
A new electrochemical sensor for the simultaneous determination of guanine and adenine: using a NiAl-layered double hydroxide/graphene oxide-multi wall carbon nanotube modified glassy carbon electrode
Behzad Rezaei*,
Hossein Khosropour,
Ali Asghar Ensafi,
Mohammad Dinari and
Afshin Nabiyan
Department of Chemistry, Isfahan University of Technology, Isfahan 84156-83111, Islamic Republic of Iran. E-mail: rezaei@cc.iut.ac.ir; Fax: +98 31 33912350; Tel: +98 31 33912351, +98 31 33913268
First published on 3rd September 2015
Abstract
An electrochemical sensor was developed for guanine (GA) and adenine (AD) detection using multiwall carbon nanotubes (MWCNTs) with the hybrid NiAl-layered double hydroxide/graphene oxide (NiAl-LDH/GO) on a glassy carbon electrode (GCE), referred to as MWCNTs/NiAl-LDH/GO/GCE. The modified electrode was used for the simultaneous detection of GA and AD. Electrochemical performances related to the direct electrooxidation of GA and AD at the modified electrode were investigated, showing that their peak currents were greatly enhanced due to the presence of MWCNTs/NiAl-LDH/GO nanohybrids. Also, it was revealed that the oxidation peak potentials of GA and AD on the modified electrode were negatively shifted, leading to the increase of their electrocatalytic activity at the surface of MWCNTs/NiAl-LDH/GO/GCE. The effects of different parameters such as pH, accumulation time, accumulation potential, and scan rate on the sensitivity were investigated too. Determination of purine bases was done by a linear sweep voltammetric (LSV) technique. The linear ranges of 0.010–45 μM, with the detection limit of 3 nM for GA, and 0.08–45 μM, with the detection limit of 20 nM for AD, were achieved. Finally, the proposed electrochemical sensor was employed for the simultaneous determination of GA and AD in single-strand deoxyribonucleic acid (ssDNA) samples.
1. Introduction
Deoxyribonucleic (DNA) acid plays the main role in the storage of genetic information and protein biosynthesis. Guanine (GA) and adenine (AD) are important components found in DNA, with critical roles in life processes.1 They have significant effects on the modulation of adenylate cyclase activity, control of blood flow, the prevention of cardiac arrhythmias and the inhibition of neurotransmitter release.2 Concentration levels of these abnormal changes can be regarded as an important parameter for HIV, myocardial cellular energy status, diagnosis of cancers, disease progress and therapy responses.3 Therefore, selective and sensitive detection methods are needed for the analysis of these compounds. Accordingly, different analytical methods such as liquid chromatography or electrophoresis combined with different detection techniques have been employed for the analysis of purine bases in nucleic acids.4–10 While these methods can be useful, they suffer from some disadvantages due to their complicated instruments, time-consuming nature and sample pretreatment requirements.On the other hand, rapidity, high sensitivity, and low cost are the main advantages of electrochemical sensors for the analysis of biological compounds.11–14 Several electrochemical detection methods are mostly based on the reduction of purine bases on a hanging mercury drop electrode15 or the oxidation of both GA and AD on various modified electrodes.16–23
Nanotechnology has become one of the most interesting areas in science and technology.24–29 Carbon nanotubes (CNTs) are important kind of nanostructure with a tensile strength one hundred times more than that of steel, a thermal conductivity better than all but the purest type of diamond, and an electrical conductivity similar to that of copper, as well as the ability to carry much higher currents. Electrodes modified with CNTs for analytical sensing have been found to show low detection limits, high sensitivities, lowering of over-potentials, and resistance to surface fouling.30–35
Layered double hydroxides (LDHs), which are hydrotalcite-like materials, are a class of two-dimensional nanostructured anionic clays. The positively charged layers contain edge-shared metal M(II) and M(III) hydroxide octahedral with charges neutralized by anions located in the interlayer spacing or at the edges of the lamella. LDHs, as a family of inorganic layer materials, have recently attracted much attention in such areas as catalysis, catalyst precursors, anion exchangers, adsorbents, electro and photoactive materials, solid-state nanoreactors, polymer composites, and bioactive materials.36–44
Multiwall carbon nanotubes (MWCNTs) and NiAl-layered double hydroxide/graphene oxide (NiAl-LDH/GO) have admirable features, but their intrinsic shortages have limited their applications. For example, NiAl-LDH/GO has low electrical conductivity, though they have high chemical reactivity. For MWCNTs with high electrical conductivity, their chemical inertness, low solubility, and dispersivity in most solvents hinder the electrochemical performance. Thus, the combination of these materials for the preparation of homogeneous nanohybrid is an effective strategy to integrate their distinguishing properties.
In this paper, we have established an extremely simple approach to synthesize nanostructured multiwall carbon nanotubes/NiAl-layered double hydroxide/graphene oxide (MWCNTs/NiAl-LDH/GO), based on using ultrasonic irradiation as a simple, green and fast method on modified MWCNTs. The electrocatalytic performance of synthesized nanohybrid was also preliminarily studied for the direct electrooxidation of GA and AD. To the best of our knowledge, detailed studies of the synthesis of nanostructured MWCNTs/NiAl-LDH/GO composites and the electrochemical performance of the resulting nanocomposites have not been reported yet. The results of the experiments with both GA and AD have indicated that the electrode with MWCNTs/NiAl-LDH/GO nanohybrids can have a synergic effect on the oxidation of GA and AD, as compared to the unmodified GCE. Because MWCNTs/NiAl-LDH/GO nanohybrids can increase the electroactive area of the electrode and have a synergic effect on the oxidation of GA and AD, the over potential in the detection of GA and AD can be decreased substantially. In addition, we did several species of interference that exist in real samples. The modified electrode can also exhibit a good behavior in the simultaneous determination of GA and AD by using linear sweep voltammetry (LSV).
2. Experimental methods
2.1. Chemicals
All chemicals, which were of analytical reagent grade, were purchased from Merck (Darmstadt, Germany) unless stated otherwise. Aluminum(III) nitrate nonahydrate [Al(NO3)3·9H2O] and nickel(II) nitrate hexahydrate [Ni(NO3)2·6H2O] were purchased from Merck Chemical Co. Double distilled water was used throughout. GA, AD and salmon sperm double-strand deoxyribonucleic acid (dsDNA) were purchased from Sigma-Aldrich.Stock solutions of GA and AD (0.01 M) were prepared by dissolving appropriate amounts of analytes in a diluted (0.2 M) NaOH solution prepared by dilution with water to mark. The solutions were stored at 4 °C when not in use.
Phosphate buffer solutions (PBS) prepared with using sodium dihydrogen phosphate and disodium monohydrogen phosphate, 0.1 M and appropriate amounts of hydrochloric acid or sodium hydroxide.
MWCNTs with a diameter of 10–30 nm, a length of 5–15 μm, and purity of >95% was procured from Aldrich.
2.2. Apparatus
A conventional three-electrode cell was used for all experiments. A GCE modified with MWCNTs/NiAl-LDH/GO as working electrodes, platinum wire as an auxiliary electrode, and an Ag/AgCl/KClsat as a reference electrode were used.Electrochemical measurements were carried out using Autolab model PGSTAT 12 potentiostat/galvanostat system (Eco Chemie B.V., Utrecht, The Netherlands) connected to a three-electrode cell plus the GPES and FRA 4.9 software.
A pH-meter (Corning, Model 140) with a double junction glass electrode was used to check pH of the solutions.
Fourier transform infrared spectroscopy (FT-IR) was carried out on Jasco-680 (Japan) spectrophotometer in the range of 4000–400 cm−1.
X-ray diffraction analysis (XRD) was conducted using a Philips Xpert MPD X-ray diffractometer with Cu-Kα radiation (λ = 1.51418 Å) at a voltage of 40 kV.
Morphologies of the as-obtained products were observed using a field-emission scanning electron microscope [(FE-SEM), Hitachi, S-4160] and a transmission electron microscope [(TEM), Philips CM 120, Netherlands].
The sono-chemical reaction was carried out on a MISONIX ultrasonic liquid processor, XL-2000 SERIES (Raleigh, North Carolina, USA). Ultrasound was a wave of frequency 2.25 × 104 Hz and the power of 100 W.
2.3. Preparation of the reduced GO and NiAl-LDH/GO hybrids
Graphene oxide (GO) was synthesized from graphite powder (with a particle size of 70 μm and the purity of 99.99%) using the modified Hummer's method.45 In brief, 1 g of graphite and 0.5 g of sodium nitrate were mixed together and this was followed by the addition of 23 mL of conc. sulphuric acid under constant stirring. After 1 h, 3 g of KMnO4 was added gradually to the above solution while keeping the temperature to less than 20 °C to prevent overheating and explosion. The mixture was stirred at 35 °C for 12 h and the resulting solution was diluted by adding 500 mL of water under vigorous stirring. To ensure the completion of reaction with KMnO4, the suspension was further treated with 30% H2O2 solution (5 mL). The resulting mixture was washed with HCl and H2O, respectively. This was followed by filtration and drying, and graphene oxide sheets were thus obtained. Next, the GO thus obtained was dispersed in water and sonicated for 2 h to form exfoliated GO. Finally, the reduction of GO was performed as follows: 100 mL of exfoliated GO suspension (2.5 mg mL−1) was prepared in distillate water. The suspension was kept at 0 °C and NaBH4 (five times ratio of GO w/w) was added to the mixture dropwise to reduce the GO. The obtained products were filtered and washed in distillate water several times before drying at room temperature.NiAl-LDH/GO was synthesized using ultrasonic irradiation as a simple, green and fast method. For the preparation of the NiAl-LDH/GO, first, 0.07 g of reduced GO was dispersed in an aqueous solution (2.06 mL) containing 9.9 mmol of NaOH. Then, 1.4 mL of a salt solution of 2.8 mmol of Ni(NO3)2·6H2O and 1.4 mmol of Al(NO3)3·9H2O were added. The resulting black suspension was stirred at 60 °C for 2 h under nitrogen atmosphere. Subsequently, the resulting black suspension was sonicated for 2 h under nitrogen conditions. The solid was recovered and washed with deionized water several times and finally, dried under vacuum at 80 °C for 12 h.
2.4. Preparation of the working electrode
A bare GCE was polished with 0.3 μm alumina slurry for 5 min to a mirror finish. Then, it was washed with double distillate water and ethanol in an ultrasonic bath for 5 min and dried at room temperature. 3.0 mg portion of MWCNTs and NiAl-LDH/GO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was dispersed in 1.0 mL ethanol and homogenized ultrasonically for 15 min. An appropriate amount of this solution was deposited on the freshly prepared GCE surface. After the evaporation of the solvent, the electrode was thoroughly rinsed with water.
:2) was dispersed in 1.0 mL ethanol and homogenized ultrasonically for 15 min. An appropriate amount of this solution was deposited on the freshly prepared GCE surface. After the evaporation of the solvent, the electrode was thoroughly rinsed with water.
The active areas of the modified electrodes were obtained by cyclic voltammetry (CV) using 1.0 mM hexacyanoferrate [K3Fe(CN)6] as the probe at different scan rates. For a reversible process, the Randles–Sevcik formula was used at room temperature:
| Ip = 2.69 × 105n2/3ACoD1/2ν1/2, | (1) |
For 1.0 mM [K3Fe(CN)6] in the 0.1 M KCl electrolyte: n = 1, D = 7.6 × 10−6 cm2 s−1,46 then from the slope of the Ip − ν1/2 relation, the microscopic areas can be calculated. In the bare GCE, NiAl-LDH/GO-GCE, MWCNTs-GCE and MWCNTs/NiAl-LDH/GO-GCE the active surface was 0.023, 0.082, 0.119 and 0.141 cm2. Then the modified electrode surface was nearly 6.1 times greater than the bare GCE.
2.5. Preparation of DNA samples
Thermally denatured dsDNA was produced according to a previous report.47 To describe it briefly, native herring sperm dsDNA samples were dissolved in water and then the solution was heated in a boiling water bath (100 °C) for about 10 min. Finally, the solution was rapidly cooled in an ice bath. Generally, thermal denaturation involves the rupture of hydrogen bonds, the disturbance of stacking interaction, but not any breakage of covalent bond. So thermally denatured dsDNA could act as a ssDNA.3. Results and discussion
3.1. Characterization of the MWCNTs/NiAl-LDH/GO
Different methods including FT-IR, XRD, FE-SEM and TEM were used to investigate the characteristics of NiAl-LDH/GO hybrids.Fig. 1 show the FT-IR spectra of GO and NiAl-LDH/GO hybrids. In the FT-IR spectra of the GO, the characteristic peaks at 3412, 1717, 1584, 1231 and 1050 cm−1 could be assigned to the O–H stretch, carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, aromatic CC stretch, epoxy C–O stretch and alkoxy C–O stretch, respectively. For the pure NiAl-LDH/GO hybrids, the broad peak centered at about 3469 cm−1 was attributed to the O–H stretching vibration of water molecules in the interlayer and hydrogen-bonded OH groups, which was accompanied with the bending mode at 1637 cm−1. The peak at around 1420–1485 cm−1 was assigned to carboxylate anion on the surface of GO intercalated LDH. Furthermore, the absorption band below 700 cm−1 was ascribed to the characteristic peak of metal oxide like Ni–O and Al–O vibrations in the lattice of LDH (Fig. 1).
O stretch, aromatic CC stretch, epoxy C–O stretch and alkoxy C–O stretch, respectively. For the pure NiAl-LDH/GO hybrids, the broad peak centered at about 3469 cm−1 was attributed to the O–H stretching vibration of water molecules in the interlayer and hydrogen-bonded OH groups, which was accompanied with the bending mode at 1637 cm−1. The peak at around 1420–1485 cm−1 was assigned to carboxylate anion on the surface of GO intercalated LDH. Furthermore, the absorption band below 700 cm−1 was ascribed to the characteristic peak of metal oxide like Ni–O and Al–O vibrations in the lattice of LDH (Fig. 1).
 | ||
| Fig. 1 FT-IR image of GO (a) and NiAl-LDH/GO (b) nanohybrid. | ||
XRD patterns of reduced GO and NiAl-LDH/GO are presented in Fig. 2. In the XRD pattern of the reduced GO, the peak for (002) plane was 2θ = 24.52° with the d-spacing of 3.63 Å. The width of the peak was attributed to a turbo static arrangement of graphene stacked sheets.48 The diffraction peaks for the (003), (006), (012), (015), (018), (110) and (113) planes of typical hydrotalcite-like LDH can be observed in Fig. 2. In the XRD pattern of, NiAl-LDH/GO hybrids no characteristic peak of reduced GO was found. These results showed that the LDH was well anchored on the reduced GO sheets, thereby effectively preventing the restacking of the as reduced graphene.47
 | ||
| Fig. 2 XRD pattern of reduced GO (a) and NiAl-LDH/GO (b) nanohybrid. | ||
The morphologies and structures of the NiAl-LDH/GO and MWCNTs/NiAl-LDH/GO were investigated by FE-SEM and TEM Techniques. The FE-SEM investigation of the NiAl-LDH/GO showed that the LDH prepared by the ultrasonic method roughly consisted of plate-like shapes stacked on the top of each of GOs sheets and also, the opening sheets of GOs for the rapture of LDH layers and vice versa, thereby forming a 3D porous structure all over the hybrid domain (Fig. 3a and b). In comparison with these two images, MWCNTs/NiAl-LDH/GO hybrid showed the presence of MWCNTs among the LDH/GO structure (Fig. 3c and d).
 | ||
| Fig. 3 FE-SEM images of NiAl-LDH/GO (a and b) and MWCNTs/NiAl-LDH/GO-GCE (c and d). | ||
Fig. 4 displays the TEM images of the NiAl-LDH/GO and MWCNTs/NiAl-LDH/GO. For LDH/GO, the TEM results revealed that typical small platelets of LDH were attached to the GO nanosheets and most LDH platelets were observed to be oriented face-on to the graphene substrate. It seemed that the interaction between the positively charged LDH sheets and the negatively charged GO successfully resulted in the deposition of LDHs on the support during the synthesis (Fig. 4a and b). In the TEM images of the MWCNTs/NiAl-LDH/GO hybrid, it could be evidently observed that that most LDH/GO was adjacent to the surface of MWCNTs, thereby suggesting the affinity between LDH/GO and MWCNTs matrix (Fig. 4c and d).
 | ||
| Fig. 4 TEM images of NiAl-LDH/GO (a and b) and MWCNTs/NiAl-LDH/GO-GCE (c and d). | ||
3.2. Electrochemistry of the biosensors
Electrochemical impedance spectroscopy (EIS) could provide detailed information on the change in the surface property of the modified electrodes. The impedance spectra included a semicircle portion and a linear portion. The semicircle diameter at higher frequencies corresponded to the electron-transfer resistance (Ret), and the linear part at lower frequencies related to the diffusion process. Fig. 5 shows the impedance spectra corresponding to the different electrodes. | ||
| Fig. 5 Nyquist plots of the different electrodes in a PBS (pH 7.0) solution containing 0.1 M KCl and 5.0 mM [K3Fe(CN)6]. The frequency range was from 10−1 to 104 Hz with perturbation amplitude of 5 mV. (a) GCE; (b) NiAl-LDH/GO-GCE; (c) MWCNTs-GCE; (d) MWCNTs/NiAl-LDH/GO-GCE. | ||
The results showed that the diameter of the semicircle was highly decreased in the presence of NiAl-LDH/GO, MWCNTs and MWCNTs/NiAl-LDH/GO, respectively. This confirmed the electrocatalytic capability of the modifier for the oxidation of 5 mM [K3Fe(CN)6] as a probe. Because MWCNTs/NiAl-LDH/GO was an excellent electric conducting material, it could accelerate the electron transfer, resulting in the reduction of Ret. The EIS change of the modified electrode, also indicated that the MWCNTs/NiAl-LDH/GO was immobilized on the modified electrode surface.
In order to study the nature of the modified electrode process on the electrode surface, LSV was performed and recorded. Linear sweep voltammograms of bare GCE (a), MWCNTs-GCE (b), NiAl-LDH/GO-GCE (c) and MWCNTs/NiAl-LDH/GO-GCE (d) in a solution containing 10.0 μM GA and AD are shown in Fig. 6. At the modified electrode, GA and AD exhibited anodic peaks potential at 0.72 and 0.96 V, respectively; on the other hand, similar peaks at the unmodified GCE were obtained at 0.78 and 1.06 V, respectively. In addition, in the case of the unmodified GCE, the voltammograms of GA and AD only exhibited a small peak, whereas the peak currents were significantly enhanced on the modified electrode. The above results indicated that the presence of NiAl-LDH/GO on GCE surface had great improvement with the electrochemical response, which was partly due to excellent characteristics of NiAl-LDH/GO such as good electrical conductivity, high chemical stability, and high surface area. The suitable electronic properties of NiAl-LDH/GO together with the MWCNTs gave the ability to promote charge transfer reactions, good anti-fouling properties, especially when mixed with a higher conductive compound such as MWCNTs when used as an electrode.
 | ||
| Fig. 6 Linear sweep voltammograms of 10.0 μM of GA and AD at the bare GCE (a), at the NiAl-LDH/GO-GCE (b), at the MWCNTs-GCE (c) and MWCNTs/NiAl-LDH/GO-GCE (d) in the scan rate of 50 mV s−1, pH 7.0 and accumulation time of 180 s. Inset shows linear sweep voltammogram of the bare GCE. | ||
3.3. Optimization of the experimental parameters
The peak current and the potential of purine bases were electrons and protons transfer steps dependent on the pH of solution. To find the optimum pH, the influence of pH over the range of 3.0–9.0 for 10.0 μM GA and AD on the performance of the sensor was investigated (Fig. 7). It could be seen in the Fig. 7A and B that the anodic peak current of GA and AD was increased by pH, reaching the maximum value at pH 7.0. Therefore, pH 7.0 was selected as the optimum pH for the determination of GA and AD.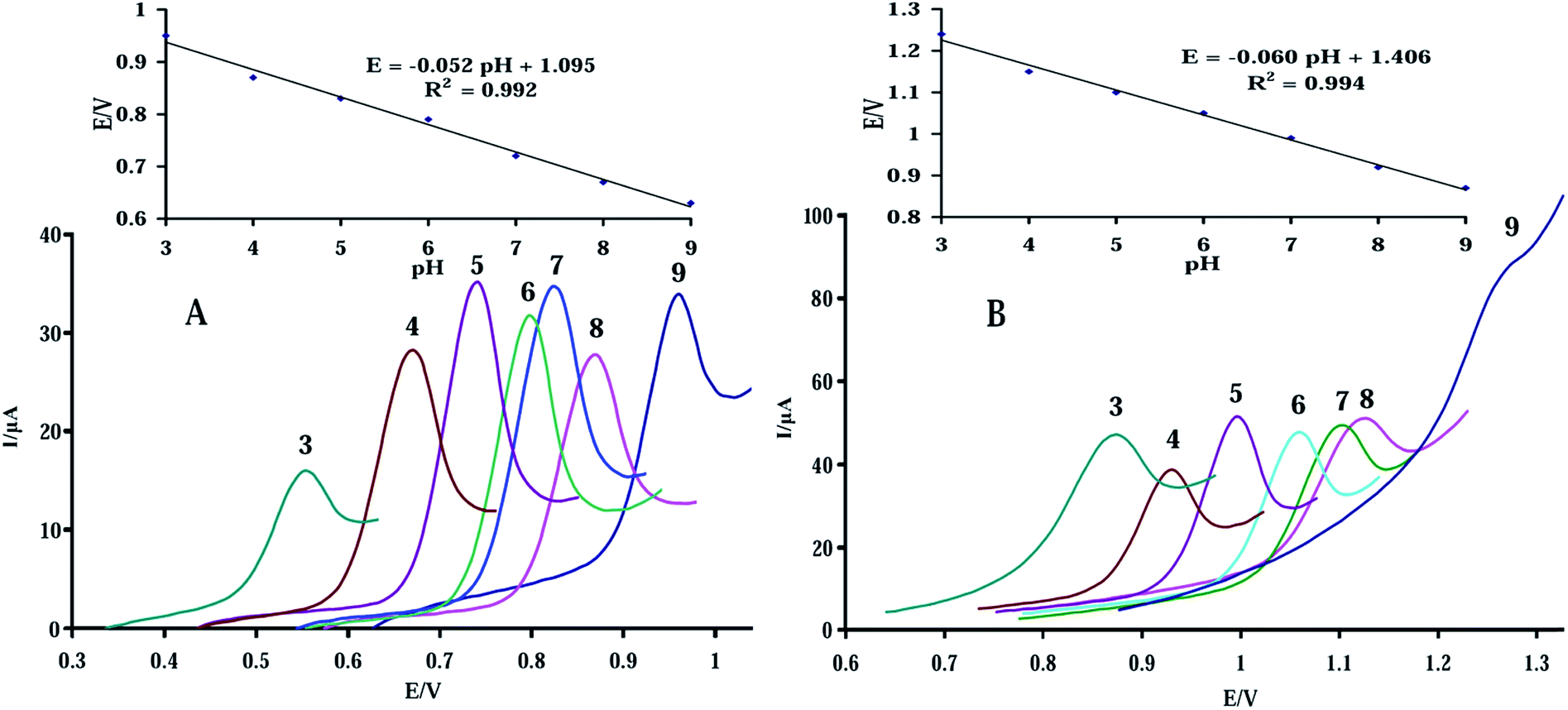 | ||
| Fig. 7 Effect of pH on electrochemical oxidation of 10.0 μM (A) GA, and (B) AD at the surface of MWCNTs/NiAl-LDH/GO-GCE (with accumulation time 180 s). Insets: plot of the oxidation peaks potential vs. the solution pH. | ||
The results also revealed that the oxidation peak potentials of GA and AD were pH dependent, shifting toward more negative potentials with increments in solution pH. The linear regression equations of Epa (V) = −0.052pH + 1.095 (R2 = 0.992) and Epa (V) = −0.060pH + 1.406 (R2 = 0.994) were obtained from the inset pictures of Fig. 7A and B for GA and AD, respectively. The slopes of 52.0 and 60.0 mV pH−1 indicated that equal numbers of protons and electrons were involved in the electrode reaction process.49
The peak currents of both GA and AD were increased with the growth of accumulation time at the GCE, but after 180 s, they were kept almost constant. The peak currents did not vary with accumulation potential ranging from 0 to +0.6 V. The accumulation potential of GA and AD was therefore carried out on open-circuit.
The amount of modifier was found to have an important effect on the response of electrode. However, the electrochemical behavior of the GA and AD was studied by casting different amounts of 3 mg mL−1 dispersed MWCNTs/NiAl-LDH/GO solution (4–16 μL) on the GCE surface. It was observed that the oxidation peak current for GA and AD was increased when the volume of the modifier suspension deposited on the surface of the electrode was increased up to 8 μL. Beyond this point, the peak current was decreased and the electrode became unstable. In all subsequent experiments, 8 μL of modifier suspension was selected as the optimum amount.
The effects of scan rate (ν) on the peak currents of GA and AD (Fig. 8A and B) were studied in the presence of 10.0 μM GA and AD. The results showed that the relevant oxidation peak currents were proportional to the scan rate in the range of 15–150 mV s−1 by following the linear regression equation of Ipa = 0.274ν + 3.006 (R2 = 0.993) for GA and Ipa = 0.235ν + 7.832 (R2 = 0.992) for AD. These results indicated that these two electrochemical processes were adsorption controlled.
 | ||
| Fig. 8 Cyclic voltammograms of 10.0 μM (A) GA and (B) AD at various scan rates of (a) 15; (b) 30; (c) 50; (d) 75; (e) 100 and (f) 150 m V s−1 in 0.1 M PBS (pH 7.0, accumulation time 180 s), inset plot of Ipa versus ν for the oxidation of GA and AD. | ||
The adsorbed amounts of GA and AD on the surface of MWCNTs/NiAl-LDH/GO-GCE were further calculated based on the following equation:46
| Ip = nFQν/4RT = n2F2AΓcν/4RT, | (2) |
Based on the relationship between Ip and ν, the values of the electron transfer number (n) of the GA and AD were calculated to be 2.09 and 2.17, respectively. Also, the obtained surface concentration of GA and AD (Γc) was 3.19 × 10−10 mol cm−2 and 3.41 × 10−10 mol cm−2, respectively.
3.4. Individual and simultaneous determination of GA and AD
Under the optimal experiment conditions established above, the calibration curve of GA and AD in PBS was measured by LSV. As shown in Fig. 9, the anodic peak current of GA was linearly related to the concentration over the range of 0.010–45 μM. The linear regression equation was Ipa (μA) = 1.461C (μM) + 1.471, with a correlation coefficient of R2 = 0.996 (Fig. 9A). The detection limit (S/N = 3)45 was 0.003 μM. Regarding AD, similar studies were also carried out in the case of GA and the calibration curve yielded a linear range from 0.08–45 μM. The linear regression equation was Ipa (μA) = 2.008C (μM) + 4.778, with a correlation coefficient of R2 = 0.997 (Fig. 9B). The detection limit (S/N = 3) was 0.02 μM. | ||
| Fig. 9 LSVs of (A) different concentrations of GA (0.01, 0.3, 1.0, 2.0, 5.0, 10.0, 14.0, 19.0, 23.0, 37.0 and 45.0 μM); (B) different concentrations of AD (0.08, 0.5, 2.0, 4.0, 10.0, 14.0, 23.0, 35.0 and 45.0 μM); and (C) simultaneous determination of GA and AD (from 0.1, 1.0, 3.0, 5.0, 10.0, 15.0, 20.0 and 25.0 μM), insets (a–c) plots of Ip vs. GA and AD concentrations in individual and simultaneous determination (accumulation time 180 s; pH 7.0; scan rate 50 mV s−1). | ||
Simultaneous determination of GA and AD is very important since these two purine bases coexist in DNA. Fig. 9C shows the LSVs of GA and AD when the concentrations of these two purine bases were simultaneously increased at the surface of the MWCNTs/NiAl-LDH/GO-GCE. The oxidation peak currents of GA and AD were increased linearly over a concentration range of 0.10–25.0 μM GA and AD by following the regression equations of Ip (μA) = 1.418CGA (μM) + 5.586 (R2 = 0.991) and Ip (μA) = 1.859CAD (μM) + 7.215 (R2 = 0.991), respectively. Thus, this proposed method allowed simultaneous and sensitive determination of GA and AD. The detection limits of GA and AD (S/N = 3) were calculated to be 0.02 and 0.04 μM, respectively.
Table 1 shows a comparison of the proposed electrochemical method and the other modified electrodes in the previously reported methods50–57 for GA and AD determination. The proposed sensor was found to have a good detection limit and linear response range.
| Electrode | Guanine | Adenine | Technique | Reference | ||
|---|---|---|---|---|---|---|
| Linear range (μmol L−1) | Detection limit (μmol L−1) | Linear range (μmol L−1) | Detection limit (μmol L−1) | |||
| a Boron doped diamond electrode.b Cyclodextrin-carbon nanotubes electrodes.c Poly(2,6-pyridinedicarboxylic acid)/chemically reduced graphene oxide modified electrode.d Polyaniline-MnO2-glassy carbon electrode.e Carbon nanotubes-poly(new fuchsin)/glassy carbon electrode.f Graphene/glassy carbon electrode.g CdS microspheres-chitosan/glassy carbon electrode.h Pt-Pd/porous silicon-carbon nanotube paste electrode.i Carbon nanotube decorated with NiFe2O4 magnetic nanoparticles-glassy carbon electrode. | ||||||
| BDDEa | 0.3–19 | 0.037 | 0.3–19 | 0.019 | DPV | 51 |
| CD-CNT/Eb | 1.2–10 | 0.2 | 1.0–25 | 0.025 | DPV | 52 |
| PPDA/CRGO/GCEc | 0.05–4.5 | 0.01 | 0.1–6 | 0.02 | DPV | 53 |
| PANI/MnO2/GCEd | 10–100 | 4.8 | 10–100 | 2.9 | DPV | 54 |
| CNT-PNF/GCEe | 20–3000 | 18.2 | 100–1800 | 8.6 | CV | 55 |
| Graphene/GCEf | 2–200 | 0.58 | 5–200 | 0.75 | DPV | 56 |
| CdS-CHIT/GCEg | 0.001–1.6 | 0.002 | 0.02–5.0 | 0.04 | DPV | 57 |
| Pt-Pd/PSi-CNTPEh | 0.1–10.0 | 0.02 | 0.1–10.0 | 0.03 | DPV | 20 |
| CNT-NiFe2O4/GCEi | 0.05–3 | 0.006 | 0.1–4 | 0.01 | LSV | 21 |
| MWCNTs/Ni-Al-GO/LDH-GCE | 0.010–45 | 0.003 | 0.08–45 | 0.02 | LSV | This work |
3.5. Stability of sensor
The stability of MWCNTs/NiAl-LDH/GO-GCE was investigated by recording the electrode response in 0.1 M PBS at pH 7.0 with 10.0 μM GA and AD. The relative standard deviations (RSD%) values after 50 cycles for determination of GA and AD were less than 6%. Also, the modified electrode was stored for 15 days, and only a small decrease (less than 3%) was observed in the oxidation peak current of GA and AD.These examinations revealed the high stability of the modified electrode response.
3.6. Selectivity of the sensor
To evaluate the selectivity of the MWCNTs/NiAl-LDH/GO-GCE, the influence of some possible interfering substances existing in the real samples was investigated in the presence 10.0 μM of GA and AD. The interfering effect was defined as the concentration of interfering species that could change the modified electrode response toward the analyte by more than 3s, where s was the standard deviation of the replicate (n = 3). The criterion used for the presence of interference was the t-test at 95% confidence level. The results showed that neither 1000-fold of K+, Na+, Ca2+, Mg2+, NH4+, Br−, ClO4−, SO42−, F−, NO3−, C2O42−, CH3COO−, CO32− and citric acid nor 500-fold of glucose, fructose, lactose, sucrose, tartaric acid, salicylic acid, methionine, valine, lysine and aspartic acid could affect the selectivity.In addition, neither did 300-fold of alanine, phenylalanine, glycine, urea, thiourea, L-cystine, ascorbic acid and Fe2+ species did not interfere with the determination of GA and AD nor did 100-fold tryptophan, Cu2+ and vitamin B2. So the proposed electrode exhibited good selectivity in the electrochemical detection.
3.7. Analytical application
The practical analytical capability of the sensor was examined by the measurement of GA and AD content of thermally denatured DNA, using the standard addition method. The modified electrode gave two well-defined oxidation peaks in the presence of thermally denatured DNA due to the oxidation of GA and AD residues. In a typical procedure, 200 μL of the thermally denatured DNA solution was added into 5 mL of the PBS to measure the oxidation peaks of GA and AD. Subsequently, 1.0, 3.0, and 5.0 μM of GA and AD were added to the above mixture and their peak currents were recorded again. The concentrations of GA and AD in the thermally denatured DNA were obtained. These results are shown in Table 2. In addition, the molar ratios of GA and AD in the thermally denatured DNA were calculated to be 22.4 mol% and 27.5 mol%, respectively. The value of (G + C)/(A + T) was equal to 0.81 ± 0.04 (n = 3) for the thermally denatured DNA sample, which was close to the standard value of 0.78.58| Samples | Guanine | Adenine | ||||||
|---|---|---|---|---|---|---|---|---|
| Added (μmol L−1) | Expected (μmol L−1) | Found (μmol L−1) | Recovery (%) | Added (μmol L−1) | Expected (μmol L−1) | Found (μmol L−1) | Recovery (%) | |
| ss-DNA | — | — | 0.66 ± 0.05 | — | — | — | 0.81 ± 0.08 | — |
| Spiked 1 | 1.00 | 1.66 | 1.70 ± 0.04 | 102.4 | 1.00 | 1.81 | 1.88 ± 0.09 | 103.8 |
| Spiked 2 | 3.00 | 3.66 | 3.59 ± 0.09 | 98.1 | 3.00 | 3.81 | 3.86 ± 0.07 | 101.3 |
4. Conclusions
In this paper, a GCE with MWCNTs/NiAl-LDH/GO nanohybrids as the modifier was fabricated and used for the investigation of the electrochemical behaviors of GA and AD. The remarkable enhancement of the oxidation peak currents was observed on the modified electrode with the negative shift of the oxidation peak potentials, indicating the typical electrocatalytic ability of the analyte. Nanostructure NiAl-LDH/GO hybrid was successfully synthesized by a simple technique through the electrostatic interaction between positively charged layers of NiAl-LDH and negatively-charged functional groups on GO. The results were attributed to the specific characteristics and synergistic effects of MWCNTs/NiAl-LDH/GO nanohybrids presented on the electrode surface. In addition, the modified electrode exhibited good reproducibility, long-term stability, and simplicity of construction and operation. The proposed method was further applied to the simultaneous determination of GA and AD in thermally denatured DNA with a good recovery, indicating the potential applications of MWCNTs/NiAl-LDH/GO nanohybrids in the electrochemical sensor.Acknowledgements
The authors express their appreciation to the Isfahan University of Technology Research Councils and Center of Excellence in Sensor for financial support of this work.References
- W. Saenger and Ch. R. Cantor, Principles of Nucleic Acid Structure, Springer, New York, 1984 Search PubMed.
- S. P. Li, P. Li, T. T. X. Dong and K. W. K. Tsim, Electrophoresis, 2001, 22, 144–150 CrossRef CAS.
- F. Q. Yang, J. Guan and S. P. Li, Talanta, 2007, 73, 269–273 CrossRef CAS PubMed.
- B. D. Gill and H. E. Indyk, Int. Dairy J., 2007, 17, 596–605 CrossRef CAS PubMed.
- M. Ganzera, P. Vrabl, E. Worle, W. Burgstaller and H. Stuppner, Anal. Biochem., 2006, 359, 132–140 CrossRef CAS PubMed.
- C. F. Yeh and S. J. Jiang, Analyst, 2002, 127, 1324–1327 RSC.
- E. B. Liu and B. C. Xue, J. Pharm. Biomed. Anal., 2006, 41, 649–653 CrossRef CAS PubMed.
- I. Heisler, J. Keller, R. Tauber, M. Sutherland and H. Fuchs, Anal. Biochem., 2002, 302, 114–122 CrossRef CAS PubMed.
- C. E. Amri, M. H. Baron and M. C. Maurel, Spectrochim. Acta, Part A, 2003, 59, 2645–2654 CrossRef.
- F. Zhang, O. Kambara, K. Tominaga, J. Nishizawa, T. Sasaki, H. Wang and M. Hayashi, RSC Adv., 2014, 4, 269–278 RSC.
- J. Wang, Analytical Electrochemistry, Wiley-VCH, New York, 3th edn, 2006 Search PubMed.
- T. N. Narayanan, C. S. R. Vusa and S. Alwarappan, Nanotechnology, 2014, 25, 335702 CrossRef PubMed.
- C. S. R. Vusa, S. Berchmans and S. Alwarappan, RSC Adv., 2014, 4, 22470–22475 RSC.
- M. Mishra, S. Alwarappan, R. K. Joshi and T. Mohanty, J. Nanosci. Nanotechnol., 2013, 13, 4040–4044 CrossRef CAS PubMed.
- E. Palecek, Nature, 1960, 188, 656–657 CrossRef CAS PubMed.
- H. S. Wang, H. X. Ju and H. Y. Chen, Electroanalysis, 2001, 13, 1105–1109 CrossRef CAS.
- K. J. Huang, D. J. Niu, J. Y. Sun, C. H. Han, Z. W. Wu, Y. L. Li and X. Q. Xiong, Colloids Surf., B, 2011, 82, 543–549 CrossRef CAS PubMed.
- E. P. Randviir and C. E. Banks, RSC Adv., 2012, 2, 5800–5805 RSC.
- K. Barman and Sk. Jasimuddin, RSC Adv., 2014, 4, 49819–49826 RSC.
- A. A. Ensafi, M. Mokhtari Abarghoui and B. Rezaei, Sens. Actuators, B, 2014, 204, 528–535 CrossRef CAS PubMed.
- A. A. Ensafi, M. Jafari-Asl, B. Rezaei and A. R. Allafchian, Sens. Actuators, B, 2013, 177, 634–642 CrossRef CAS PubMed.
- V. Raj, J. Silambarasan and P. Rajakumar, RSC Adv., 2014, 4, 33874–33882 RSC.
- H. Sharma, N. Singh and D. O. Jang, RSC Adv., 2015, 5, 6962–6969 RSC.
- M. Mazloum-Ardakani, H. Beitollahi, M. K. Amini, B. B. F. Mirjalili and F. Mirkhalaf, J. Electroanal. Chem., 2011, 651, 243–249 CrossRef CAS PubMed.
- H. Karimi-Maleh, M. Keyvanfard, K. Alizad, M. Fouladgar, H. Beitollahi, A. Mokhtari and F. Gholami-Orimi, Int. J. Electrochem. Sci., 2011, 6, 6141–6150 CAS.
- M. Mazloum-Ardakani, B. Ganjipour, H. Beitollahi, M. K. Amini, F. Mirkhalaf, H. Naeimi and M. Nejati-Barzoki, Electrochim. Acta, 2011, 56, 9113–9120 CrossRef CAS PubMed.
- H. Beitollahi, A. Mohadesi, S. Mohammadi, A. Pahlavan, H. Karimi-Maleh and A. Akbari, J. Mol. Liq., 2012, 169, 130–135 CrossRef CAS PubMed.
- H. Karimi-Maleh, M. Keyvanfard, K. Alizad, V. Khosravi and M. Asnaashari isfahani, Int. J. Electrochem. Sci., 2012, 7, 6816–6830 CAS.
- T. Tavana, M. A. Khalilzadeh, H. Karimi-Maleh, A. A. Ensafi, H. Beitollahi and D. Zareyee, J. Mol. Liq., 2012, 168, 69–74 CrossRef CAS PubMed.
- S. Alwarappan, S. Prabhulkar, A. Durygin and C. Z. Li, J. Nanosci. Nanotechnol., 2009, 9, 2991–2996 CrossRef CAS PubMed.
- F. Mirrahimi, M. A. Taher, H. Beitollahi and R. Hosseinzadeh, Appl. Organomet. Chem., 2012, 26, 194–198 CrossRef CAS PubMed.
- M. Mazloum-Ardakani, H. Beitollahi, M. K. Amini, F. Mirkhalaf and B. B. F. Mirjalili, Biosens. Bioelectron., 2011, 26, 2102–2106 CrossRef CAS PubMed.
- M. Mazloum-Ardakani, H. Beitollahi, M. A. Sheikh-Mohseni, H. Naeimi and N. Taghavinia, Appl. Catal., A, 2010, 378, 195–201 CrossRef CAS PubMed.
- Z. Taleat, M. Mazloum Ardakani, H. Naeimi, H. Beitollahi, M. Nejati and H. R. Zare, Anal. Sci., 2008, 24, 1039–1044 CrossRef CAS.
- H. Beitollahi, M. Mazloum Ardakani, B. Ganjipour and H. Naeimi, Biosens. Bioelectron., 2008, 24, 362–368 CrossRef CAS PubMed.
- B. Sels, D. de Vos, M. Buntinx, F. Pierard, A. K. Mesmaeker and P. Jacobs, Nature, 1999, 400, 855–857 CrossRef CAS.
- F. Li, Q. Tan, D. G. Evans and X. Duan, Catal. Lett., 2005, 99, 151–156 CrossRef CAS.
- D. L. Bish, Bull. Mineral., 1980, 103, 170–175 CAS.
- P. C. Pavan, G. A. Gomes and J. B. Valim, Microporous Mesoporous Mater., 1998, 21, 659–665 CrossRef CAS.
- A. Sugimoto, S. Ishida and K. Hanawa, J. Electrochem. Soc., 1999, 146, 1251–1255 CrossRef CAS PubMed.
- C. Gerardin, D. Kostadinova, N. Sanson, B. Coq and D. Tichit, Chem. Mater., 2005, 17, 6473–6478 CrossRef CAS.
- Z. Liu, R. Ma, M. Osada, N. Iyi, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2006, 128, 4872–4880 CrossRef CAS PubMed.
- J. H. Choy, S. Y. Kwak, Y. J. Jeong and J. S. Park, Angew. Chem., Int. Ed., 2000, 39, 4042–4045 CrossRef CAS.
- L. Desigaux, M. B. Belkacem, P. Richard, J. Cellier, P. Leone, L. Cario, F. Leroux, C. Taviot-Gueho and B. Pitard, Nano Lett., 2006, 6, 199–204 CrossRef CAS PubMed.
- W. Hummers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- R. N. Adams, Electrochemistry at Solid Electrodes, Marcel-Dekker, New York, 1969 Search PubMed.
- J. Marmur, R. Rownd and C. L. Schildkraut, Progress in Nucleic Acid Research, Academic Press, New York, 1963, p. 232 Search PubMed.
- A. Reyhani, S. Z. Mortazavi, S. Mirershadi, A. Z. Moshfegh, P. Parvin and A. N. Golikand, J. Phys. Chem. C, 2011, 115, 6994–7001 CAS.
- E. Laviron, J. Electroanal. Chem., 1974, 52, 355–393 CrossRef CAS.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson Education Limited, 5th edn, 2005 Search PubMed.
- L. Svorce and K. Klacher, Sens. Actuators, B, 2014, 194, 332–342 CrossRef PubMed.
- Z. H. Wang, S. F. Xiao and Y. Chen, J. Electroanal. Chem., 2006, 589, 237–242 CrossRef CAS PubMed.
- C. Li, X. Qiu and Y. Ling, J. Electroanal. Chem., 2013, 704, 44–49 CrossRef CAS PubMed.
- M. U. Prathap, R. Srivastava and B. Satpati, Electrochim. Acta, 2013, 114, 285–295 CrossRef PubMed.
- C. Tang, U. Yogeswaran and Sh. Chen, Anal. Chim. Acta, 2009, 636, 19–27 CrossRef CAS PubMed.
- H. Yin, Y. Zhou, Q. Ma, Sh. Ai, P. Ju, L. Zhu and L. Lu, Process Biochem., 2010, 45, 1707–1712 CrossRef CAS PubMed.
- A. Ferancová, S. Rengaraj, Y. Kim, J. Labuda and M. Sillanpää, Biosens. Bioelectron., 2010, 26, 314–320 CrossRef PubMed.
- N. Davidson, The Biochemistry of the Nucleic Acids, Cox & Nyman, Norfolk, UK, 7th edn, 1972 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |