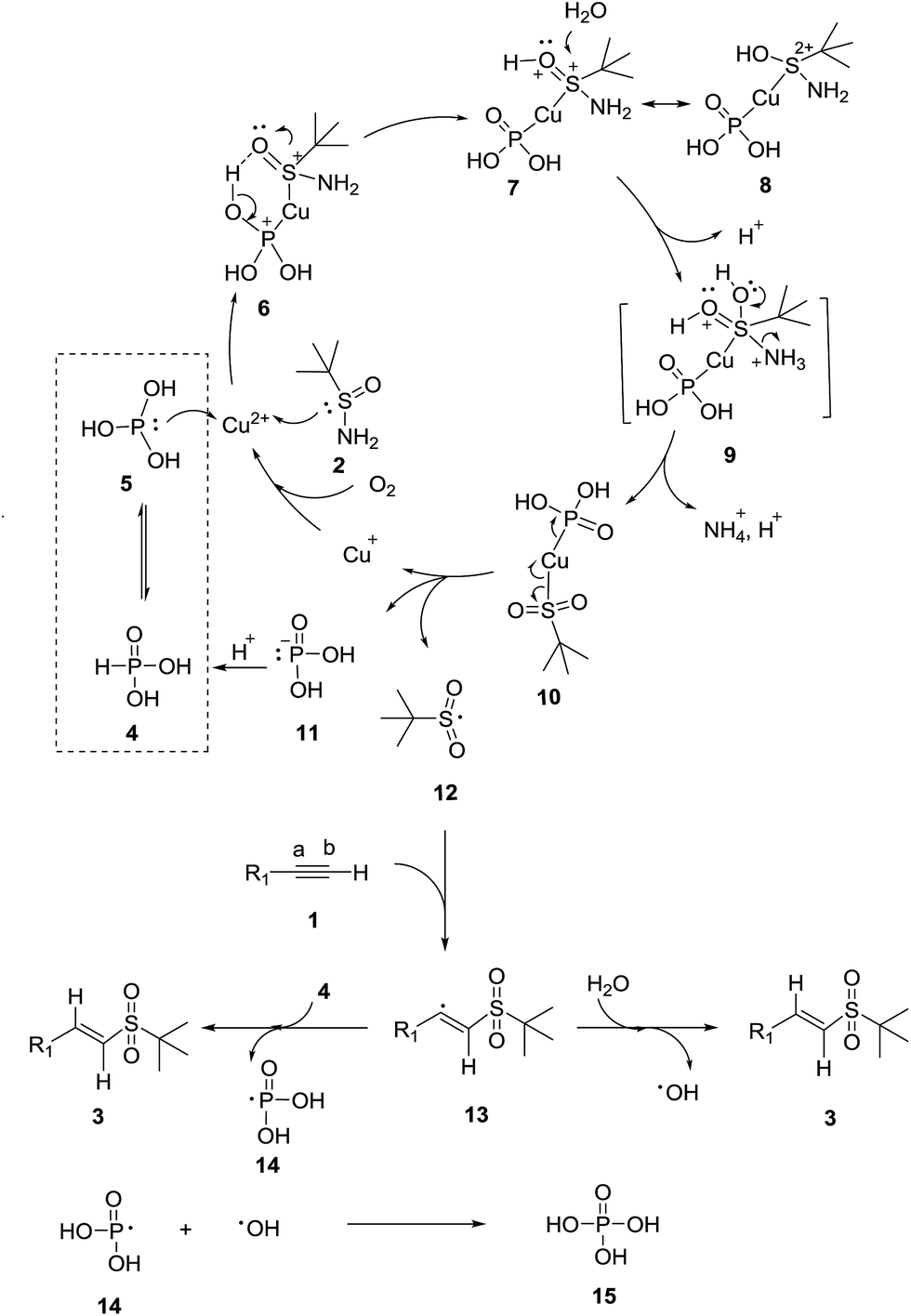
Although the reaction mechanism is not fully understood, a plausible mechanism for the reaction is proposed in Scheme 3. Herein, the cheap and easily available phosphorous acid 4 (tetracoordinated form) existed with its tautomer 5 (tricoordinated form). 5 and t-butylsulfinamide 2 initially coordinated with Cu2+ to form complex 6. Moreover, a six-membered ring was generated inside 6 through an intramolecular hydrogen bond (O–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S), and subsequently a proton transfer from the OH group to the oxygen of the SO group occurs, thus giving copper complex 7. It can be noted that complex 7 could resonate with 8. The two positive charges at the sulfur atom of the resonance structure 8 implies the high sensitivity of the protonated sulfinyl group to nucleophilic attack. Subsequently, a molecule of water attacked the protonated sulfinyl group of 7 to afford the pentavalent intermediate 9. Then, intermediate 9 immediately lost an amino group, to yield copper complex 10. Copper complex 10 then homolytically cleaved to complex 11, Cu+ and tert-butyl sulfonyl radical 12. Cu+ was oxidized back to Cu2+ by oxygen in air, and phosphonate ion 11 was protonated to form phosphorous acid 4. Herein, the selective addition of radical 12 to the terminal alkyne 1 led to the formation of vinyl radical 13. Then, vinyl radical 13 interacted with water to yield the final vinyl sulfone 3 and the hydroxyl radical. Moreover, the vinyl radical 13 could react with another phosphorous acid 4 molecule to produce the final product 3 together with phosphite radical 14. Then, phosphoric acid 15 might possibly be formed via the termination reaction of the phosphite radical 14 and hydroxyl radical in a related reaction process.
S), and subsequently a proton transfer from the OH group to the oxygen of the SO group occurs, thus giving copper complex 7. It can be noted that complex 7 could resonate with 8. The two positive charges at the sulfur atom of the resonance structure 8 implies the high sensitivity of the protonated sulfinyl group to nucleophilic attack. Subsequently, a molecule of water attacked the protonated sulfinyl group of 7 to afford the pentavalent intermediate 9. Then, intermediate 9 immediately lost an amino group, to yield copper complex 10. Copper complex 10 then homolytically cleaved to complex 11, Cu+ and tert-butyl sulfonyl radical 12. Cu+ was oxidized back to Cu2+ by oxygen in air, and phosphonate ion 11 was protonated to form phosphorous acid 4. Herein, the selective addition of radical 12 to the terminal alkyne 1 led to the formation of vinyl radical 13. Then, vinyl radical 13 interacted with water to yield the final vinyl sulfone 3 and the hydroxyl radical. Moreover, the vinyl radical 13 could react with another phosphorous acid 4 molecule to produce the final product 3 together with phosphite radical 14. Then, phosphoric acid 15 might possibly be formed via the termination reaction of the phosphite radical 14 and hydroxyl radical in a related reaction process.
 | ||
| Scheme 3 Plausible mechanism. | ||
Conclusions
In conclusion, the practical CuSO4·5H2O catalyzed synthesis of (E)-vinyl sulfones from alkynes and t-butylsulfinamide was developed. The cleavage of the N–S bond of t-butylsulfinamide, which was catalyzed by an inexpensive and easily available CuSO4·5H2O-phosphorous acid catalytic system, was the key step for this transformation. The method described in this study opens a new door to the construction of vinyl sulfones. Studies of the detailed mechanism of this process and its application are currently underway in our laboratory.Acknowledgements
This study was financially supported by the NSFC (no. 21072178), the Innovation Specialist Projects of Henan Province (no. 114200510023), and the Technology and Science talent Cultivation Project in Zhengzhou City (no. 112PLJRC359). The Special Fund for the Doctoral Program of Higher Education (no. 20124101110003) and Foundational and Frontier Technology Research Foundation of Henan Province are acknowledged for their financial support (no. 132300410027).Notes and references
- (a) C. M. Kam, M. G. Gotz, G. Koot, M. McGuire, D. Thiele, D. Hudig and J. C. Powers, Arch. Biochem. Biophys., 2004, 427, 123 CrossRef CAS PubMed; (b) L. M. Ni, S. Zheng, P. K. Somers, L. K. Hoong, R. R. Hill, E. M. Marino, K. L. Suen, U. Saxena and C. Q. Meng, Bioorg. Med. Chem. Lett., 2003, 13, 745 CrossRef CAS; (c) G. E. Korver, C. M. Kam, J. C. Powers and D. Hudig, Int. Immunopharmacol., 2001, 1, 21 CrossRef CAS; (d) J. T. Palmer, D. Rasnick, J. L. Klaus and D. Bromme, J. Med. Chem., 1995, 38, 3193 CrossRef CAS; (e) B. A. Frankel, M. Bentley, R. G. Kruger and D. G. McCafferty, J. Am. Chem. Soc., 2004, 126, 3404 CrossRef CAS PubMed; (f) M. M. M. Santos and R. Moreira, Mini-Rev. Med. Chem., 2007, 7, 1040 CrossRef CAS.
- I. D. Kerr, J. H. Lee, C. J. Farady, R. Marion, M. Rickert, M. Sajid, K. C. Pandey, C. R. Caffrey, J. Legac, E. Hansell, J. H. McKerrow, C. S. Craik, P. J. Rosenthal and L. S. Brinen, J. Biol. Chem., 2009, 284, 25697 CrossRef CAS PubMed.
- S. Y. Woo, J. H. Kim, M. K. Moon, S. H. Han, S. K. Yeon, J. W. Choi, B. K. Jang, H. J. Song, Y. G. Kang, J. W. Kim, J. Lee, D. J. Kim, O. Hwang and K. D. Park, J. Med. Chem., 2014, 57, 1473 CrossRef CAS PubMed.
- (a) S. Chodroff and W. F. Whitmore, J. Am. Chem. Soc., 1950, 72, 1073 CrossRef CAS; (b) D. A. R. Happer and B. E. Steenson, Synthesis, 1980, 806 CrossRef CAS.
- I. C. Popoff, J. L. Dever and G. R. Leader, J. Org. Chem., 1969, 34, 1128 CrossRef CAS.
- (a) T. Back and G. C. Scott, J. Org. Chem., 1981, 46, 3249 CrossRef CAS; (b) H. Qian and X. Huang, Synlett, 2001, 1913 CrossRef CAS; (c) V. Nair, A. Augustine, T. G. George and L. G. Nair, Tetrahedron Lett., 2001, 42, 6763 CrossRef CAS.
- (a) Q. Xue, Z. Mao, Y. Shi, H. Mao, Y. Cheng and C. Zhu, Tetrahedron Lett., 2012, 53, 1851 CrossRef CAS PubMed; (b) M. Kirihara, J. Yamamoto, T. Noguchi and Y. Hirai, Tetrahedron Lett., 2009, 50, 1180 CrossRef CAS PubMed; (c) X. Huang, D.-H. Duan and W.-X. Zheng, J. Org. Chem., 2003, 68, 1958 CrossRef CAS PubMed.
- Y. L. Xu, J. W. Zhao, X. D. Tang, W. Q. Wu and H. F. Jiang, Adv. Synth. Catal., 2014, 356, 2029 CrossRef CAS PubMed.
- (a) N. Taniguchi, Synlett, 2011, 1308 CrossRef CAS; (b) N. Zhang, D. S. Yang, W. Wei, L. Yuan, Y. J. Cao and H. Wang, RSC Adv., 2015, 5, 37013 RSC; (c) Y. C. Luo, X. J. Pan and G.-Q Yuan, Tetrahedron, 2015, 71, 2119 CrossRef CAS PubMed.
- S. Cacchi, G. Fabrizi, A. Goggiamani, L. M. Parisi and R. Bernini, J. Org. Chem., 2004, 69, 5608 CrossRef CAS PubMed.
- F. Huang and R. A. Batey, Tetrahedron, 2007, 63, 7667 CrossRef CAS PubMed.
- (a) N. Taniguchi, Synlett, 2012, 23, 1245 CrossRef CAS; (b) W. Wei, J. Li, D. Yang, J. Wen, Y. Jiao, J. You and H. Wang, Org. Biomol. Chem., 2014, 12, 1861 RSC; (c) R. J. Song, Y. Liu, Y. Y. Liu and J. H. Li, J. Org. Chem., 2011, 76, 1001 CrossRef CAS PubMed.
- X. Li, Y. Xu, W. Wu, C. Jiang, C. Qi and H. Jiang, Chem.–Eur. J., 2014, 20, 7911 CrossRef CAS PubMed.
- S. Tang, Y. Wu, W. Liao, R. Bai, C. Liu and A. Lei, Chem. Commun., 2014, 50, 4496 RSC.
- Y. Xi, B. Dong, E. J. McClain, Q. Wang, T. L. Gregg, N. G. Akhmedov, J. L. Petersen and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 4657 CrossRef CAS PubMed.
- Q. Jiang, B. Xu, J. Jia, A. Zhao, Y.-R. Zhao, Y.-Y. Li, N.-N. He and C.-C. Guo, J. Org. Chem., 2014, 79, 7372 CrossRef CAS PubMed.
- Y. Jiang and T.-P. Loh, Chem. Sci., 2014, 5, 4939 RSC.
- X. F. Gao, X. J. Pan, J. Gao, H. W. Huang, G. Q. Yuan and Y. W. Li, Chem. Commun., 2015, 51, 210 RSC.
- J. Y. Chen, X. L. Chen, X. Li, L. B. Qu, Q. Zhang, L. K. Duan, Y. Y. Xia, X. Chen, K. Sun, Z. D. Liu and Y. F. Zhao, Eur. J. Org. Chem., 2015, 2, 314 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra13474a |
| This journal is © The Royal Society of Chemistry 2015 |