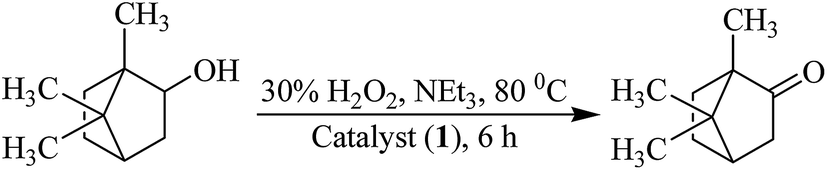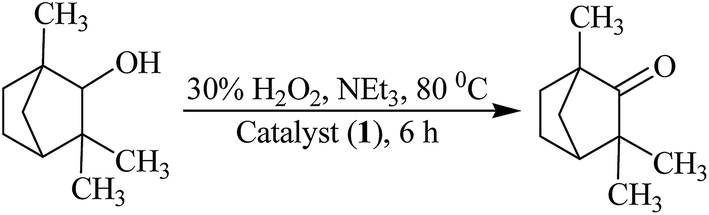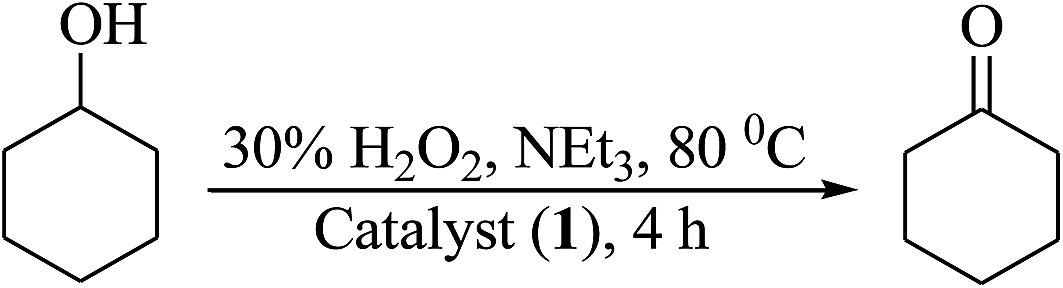DOI:
10.1039/C5RA16490G
(Paper)
RSC Adv., 2015,
5, 101076-101088
Effect of N-based additive on the optimization of liquid phase oxidation of bicyclic, cyclic and aromatic alcohols catalyzed by dioxidomolybdenum(VI) and oxidoperoxidomolybdenum(VI) complexes†
Received
16th August 2015
, Accepted 16th November 2015
First published on 17th November 2015
Abstract
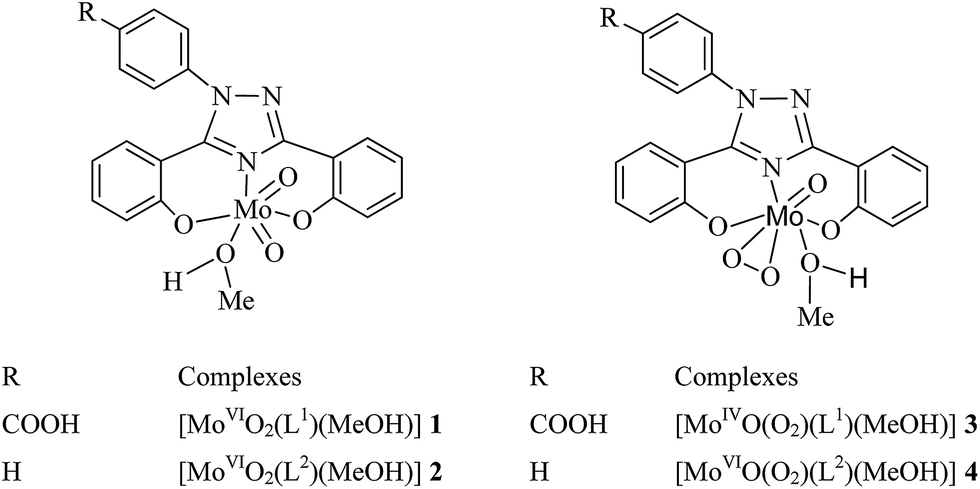
Two dioxidomolybdenum(VI) complexes, [MoVIO2(L1)(MeOH)] (1) and [MoVIO2(L2)(MeOH)] (2) and their corresponding oxidoperoxidomolybdenum(VI) complexes, [MoVIO(O2)(L1)(MeOH)] (3) and [MoVIO(O2)(L2)(MeOH)] (4) with ONO tridentate ligands, 4-[3,5-bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]benzoic acid (H2L1, I) and 3,5-bis(2-hydroxyphenyl)-1-phenyl-1,2,4-triazole (H2L2, II) have been synthesized and characterized by elemental analysis, spectroscopic techniques (infrared, UV-Vis, 1H and 13C NMR) and thermogravimetric analysis. Structures of 1a (DMSO coordinated) and 2 (methanol coordinated) confirmed by single crystal X-ray study reveal that the tridentate ligands bind to the metal center through two oxygen atoms and a ring nitrogen atom. These complexes have been tested as catalysts for the homogeneous oxidation of bicyclic (isoborneol and fenchyl alcohol), aromatic (benzyl alcohol and cumic alcohol) and cyclic (cyclohexanol) alcohols, using 30% H2O2 as an oxidant. Various parameters such as amounts of catalyst, oxidant, solvent and temperature of the reaction mixture have been taken into consideration for the maximum conversion of substrates. Effect of N-based additive (NEt3) on the conversion of substrates as well as selectivity of the corresponding product(s) under the optimized reaction conditions has also been checked and obtained results suggest that addition of an additive reduces time to achieve equilibrium and increases conversion of alcohols.
Introduction
Catalytic oxidation has been identified as a most efficient method to generate products that are commercially valuable.1 Catalytic oxidation of alcohols to corresponding carbonyl compounds is of great interest for both laboratory and synthetic industrial diligences.2 However, developing green oxidation process of alcohols is still a challenging task in catalysis.3 Homogeneous catalysts are often much active and selective, producing excellent yields, from economic and environmental point of view.4 The development of transition metal complexes as catalysts for oxidation reactions,5–8 in general, and less toxic dioxidomolybdenum(VI) complexes, in particular, are under extensive study.9 Efficient and selective catalytic systems for oxidation of isoborneol to camphor and fenchyl alcohol to fenchone are continuously sought after.10 Further, fenchone is a constituent of absinthe and the essential oil of fennel. Fenchone is also used as a flavor in foods and in perfumery. Oxidation of cyclohexanol is significant as it renders intermediate that has usages as plasticizers, food additives and solvent system in coating industry.11,12 Cyclohexanone was primarily used in the production of precursors for Nylon 6 and Nylon 6,6.11 On the other hand oxidation of aromatic alcohols such as benzyl alcohol and cumic alcohol give way to products that have both commercial and biological implications. Benzaldehyde and benzoic acid, distinctive products obtained by oxidation of benzyl alcohol, have crucial role as intermediates in fragrances, perfumes, flame retardants and pharmaceutical industry.13 Cuminaldehyde, an oxidation product of cumic alcohols, due to its very pleasant smell, find applications in perfume and other cosmetic industry. In biological prospect, cuminaldehyde, as a small molecule, inhibits the fibrillation of alpha-synuclein and thus controls the progress of parkinson's disease, dementia and multiple system atrophy.
Recently, we prepared homogeneous as well as heterogeneous catalysts based on dioxidomolybdenum(VI) complexes and tested them for the oxidative bromination of salicylaldehyde, styrene, trans-stilbene and thymol, and oxidation of methyl phenyl sulfide, styrene, cyclohexene, benzoin and different secondary alcohols.14 Considering the catalytic potential of such complexes and in search of catalytically active new molybdenum complexes, we have synthesized two dioxido- and two oxidoperoxidomolybdenum(VI) complexes employing dibasic tridentate ONO donor ligand systems; Scheme 1. These complexes have been tested as catalysts for the homogenous oxidation of bicyclic (isoborneol and fenchyl alcohol), aromatic (benzyl alcohol and cumic alcohol) and cyclic (cyclohexanol) alcohols under atmospheric conditions. Various parameters such as amount of catalyst, oxidant, solvent and temperature of the reaction mixture have been taken into consideration for the maximum conversion of substrates. Addition of N-based additive to the reaction mixture reduces time considerably and increases conversion of secondary alcohols.
 |
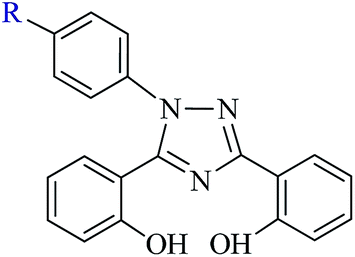
| | Scheme 1 Ligands, 4-[3,5-bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]benzoic acid (R = COOH: H2L1, I) and 3,5-bis(2-hydroxyphenyl)-1-phenyl-1,2,4-triazole (R = H: H2L2, II) used in this study. | |
Experimental
Material
Salicylic acid, salicylamide, 4-hydrazino-benzoic acid, phenylhydrazine hydrochloride (Aldrich, USA), ammonium molybdate tetrahydrate (Loba Chemie, India), thionyl chloride (SRL, India) and 30% H2O2 (Rankem, India) were used as obtained. Catalytic substrates i.e. isoborneol (1,7,7-trimethylbicyclo[2.2.1]heptan-2-ol), fenchyl alcohol (1,3,3-trimethylbicyclo[2.2.1]heptan-2-ol) and cumic alcohol (4-isopropylphenyl)methanol were purchased from Aldrich, USA. Benzyl alcohol (phenylmethanol) and cyclohexanol were incurred from Qualigens, India and used as such without further purification. Ethanol (reagent grade) was purified by distillation prior to use. Other solvents used were of analytical reagent (AR) grade. [MoVIO2(acac)2],15 4-[3,5-bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]benzoic acid (H2L1, I) and 3,5-bis(2-hydroxyphenyl)-1-phenyl-1,2,4-triazole (H2L2, II)16 were synthesized according to the methods reported in literature.
Instrumentation and characterization procedures
Elemental analysis (C, H and N) of the ligands and complexes were carried out on Elementar Analyser Vario-El-III. Thermogravimetric analysis (TGA) of the complexes was carried out using TG Stanton Redcroft STA 780. Infrared spectra were recorded as KBr pellets on a Nicolet 1100 FT-IR spectrometer after grinding the sample with KBr. UV-Vis spectra of ligands and complexes were recorded on a Shimadzu 1601 single beam spectrophotometer in methanol. 1H NMR spectra was obtained in DMSO-d6 on a Bruker Avance 400 MHz spectrometer. Electrochemical experiment has been carried out in a classic three-electrode cell having platinum disc working electrode, platinum wire as counter electrode, and Ag/AgCl as reference electrode on CHI760 electrochemical workstation. Tetra-n-butylammonium hexafluorophosphate (0.1 M) was used as supporting electrolyte. Redox analysis of the compounds was carried out with scan rate 0.1 V s−1 in dry and degassed DMF with no trace of decomposition as reflected in smooth curve. The solutions were purged with N2 for ca. 15 min before conducting experimental analysis in order to remove the dissolved O2. The oxidation products were analyzed with a Shimadzu 2010 plus gas-chromatograph fitted with an Rtx-1 capillary column (30 m × 0.25 mm × 0.25 μm) and a FID detector and the identity of the products confirmed using the GC-MS Shimadzu QP-5000.
X-ray crystal structure determination
Three-dimensional X-ray data were collected on a Bruker Kappa Apex CCD diffractometer at room temperature for 1a and low temperature for 2 by the ϕ–ω scan method. Reflections were measured from a hemisphere of data collected from frames, each of them covering 0.3° in ω. A total of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 754 and 56836 reflections measured, all were corrected for Lorentz and polarization effects and for absorption by multi-scan methods based on symmetry-equivalent and repeated reflections. Of them, 5145 for 1a and 7391 for 2 independent reflections exceeded the significance level (∣F∣/σ∣F∣) > 4.0. After data collection, a multi-scan absorption correction (SADABS)17 was applied, and the structure was solved by direct methods and refined by full matrix least-squares on F2 data using SHELX suite of programs.18 Hydrogen atoms were located in a difference Fourier map and left to refine freely, except for C(24) in 1a and for C(1M) and C(2M) in 2, which were included in calculated positions and refined in the riding mode. Hydrogen atoms of C(1S) and C(2S) in compound 1a were located in a difference Fourier map and fixed to the carbon atoms. Refinements were done with allowance for thermal anisotropy of all non-hydrogen atoms. A final difference Fourier map showed no residual density: 0.787 and −0.362 e Å−3 for 1a and 0.702 and −0.828 e Å−3 for 2. A weighting scheme w = 1/[σ2(Fo2) + (0.039600P)2 + 0.498600P] for 1a and w = 1/[σ2(Fo2) + (0.033600P)2 + 0.322500P] for 2, where P = (|Fo|2 + 2|Fc|2)/3, were used in the latter stages of refinement. The crystal of 1a presents important disorders on DMSO molecules. These disorders were resolved and the atomic sites were observed and refined with anisotropic atomic displacement parameters. More specifically these disorders were refined using 75 restraints (SADI, SIMU and DELU restraints were used). The site occupancy factors were 0.91517 for S(1A) and 0.22651 for S(2A). Further details of the crystal structure determination are given in Table 1.
754 and 56836 reflections measured, all were corrected for Lorentz and polarization effects and for absorption by multi-scan methods based on symmetry-equivalent and repeated reflections. Of them, 5145 for 1a and 7391 for 2 independent reflections exceeded the significance level (∣F∣/σ∣F∣) > 4.0. After data collection, a multi-scan absorption correction (SADABS)17 was applied, and the structure was solved by direct methods and refined by full matrix least-squares on F2 data using SHELX suite of programs.18 Hydrogen atoms were located in a difference Fourier map and left to refine freely, except for C(24) in 1a and for C(1M) and C(2M) in 2, which were included in calculated positions and refined in the riding mode. Hydrogen atoms of C(1S) and C(2S) in compound 1a were located in a difference Fourier map and fixed to the carbon atoms. Refinements were done with allowance for thermal anisotropy of all non-hydrogen atoms. A final difference Fourier map showed no residual density: 0.787 and −0.362 e Å−3 for 1a and 0.702 and −0.828 e Å−3 for 2. A weighting scheme w = 1/[σ2(Fo2) + (0.039600P)2 + 0.498600P] for 1a and w = 1/[σ2(Fo2) + (0.033600P)2 + 0.322500P] for 2, where P = (|Fo|2 + 2|Fc|2)/3, were used in the latter stages of refinement. The crystal of 1a presents important disorders on DMSO molecules. These disorders were resolved and the atomic sites were observed and refined with anisotropic atomic displacement parameters. More specifically these disorders were refined using 75 restraints (SADI, SIMU and DELU restraints were used). The site occupancy factors were 0.91517 for S(1A) and 0.22651 for S(2A). Further details of the crystal structure determination are given in Table 1.
Table 1 Crystal data and structure refinement for [MoVIO2(L1)(DMSO)]·DMSO 1a and for [MoVIO2(L2)(MeOH)]·MeOH 2
| |
1a |
2 |
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = {∑[w(||Fo|2 − |Fc|2|)2]|/∑[w(Fo2)2]}1/2. |
| Formula |
C26H27MoN3O8S2 |
C22H21MoN3O6 |
| Formula weight |
669.57 |
519.36 |
| T, K |
293(2) |
100(2) |
| Wavelength, Å |
0.71073 |
0.71073 |
| Crystal system |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å |
11.771(2) |
7.7600(3) |
| b/Å |
11.984(2) |
9.9273(4) |
| c/Å |
12.499(2) |
14.1328(6) |
| α/° |
89.199(5) |
90.030(2) |
| β/° |
69.782(4) |
100.548(2) |
| γ/° |
61.016(4) |
99.031(2) |
| V/Å3 |
1421.8(4) |
1056.59(7) |
| Z |
2 |
2 |
| F000 |
684 |
528 |
| Dcalc/g cm−3 |
1.564 |
1.632 |
| μ/mm−1 |
0.661 |
0.666 |
| θ/(°) |
2.19 to 27.89 |
1.47 to 34.79 |
| Rint |
0.0327 |
0.0678 |
| Crystal size/mm3 |
0.31 × 0.21 × 0.09 |
0.34 × 0.22 × 0.12 |
| Goodness-of-fit on F2 |
1.035 |
1.061 |
| R1[I > 2σ(I)]a |
0.0377 |
0.0362 |
| wR2 (all data)b |
0.0913 |
0.0829 |
| Largest differences peak and hole (e Å−3) |
0.787 and −0.362 |
0.702 and −0.828 |
Preparations
[MoVIO2(L1)(MeOH)] 1. Ligand H2L1 (1.865 g, 0.005 mol) was dissolved in 50 mL of methanol. A methanolic solution (30 mL) of [MoVIO2(acac)2] (1.63 g, 0.005 mol) was added drop wise to the above solution with stirring and the final reaction mixture was refluxed for 2 h on a water bath. The obtained yellow colored solution was concentrated on a water bath to 20 mL and kept at room temperature for overnight. The separated crystalline yellow 1 was filtered, washed with cold methanol and dried in vacuum desiccator over silica gel. Yield: 2.15 g (81%). (Found: C, 46.79; H, 3.31; N, 7.54%. Calc'd. for C22H17N3O7Mo (533.01): C, 48.73; H, 3.22; N, 7.91%). Selected IR bands (KBr, νmax/cm−1): 3427 (OH), 1711 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1604 (C–C), 1573 (CN), 1527 (CC), 1310 (C–O), 1266 (C–N), 1038 (NN), 937 (OMoOantisym), 906 (OMoOsym), 754 (C–H). X-ray diffraction quality crystals for [MoVIO2(L1)(DMSO)]·DMSO 1a were obtained from its DMSO solution at room temperature.
O), 1604 (C–C), 1573 (CN), 1527 (CC), 1310 (C–O), 1266 (C–N), 1038 (NN), 937 (OMoOantisym), 906 (OMoOsym), 754 (C–H). X-ray diffraction quality crystals for [MoVIO2(L1)(DMSO)]·DMSO 1a were obtained from its DMSO solution at room temperature.
[MoVIO2(L2)(MeOH)] 2. Complex 2 was prepared analogously to 1 considering ligand II (1.645 g, 0.005 mol). Yield: 2.03 g (83%). (Found: C, 50.99; H, 3.62; N, 8.39%. Calc'd. for C21H17MoN3O5 (489.02): C, 51.76; H, 3.52; N, 8.62%). Selected IR bands (KBr, νmax/cm−1): 3435 (OH), 1601 (C–C), 1574 (CN), 1524 (CC), 1306 (C–O), 1265 (C–N), 1000 (NN), 924 (OMoOantisym), 904 (OMoOsym), 753 (C–H). X-ray diffraction quality crystals for 2 were obtained by slow evaporation of its methanolic solution in air.
[MoVIO(O2)(L1)(MeOH)] 3. The oxidoperoxidomolybdenum(VI) precursor was prepared in situ by stirring MoO3 (0.720 g, 0.005 mol) in 10 mL of 30% H2O2 for 4 h at room temperature and filtered. This was added drop wise to a filtered solution of H2L1 (I, 1.865 g, 0.005 mol) dissolved in methanol (50 mL) with slow stirring. The stirring was continued for additional 6 h. Reaction mixture was then aerated to reduce the volume to ca. 15 mL and simultaneously light yellow solid separated out. This was filtered off, washed with cold methanol and dried in vacuum desiccator over silica gel. Yield: 2.10 g (77%). (Found: C, 47.01; H, 2.89; N, 7.55%. Calc'd. for C22H17MoN3O8 (547.33): C, 48.28; H, 3.13; N, 7.68%). Selected IR bands (KBr, νmax/cm−1): 3255 (OH), 1716 (CO), 1608 (C–C), 1588 (CN), 1515 (CC), 1325 (C–O), 1290 (C–N), 1049 (NN), 949 (MoO), 853 (O–O), 754 (C–H), 624, 568 (Mo(O2)antisym and sym).
[MoVIO(O2)(L2)(MeOH)] 4. Complex 4 was prepared similarly as outlined for 3 using 1.645 g (0.005 mol) of II. Yield: 1.99 g (79%). (Found: C, 49.51; H, 3.23; N, 7.95%. Calc'd for C21H17MoN3O6 (503.32): C, 50.11; H, 3.40; N, 8.35%). Selected IR bands (KBr, νmax/cm−1): 3261 (OH), 1622 (C–C), 1590 (CN), 1497 (CC), 1359 (C–O), 1294 (C–N), 1029 (NN), 936 (MoO), 855 (O–O), 749 (C–H), 616, 587 (Mo(O2)antisym and sym).
Catalytic activity study-oxidation of alcohols
Conventional liquid phase method was employed for the oxidation of bicyclic, aromatic and cyclic alcohols. Catalytic reactions were carried out in a 50 mL round-bottom flask equipped with a reflux condenser. Under typical conditions, a relevant alcohol (10 mmol), aqueous 30% H2O2 (1.69 g, 15 mmol) and catalyst (0.002 g) were dissolved in acetonitrile (5 mL). Oxidation reactions were carried out both in the absence or presence of N-based additive i.e. NEt3 (0.05 mmol) at 80 °C under magnetic stirring for a particular time depending upon the substrates. The reaction was monitored by withdrawing small aliquots of the reaction mixture at definite time interval, extracting with hexane and analyzing quantitatively by gas chromatograph. The identities of the products were confirmed by GC-MS.
Results and discussion
Synthesis and characterization of complexes
The reaction between equimolar amounts of [MoVIO2(acac)2] and H2L1 I or H2L2 II in refluxing methanol leads to the formation of [MoVIO2]2+ complexes [MoVIO2(L1)(MeOH)] 1 and [MoVIO2(L2)(MeOH)] 2, respectively (eqn (1) considering I as a representative example). The in situ generated oxidoperoxidomolybdenum(VI) species, by reacting MoO3 with H2O2, also reacts with ligands I and II in methanol giving corresponding [MoVIO(O2)]2+ complexes, [MoVIO(O2)(L1)(MeOH)] 3 and [MoVIO(O2)(L2)(MeOH)] 4, respectively (eqn (2) considering I as representative example).| | |
[MoVIO2(acac)2] + H2L1 + MeOH → [MoO2(L1)(MeOH)] + 2Hacac
| (1) |
| | |
[MoVIO(O2)(H2O)n]2+ + H2L1 + MeOH → [MoVIO(O2)(L1)(MeOH)] + nH2O
| (2) |
Scheme 2 provides their possible structures which are based on the spectroscopic (IR, UV/Vis, 1H and 13C NMR) data, thermogravimetric and elemental analysis, and X-ray diffraction study of 1a and 2. All these complexes exist as monomer and are soluble in methanol, ethanol, DMF and DMSO.
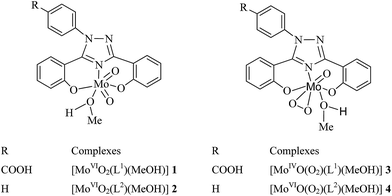 |
| | Scheme 2 Proposed structures of [MoO2]2+ and [MoO(O2)]2+ complexes. | |
Thermal study
The thermal stability of [MoO2]2+ and [MoO(O2)]2+complexes has been studied under an oxygen atmosphere. Both types of complexes are thermally stable at least up to ca. 135 °C and there after lose weight roughly equal to one methanol molecule in the temperature range 135–230 °C. In [MoO(O2)]2+ complexes, the observed weight loss is slightly more than calculated for methanol in this temperature range suggesting the decomposition of peroxido moiety partly as well along with the loss of methanol. In second step all complexes decompose in two or three overlapping steps and convert into MoO3. Detail of this study is presented in Table S1.†
Structure description
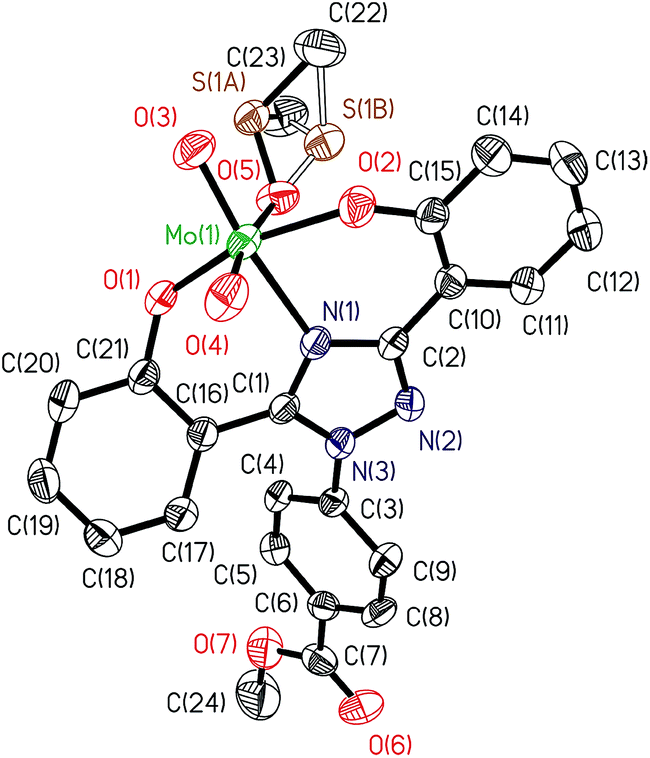
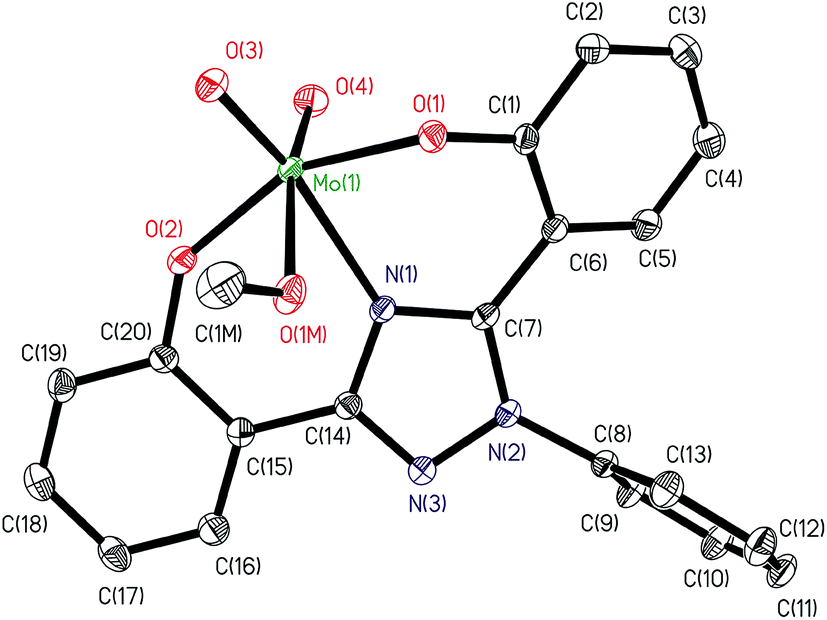
ORTEP diagram of [MoVIO2(L1)(DMSO)]·DMSO 1a and [MoVIO2(L2)(MeOH)]·MeOH 2 are shown in Fig. 1 and 2. Fig. 3 presents the interactions between the methanol molecules and the complexes in the crystal packing through the hydrogen bonds. Selected bond distances and angles are given in Table 2. Both of complexes adopt a six-coordinated structure around Mo atom in a distorted octahedral geometry. The coordination sphere is formed by the phenolic oxygen atoms and one of the triazole nitrogen atoms of the ligand. One DMSO molecule in 1a and one methanol molecule in 2 and two terminal oxido groups complete the coordination sphere. The OMoO angle are 105.24(10)° in 1a and 105.48(6)° in 2 and MoO distances are: 1.6992(18) Å and 1.695(2) Å in 1a, and 1.6998(12) Å and 1.7070(12) Å in 2. The Mo–ODMSO distance is 2.2869(19) Å in 1a. The Mo–O–C angle and Mo–O distance for Mo–OHCH3 in 2 are 124.85(11)° and 2.2831(12) Å, respectively, similar to other examples in the literature.14a,19 Other DMSO molecule in 1a and other methanol molecule in 2 are presents in the asymmetric unit.
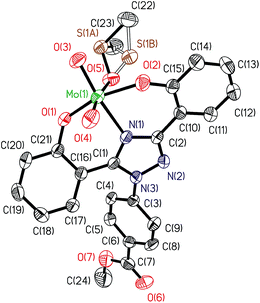 |
| | Fig. 1 ORTEP plot of complex in [MoVIO2(L1)(DMSO)]·DMSO 1a. All the non-hydrogen atoms are presented by their 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. | |
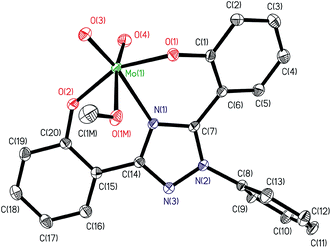 |
| | Fig. 2 ORTEP plot of complex in [MoVIO2(L2)(MeOH)]·MeOH 2. All the non-hydrogen atoms are presented by their 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. | |
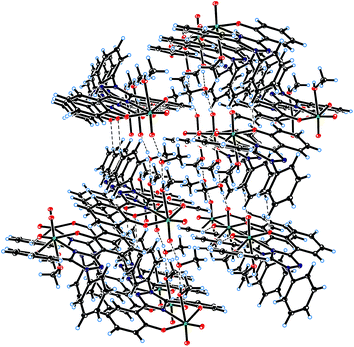 |
| | Fig. 3 Crystal packing in the complex [MoVIO2(L2)(MeOH)]·MeOH 2. Intermolecular hydrogen bonds between O(1M) and O(2M) of methanol molecules and with O(4) of coordinated oxido groups are shown in dashed lines. | |
Table 2 Bond lengths [Å] and angles [°] for [MoVIO2(L1)(DMSO)]·DMSO 1a and for [MoVIO2(L2)(MeOH)]·MeOH 2
| Bond lengths |
1a |
2 |
| Mo(1)–O(1) |
1.9616(18) |
1.9489(12) |
| Mo(1)–O(2) |
1.9473(18) |
1.9242(12) |
| Mo(1)–O(3) |
1.6992(18) |
1.6998(12) |
| Mo(1)–O(4) |
1.695(2) |
1.7070(12) |
| Mo(1)–N(1) |
2.259(2) |
2.2483(13) |
| Mo(1)–O(5) |
2.2869(19) |
Mo(1)–O(1M) 2.2831(12) |
| Bond angles |
1a |
2 |
| O(3)–Mo(1)–O(4) |
105.24(10) |
105.48(6) |
| O(3)–Mo(1)–O(2) |
99.29(9) |
98.06(6) |
| O(4)–Mo(1)–O(2) |
97.78(9) |
99.48(6) |
| O(3)–Mo(1)–O(1) |
98.37(8) |
97.83(6) |
| O(4)–Mo(1)–O(1) |
95.01(9) |
95.11(6) |
| O(2)–Mo(1)–O(1) |
154.61(8) |
154.69(5) |
| O(3)–Mo(1)–N(1) |
161.67(9) |
158.74(6) |
| O(4)–Mo(1)–N(1) |
93.07(9) |
95.76(6) |
| O(2)–Mo(1)–N(1) |
78.63(7) |
79.04(5) |
| O(1)–Mo(1)–N(1) |
78.86(7) |
79.00(5) |
| O(3)–Mo(1)–O(5)/O(1M) |
86.69(9) |
85.48(5) |
| O(4)–Mo(1)–O(5)/O(1M) |
168.02(8) |
168.59(6) |
| O(2)–Mo(1)–O(5)/O(1M) |
81.07(8) |
81.77(5) |
| O(1)–Mo(1)–O(5)/O(1M) |
81.89(7) |
79.97(5) |
| N(1)–Mo(1)–O(5)/O(1M) |
74.99(7) |
73.26(5) |
| C(1M)–O(1M)–Mo(1) |
|
124.85(11) |
Steric requirements, derived from coordination sphere around Mo and the ligand structure, prevent π–π interactions between the phenol and phenyl rings in the two compounds. Hydrogen bonds between methanol molecules and terminal oxygen atom, O(4), of the complex determine the ordering of the structure in the crystal packing of 2 (see Fig. 3 and Table 3).
Table 3 Hydrogen bonds for [MoVIO2(L2)(MeOH)]·MeOH 2a
| D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
∠(DHA) |
| Symmetry transformations used to generate equivalent atoms: #1 x + 1, y, z. |
| O(1M)–H(1M)⋯O(2M) |
0.75(2) |
1.90(2) |
2.6489(19) |
172(3) |
| O(2M)–H(2M)⋯O(4)#1 |
0.70(3) |
2.05(3) |
2.7423(18) |
172(3) |
Spectroscopic study
The [MoVIO2]2+ complexes are dominated by two prominent peaks at 924–937 and 904–906 cm−1 which are assigned to νasym(OMoO) and νsym(OMoO) modes, respectively due to cis-[MoVIO2] structure.20 The ν(MoO) in [MoVIO(O2)]2+ complexes appears at 936–949 cm−1 along with three new peaks at 853–855 cm−1 due to ν(O–O), 616–624 cm−1 due to νasym[Mo(O2)] and at 568–587 cm−1 due to νsym[Mo(O2)] mode.
The band appearing at 1583 cm−1 (in I) or 1591 cm−1 (in II) due to ν(CN) stretch moves to lower wave numbers and appears at 1573–1588 cm−1 (in 1 and 3) or 1574–1590 cm−1 (in 2 and 4), suggesting the coordination of azomethine nitrogen to the molybdenum. This is further supported by the shift of the ν(N–N) stretch appearing at 1015 cm−1 (in I) and 991 cm−1 (in II) to higher wave number due to reduced repulsion between the lone pairs of adjacent nitrogen atoms. The coordination of phenolic oxygen could not be assigned unequivocally as all complexes also exhibit a broad band in the ca. 3400 cm−1 region due to coordinated methanol.
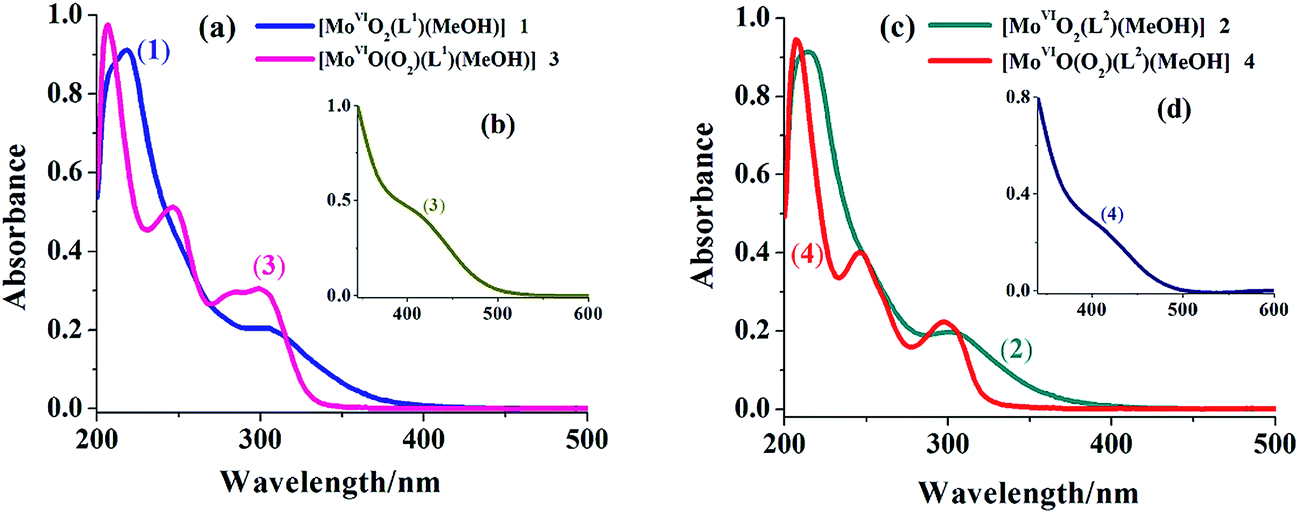
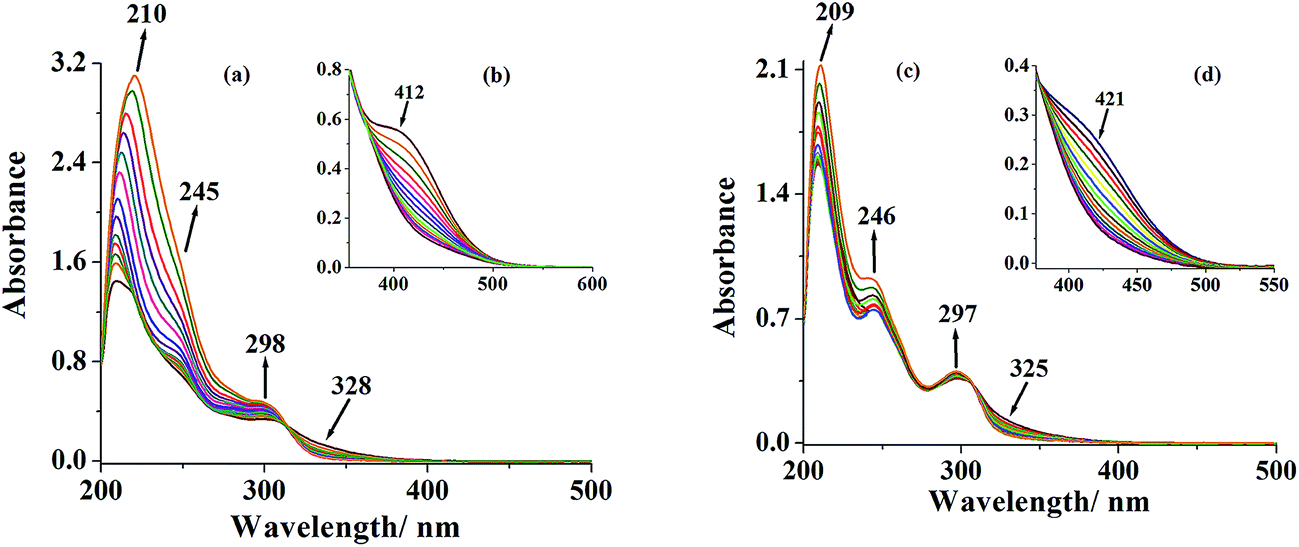
UV-vis spectral data of the triazole ring based ligands and their corresponding complexes are presented in Table 4 and spectra of complexes are presented in Fig. 4 (see Fig. S1† for spectra of ligands). Both ligand systems exhibit very similar three characteristic absorption bands at 207 (ε = 1.88 × 104), 249 (ε = 8.76 × 103) and 301 (ε = 4.87 × 103) (in I) and at 208 (ε = 1.97 × 104), 246 (ε = 8.40 × 103) and 296 (ε = 4.63 × 103) nm (in II). Based on the extinction co-efficient values, these characteristic peaks were designated as transition between electronic energy levels, φ → φ*, π → π* and n → π*, respectively. In complexes, these prominent bands are also observed with slight variations in their positions. In addition, a new band of medium intensity appears around 400 nm, which is assigned to a ligand to metal charge transfer (LMCT) band originating from electrons movement from filled p-orbital of ligand to the vacant d-orbital of the metal ion of proper symmetry.21
Table 4 UV-vis spectral data of ligands and complexes
| Compounds |
λmax/nm (ε/M−1cm−1) |
| H2L1 I |
301 (4.87 × 103), 249 (8.76 × 103), 207 (1.88 × 104) |
| H2L2 II |
296 (4.63 × 103), 246 (8.40 × 103), 208 (1.97 × 104) |
| [MoVIO2(L1)(MeOH)] 1 |
412 (2.24 × 103), 328 (9.65 × 103), 245 (2.06 × 104), 210 (4.42 × 104) |
| [MoVIO2(L2)(MeOH)] 2 |
421 (2.59 × 103), 325 (9.09 × 103), 246 (1.74 × 104), 209 (3.66 × 104) |
| [MoVIO(O2)(L1)(MeOH)] 3 |
407 (2.03 × 103), 299 (6.63 × 103), 245 (1.11 × 104), 206 (2.12 × 104) |
| [MoVIO(O2)(L2)(MeOH)] 4 |
415 (2.76 × 103), 294 (5.43 × 103), 246 (9.80 × 103), 207 (2.31 × 104) |
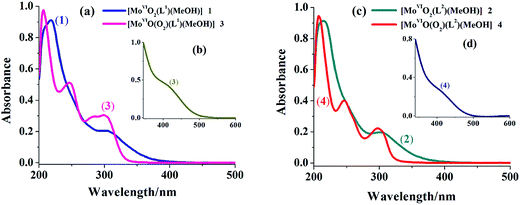 |
| | Fig. 4 (a) Electronic spectra of [MoVIO2]2+- and [MoVIO(O2)]2+-complexes with ligand (H2L1) recorded in methanol {conc. of complex 1:2.6 × 10−4 M and conc. of complex 3: 2.9 × 10−4 M}. (b) Inset shows LMCT band of complex 3 (conc. 5.7 × 10−5 M) recorded in methanol. (c) Electronic spectra of [MoVIO2]2+- and [MoVIO(O2)]2+-complexes with ligand (H2L2) recorded in methanol {conc. of complex 2: 3.4 × 10−4 M and conc. of complex 4: 2.7 × 10−4 M}. (d) Inset shows LMCT band of complex 4 (conc. 6.4 × 10−5 M) recorded in methanol. | |
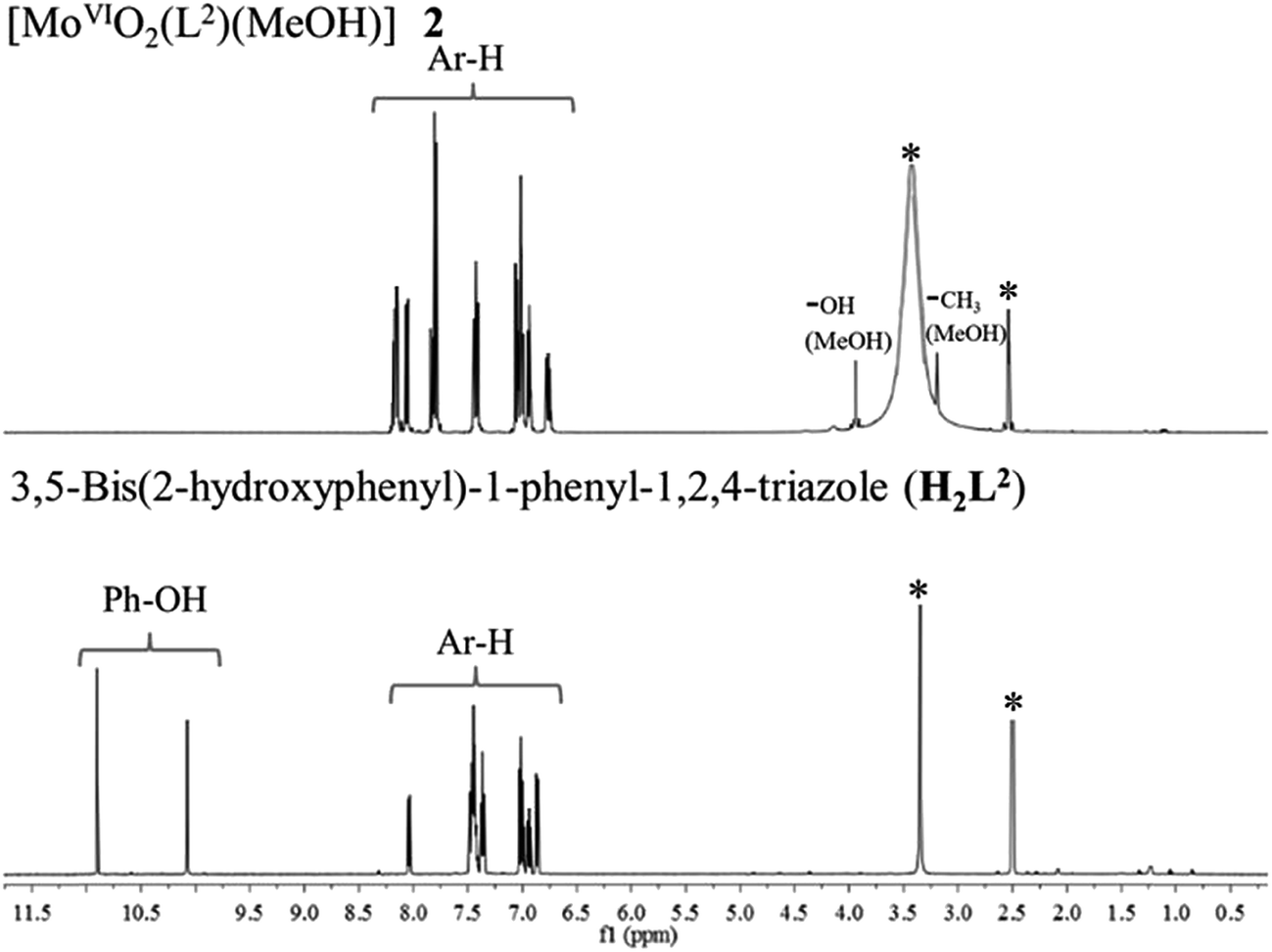
The 1H NMR spectra of the ligands (Table 5) exhibit two singlet at δ = 11.44 and 10.51 ppm (in I), and at δ = 10.90 and 10.07 ppm (in II) due to phenolic (–OH) protons. The absence of these signals in [MoVIO2]2+ complexes (Fig. 5 for representative spectra) are in agreement with the subsequent replacement of H by the metal ion. Thus, 1H NMR data supports the coordination of phenolic oxygen while IR spectral data confirms the coordination of ring nitrogen. The methyl and alcoholic protons of the coordinated methanol in complexes appear at δ = 3.21–3.23 ppm and at δ = 3.92–3.93 ppm, respectively. The characteristic broad peak for carboxylic proton was observed at δ = 12.00, 13.05 and 13.20 ppm in I, 1 and 3, respectively.
Table 5 1H NMR spectral data of ligands and complexesa
| Compounds |
–OH (phenolic) |
Aromatic protons |
| Letters given in parentheses indicate the signal structure: s = singlet, d = doublet, dd = doublet of doublet, m = multiplet and br = broad. |
| I |
11.44 (s, 1H), 10.51 (s, 1H) |
12.00 (br, 1H), 8.06–8.03 (d, 1H) (J = 12 Hz), 7.87–7.85 (d, 2H) 7.83–7.81 (t, 2H), 7.56–7.55 (d, 1H), 7.46–7.43 (t, 2H), 7.05–7.01 (m, 3H) (J = 12 Hz), 6.88–6.87 (d, 1H) |
| II |
10.90 (s,1H), 10.07 (s,1H) |
8.04–8.02 (dd, 1H) (J = 8 Hz), 7.48–7.42 (m, 6H) (J = 24 Hz), 7.37–7.34 (t, 2H), 7.02–6.98 (t, 2H), 6.95–6.92 (d, 1H), 6.87–6.85 (d, 1H) |
| 1 |
|
13.05 (br, 1H), 8.23–8.19 (d, 1H) (J = 16 Hz), 8.11–8.09 (d, 2H), 7.88–7.83 (t, 2H), 7.47–7.44 (t, 2H), 7.39–7.32 (m, 3H) (J = 28 Hz), 6.98–6.95 (d, 1H), 6.80–6.77 (d, 1H) |
| 2 |
|
8.23–8.18 (d, 2H) (J = 20 Hz), 8.12–8.09 (dd, 1H) (J = 12 Hz), 7.88–7.81 (m, 4H) (J = 28 Hz), 7.47–7.43 (t, 2H), 7.09–7.05 (t, 2H), 6.98–6.95 (d, 1H), 6.80–6.76 (d, 1H) |
| 3 |
|
13.20 (br, 1H), 8.15–8.11 (d, 1H) (J = 16 Hz), 8.02–7.98 (d, 2H), 7.89–7.84 (t, 2H), 7.51–7.54 (d, 1H), 7.41–7.36 (t, 2H), 7.03–6.97 (m, 3H) (J = 24 Hz), 6.86–6.85 (d, 1H) |
| 4 |
|
8.07–8.04 (dd, 1H) (J = 12 Hz), 7.51–7.43 (m, 6H) (J = 32 Hz), 7.40–7.37 (t, 2H), 7.05–7.01 (t, 2H), 6.97–6.94 (d, 1H), 6.85–6.84 (d, 1H) |
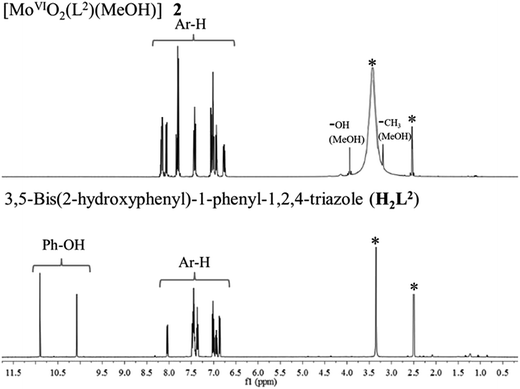 |
| | Fig. 5 1H NMR spectra of [MoVIO2(L2)(MeOH)] 2 and H2L2 II. Signal with star (*) is due to protons of water/methyl groups of DMSO. | |
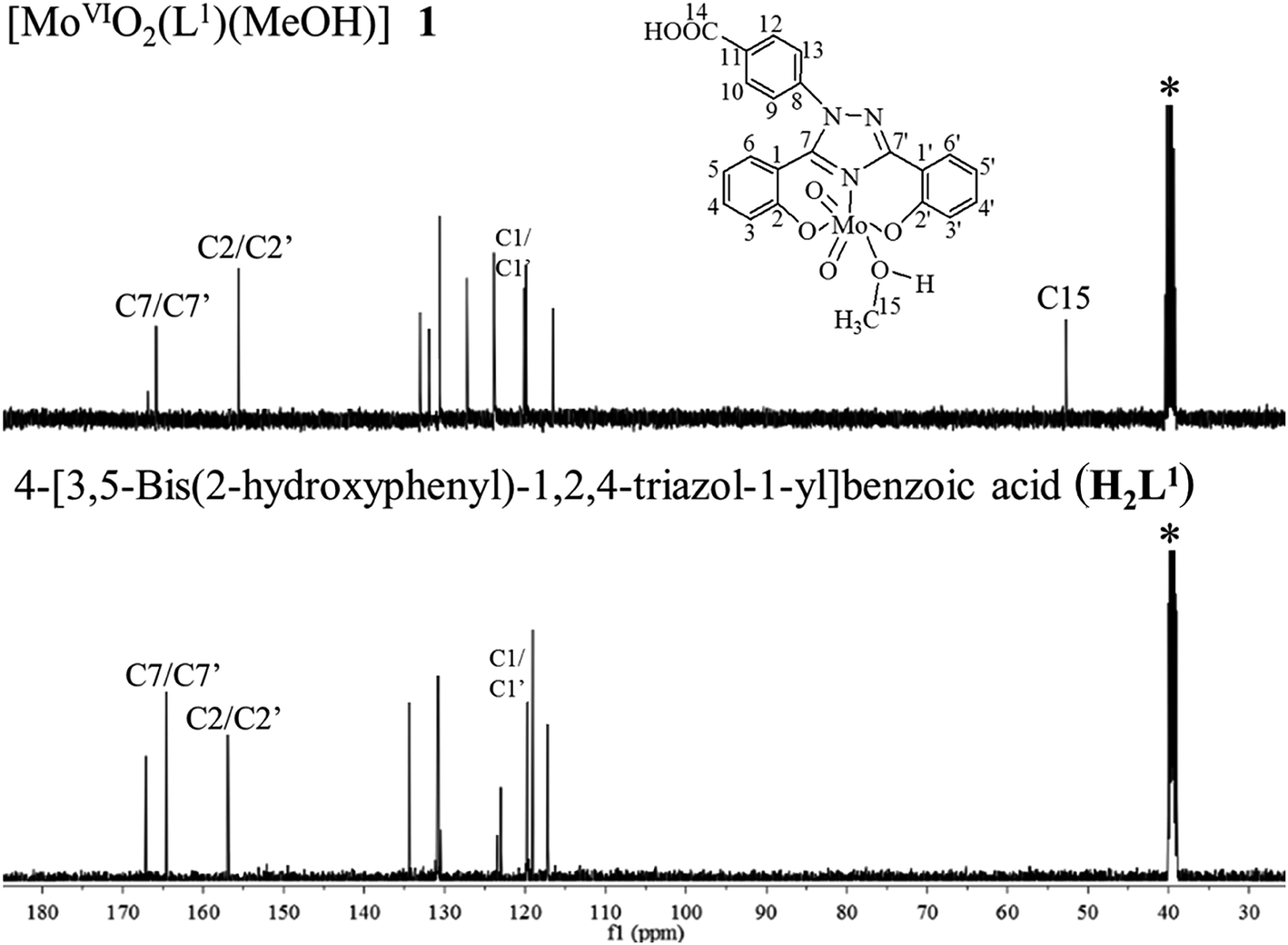
Comparison of the 13C NMR spectral data of ligand with the corresponding complexes also provides useful information for the elucidation of the structure of the complexes. Table 6 provides 13C NMR spectral data of ligands and their [MoVIO2]2+ and [MoVIO(O2)]2+ complexes; 13C NMR spectra of H2L1 and [MoVIO2(L1)(MeOH)] 1 are given in Fig. 6. Assignments of the peaks are based on the coordination-induced shifts [Δδ = δ(complex) – δ(free ligand)] of the signals for carbon atoms in the vicinity of the coordinating atoms22 and Chemdarw® program. Ligands I and II display 11 and 10 13C NMR signals corresponding to 21 and 20 carbon atoms, respectively. All expected signals are also present in complexes. A large coordination induced shift of the signals for the carbon atoms associated with phenolic oxygen (C2/C2′) and triazole nitrogen (C7/C7′) (see Table 6) confirms the coordination of these functionalities to the molybdenum. The signals for carbon atoms (C1/C1′) closure to the above carbons are only slightly affected. The signal due to methyl carbons (C14/C15) of coordinated methanol was observed at δ = 49.12–53.18 ppm in all complexes.
Table 6 13C NMR spectral data of ligands and complexes
| Compoundsa |
C15 |
C1/C1′ |
C2/C2′ |
C7/C7′ |
Ar-C |
| For numbering of carbons see Fig. 6. |
| H2L1 I |
|
119.74 |
156.93 |
164.55 |
167.09, 134.35, 130.77, 130.40, 123.43, 123.01, 119.05, 117.20 |
| [MoVIO2(L1)(MeOH)] 1 (Δδ) |
52.93 |
120.40 (0.66) |
155.30 (−1.63) |
165.61 (1.06) |
166.72, 133.66, 132.48, 130.78, 127.98, 123.61, 120.17, 116.93 |
| [MoVIO(O2)(L1)(MeOH)] 3 (Δδ) |
49.12 |
120.25 (0.51) |
155.23 (−1.70) |
165.76 (1.21) |
166.98, 133.13, 132.03, 130.73, 127.33, 123.98, 120.02, 116.65 |
| H2L2 II |
|
119.66 |
156.30 |
159.53 |
131.31, 129.19, 128.66, 126.62, 123.75, 119.23, 116.05 |
| [MoVIO2(L2)(MeOH)] 2 (Δδ) |
53.18 |
120.57 (0.91) |
154.43 (−1.87) |
160.62 (1.09) |
132.61, 131.65, 129.28, 127.20, 121.05, 120.36, 115.43 |
| [MoVIO(O2)(L2)(MeOH)] 4 (Δδ) |
51.74 |
120.39 (0.73) |
154.39 (−1.91) |
160.71 (1.18) |
132.82, 131.52, 129.71, 127.15, 121.28, 119.77, 115.08 |
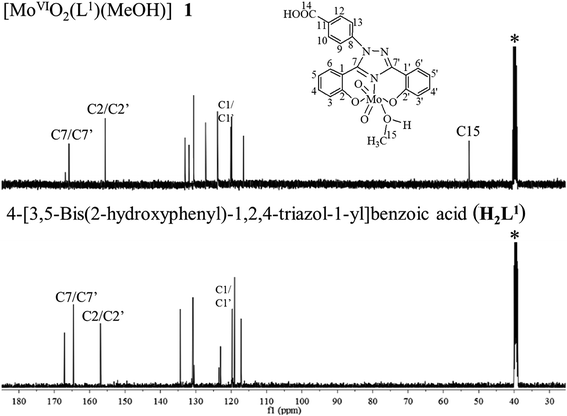 |
| | Fig. 6 13C NMR spectra of [MoVIO2(L1)(MeOH)] 1 and H2L1 I. Signal with star (*) is due to methyl carbon of DMSO. | |
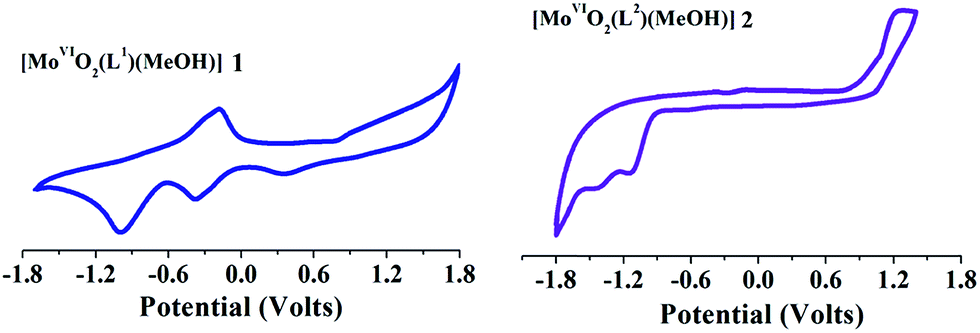
Electrochemical study
Cyclic voltammetry measurements were carried out to investigate the redox stability of complexes in solution state. Voltammograms of redox active [MoVIO2]2+- and [MoVIO(O2)]2+-complexes (1–4) were recorded in the range 1.8 V to −1.8 V vs. Ag/AgCl at room temperature. Relevant data are presented in Table 7 and representative cyclic voltammograms of [MoVIO2(L1)(MeOH)] 1 and [MoVIO2(L2)(MeOH)] 2 recorded in DMF at room temperature are presented in Fig. 7. The voltammograms of the complexes exhibit two reductive responses within the potential window −0.38 V to −1.68 V, which are ascribed to MoVI/MoV and MoV/MoIV processes, respectively.23–26 Reductive responses corresponding to MoVI/MoV and MoV/MoIV processes in the polar aprotic solvent are irreversible in nature.26 Both reduction processes may be exemplified as a metal-centered one-electron transfer involving the MoVI, MoV and MoIV oxidation states.26–28 The lack of anodic response, at a higher scan rate, is possibly due to rapid decomposition of the reduced species.23–26,28 The more negative the cathodic reduction potential, the more difficult for the [MoVIO2]2+ complexes to be reduced. The electron withdrawing group in the ligand leads to the shifting of the cathodic reduction potential in [MoVIO2]2+ complex towards less negative value.29
Table 7 Cyclic voltammetric results for dioxido- and oxidoperoxidomolybdenum(VI) complexes at 298 K
| Complexes |
Epca [V] |
| Solvent: DMF; working electrode: platinum disc; counter electrode: platinum wire; reference electrode: Ag/AgCl; supporting electrolyte: 0.1 M TBAP; scan rate: 0.1 V s−1, quiet time: 2 s, temperature: 298 K. Potential range: 1.8 to −1.8 V. Epc is the cathodic peak potential. |
| [MoVIO2(L1)(MeOH)] 1 |
−0.38, −1.01 |
| [MoVIO2(L2)(MeOH)] 2 |
−1.15, −1.44 |
| [MoVIO(O2)(L1)(MeOH)] 3 |
−1.27, −1.68 |
| [MoVIO(O2)(L2)(MeOH)] 4 |
−0.96, −1.41 |
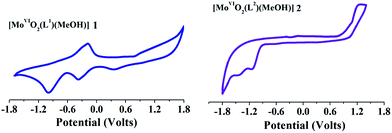 |
| | Fig. 7 Cyclic voltammograms of [MoVIO2(L1)(MeOH)] 1 and [MoVIO2(L2)(MeOH)] 2 recorded in DMF at room temperature. | |
Catalytic activity study
Using molybdenum complexes as catalyst precursors, oxidation of bicyclic alcohols (isoborneol and fenchyl alcohol), cyclic (cyclohexanol) and aromatic alcohols (benzyl alcohol and cumic alcohol) have been carried out under atmospheric conditions. Effect of N-based additive (i.e. NEt3) on the rate of reaction and conversion has also been checked.
Oxidation of isoborneol. Catalytic oxidation of isoborneol, using [MoVIO2]2+ complexes prepared here in the presence of H2O2, gives camphor (1,7,7-trimethylbicyclo[2.2.1]heptan-2-one) selectively; Scheme 3. Considering 1 as a representative catalyst precursor, parameters like, amounts of catalyst, oxidant and solvent and temperature of the reaction mixture have been optimized for the maximum oxidation of substrate.
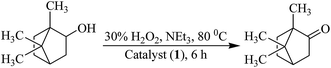 |
| | Scheme 3 Oxidation of isoborneol to camphor. | |
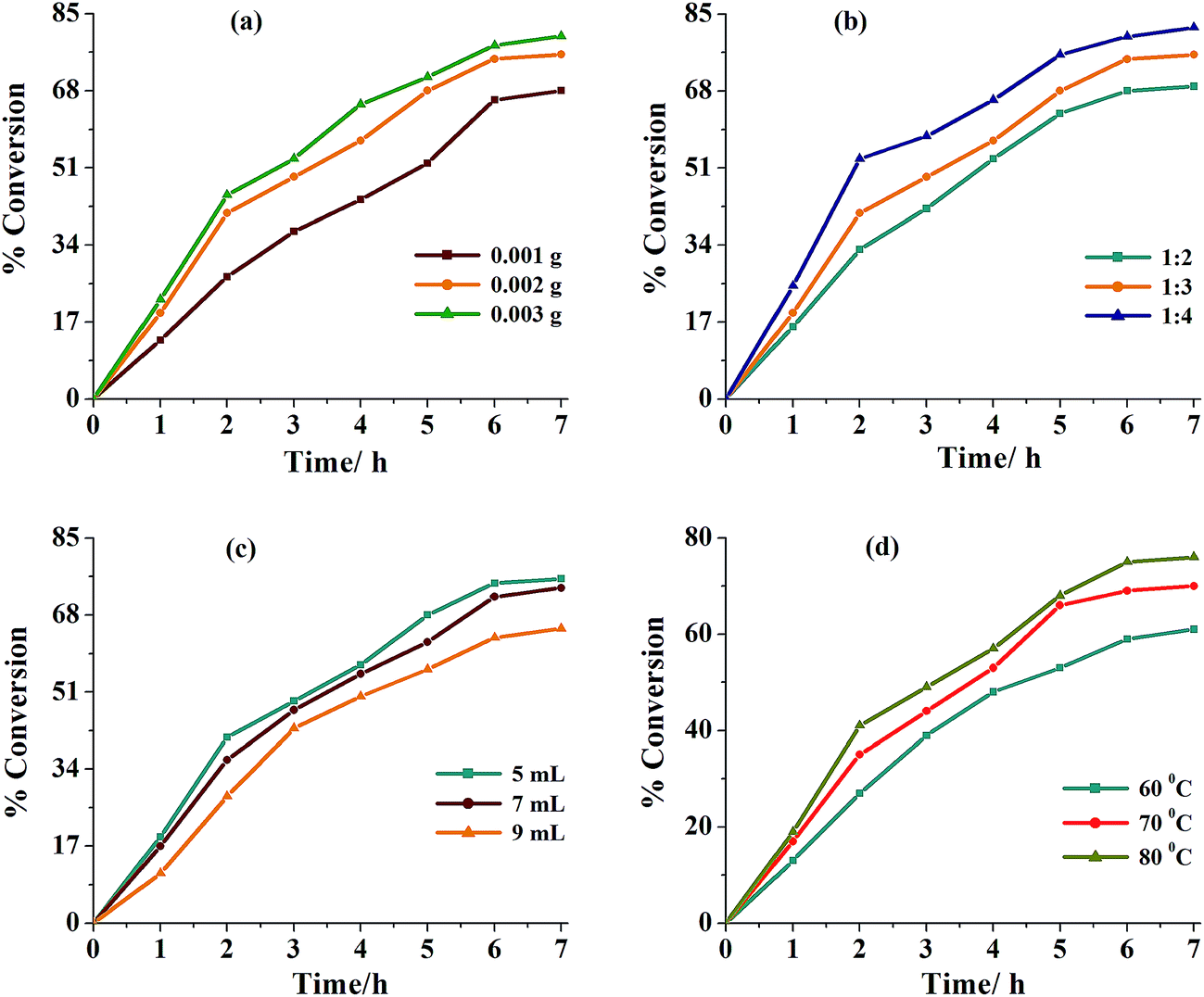
Thus, for 0.010 mol (1.54 g) of isoborneol; three different amounts of catalyst precursor (i.e. 0.001, 0.002 and 0.003 g) and aqueous 30% H2O2 (0.020, 0.030 and 0.040 mol) were taken in three different volumes of solvent (MeCN: 5, 7 and 9 mL) and the reaction was carried out in the presence of NEt3 (0.05 g, 0.0005 mol) at three different temperatures (60, 70 and 80 °C). Fig. 8 and Table 8 summarize all the conditions and the conversion obtained under a particular set of conditions. It is clear from the Table 8 that the optimized reaction conditions for 0.010 mol (1.54 g) of isoborneol are: catalyst 1 (0.002 g, 3.8 × 10−6 mol), 30% H2O2 (3.39 g, 0.030 mol), MeCN (5 mL), NEt3 (0.05 g, 0.0005 mol) and reaction temperature (80 °C) (i.e. entry no. 2 of Table 8). Under these conditions, reaction requires 6 h to attain equilibrium and gave a maximum of 75% conversion. In the absence of NEt3 under above reaction conditions, almost similar conversion (66–80%) was obtained but ca. 24 h was required to achieve the equilibrium. In the absence of catalyst as well as NEt3, only 5% conversion were achieved in 24 h while in the absence of catalyst but in the presence of NEt3, a maximum of 14% conversion was obtained in 6 h of reaction.
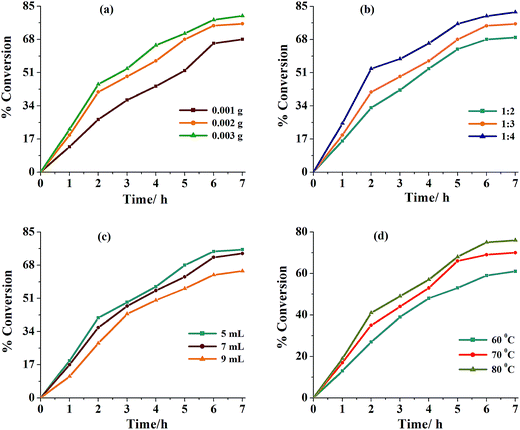 |
| | Fig. 8 (a) Effect of variation of amount (0.001, 0.002 and 0.003 g) of catalyst 1 on the oxidation of isoborneol. Reaction conditions: isoborneol (1.54 g, 0.010 mol), 30% H2O2 (3.39 g, 0.030 mol), MeCN (5 mL), NEt3 (0.05 g, 0.0005 mol) and reaction temp (80 °C) for 6 h. (b) Effect of amount of oxidant (30% H2O2) (substrate to oxidant ratio: 1:2, 1:3 and 1:4) on the oxidation of isoborneol. Reaction conditions: isoborneol (1.54 g, 0.010 mol), 1 (0.002 g), MeCN (5 mL), NEt3 (0.05 g, 0.0005 mol) and reaction temp (80 °C) for 6 h. (c) Effect of variation of amount of solvent (5, 7 and 9 mL) on the rate of the oxidation of isoborneol. Reaction conditions: isoborneol (1.54 g, 0.010 mol), 30% H2O2 (3.39 g, 0.030 mol), 1 (0.002 g), NEt3 (0.05 g, 0.0005 mol) and reaction temp (80 °C) for 6 h. (d) Effect of different temp (60, 70 and 80 °C) on the oxidation of isoborneol. Reaction conditions: isoborneol (1.54 g, 0.010 mol), 1 (0.002 g), 30% H2O2 (3.39 g, 0.030 mol), MeCN (5 mL) and NEt3 (0.05 g, 0.0005 mol) for 6 h. | |
Table 8 Conversion of isoborneol (1.54 g, 0.010 mol) using [MoVIO2(L1)(MeOH)] 1 as catalyst precursor in presence of NEt3 (0.05 g, 0.0005 mol) in 6 h of reaction time under different reaction conditions
| Entry no. |
Catalyst [g (mmol)] |
H2O2 [g (mol)] |
MeCN [mL] |
Temp. [°C] |
Conv. [%] |
| 1 |
0.001 (1.8 × 10−3) |
3.39 (0.030) |
5 |
80 |
66 |
| 2 |
0.002 (3.8 × 10−3) |
3.39 (0.030) |
5 |
80 |
75 |
| 3 |
0.003 (5.6 × 10−3) |
3.39 (0.030) |
5 |
80 |
78 |
| 4 |
0.002 (3.8 × 10−3) |
2.26 (0.020) |
5 |
80 |
68 |
| 5 |
0.002 (3.8 × 10−3) |
4.52 (0.040) |
5 |
80 |
80 |
| 6 |
0.002 (3.8 × 10−3) |
3.39 (0.030) |
7 |
80 |
72 |
| 7 |
0.002 (3.8 × 10−3) |
3.39 (0.030) |
9 |
80 |
63 |
| 8 |
0.002 (3.8 × 10−3) |
3.39 (0.030) |
5 |
70 |
69 |
| 9 |
0.002 (3.8 × 10−3) |
3.39 (0.030) |
5 |
60 |
59 |
Oxidation of fenchyl alcohol. Molybdenum complexes 1, 2, 3 and 4 also catalyze the oxidation of fenchyl alcohol to fenchone (1,3,3-trimethylbicyclo[2.2.1]heptan-2-one) selectively (Scheme 4). Again various parameters as mentioned above have been taken into account to optimize the reaction conditions while taking 1 as a catalyst precursor. Reactions were carried out in the presence as well as in the absence of additive.
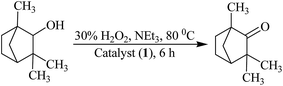 |
| | Scheme 4 Oxidation of fenchyl Alcohol to fenchone. | |
As presented in Table S2 and Fig. S2,† the optimized reaction conditions for the oxidation of 0.010 mol (1.54 g) of fenchyl alcohol are: catalyst 1 (0.003 g, 5.6 × 10−6 mol), oxidant 30% H2O2 (3.39 g, 0.030 mol), N-based additive NEt3 (0.05 g, 0.0005 mol), MeCN (5 mL) and reaction temperature (80 °C) (i.e. entry no. 3 of Table S2†). These conditions are very similar to the one already concluded for the oxidation of isoborneol. Under these conditions reaction requires 6 h to attain equilibrium and give a maximum of 74% conversion. In the absence of catalyst as well as NEt3, only 19% conversion was achieved in 24 h and in the absence of catalyst but in the presence of NEt3, only 13% conversion was obtained in 6 h.
Under the optimized reaction conditions, catalytic potentials of complexes 2, 3 and 4 have also been tested for the oxidation of isoborneol and fenchyl alcohol, and results are summarized in Table 9. With 71–82% conversion of isoborneol, the catalytic potentials of 2, 3 and 4 are equally good and comparable to 1. A slightly lower conversion (69–76%) than isoborneol has been obtained for fenchyl alcohol with 2, 3 and 4 but their catalytic potentials are again comparable to 1 for this particular reaction. However, within these complexes, catalytic potentials of [MoVIO(O2)]2+ complexes are little better than [MoVIO2]2+ complexes. This is expected as [MoVIO2]2+ complexes have to pass through an active intermediate species i.e. [MoVIO(O2)]2+ complexes by their reaction with H2O2 while peroxide complexes can start catalytic activity instantly.
Table 9 Oxidation of different alcohols and TOF's using different molybdenum(VI) complexes as catalyst precursor
| Substrates |
Catalyst (conc. in mmol) |
With additivea |
Without additive |
| Conv. [%] |
TOFb [h−1] |
Conv. [%] |
TOFc [h−1] |
| NEt3 = 0.05 mmol. TOF values calculated at 6 h of reaction time for both substrates. TOF values calculated at 24 h of reaction time. |
| Isoborneol (0.77 g, 5 mmol) |
1 (3.8 × 10−3) |
75 |
328 |
68 |
74 |
| 2 (3.8 × 10−3) |
71 |
311 |
66 |
71 |
| 3 (3.8 × 10−3) |
82 |
359 |
80 |
87 |
| 4 (3.8 × 10−3) |
79 |
346 |
73 |
79 |
| Fenchyl alcohol (0.77 g, 5 mmol) |
1 (5.6 × 10−3) |
74 |
217 |
67 |
49 |
| 2 (5.6 × 10−3) |
69 |
205 |
63 |
47 |
| 3 (5.6 × 10−3) |
76 |
226 |
70 |
52 |
| 4 (5.6 × 10−3) |
71 |
211 |
68 |
50 |
Catalytic potentials of these complexes have also been tested under optimized conditions in the absence of N-based additive (NEt3) (Table 9). In the absence of N-based additive (NEt3), 68, 66, 80 and 73% conversion of isoborneol with catalyst 1, 2, 3 and 4, respectively were obtained in 24 h of reaction time. Similarly, 63–70% conversion of fenchyl alcohol was obtained in 24 h with these catalysts in the absence of N-based additive. Thus, N-based additive reduces the reaction time for oxidation of isoborneol as well as fenchyl alcohol from 24 h to 6 h.
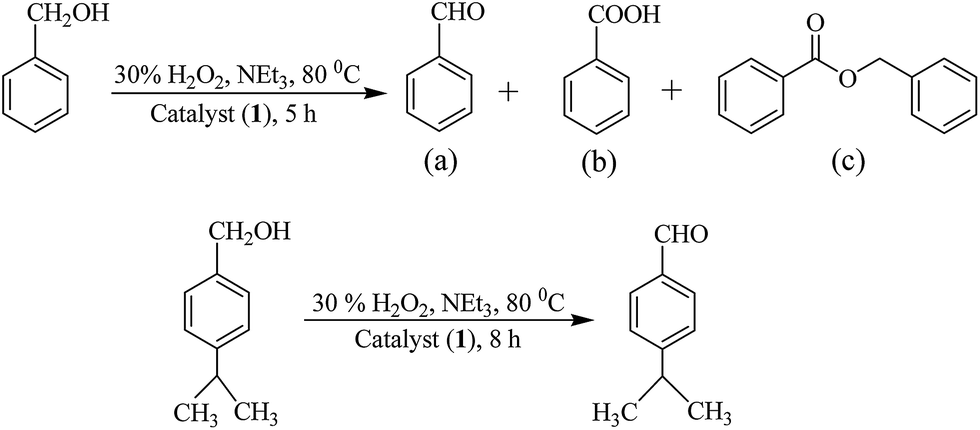
Oxidation of benzyl alcohol and cumic alcohol. We have also optimized reaction conditions for the oxidation of benzyl alcohol and cumic alcohol. While benzyl alcohol gives three products (benzaldehyde, benzoic acid and benzylbenzoate), cumic alcohol gives cuminaldehyde selectively (Scheme 5). All experimental details are presented in Tables S3, S4, Fig. S3 and S4.† The optimized reaction conditions for the maximum oxidation of both alcohols are same [i.e. for 0.005 mol (0.54 g) of benzyl alcohol or 0.005 mol (0.75 g) of cumic alcohol: catalyst 1 (0.002 g, 3.8 × 10−6 mol), 30% aqueous H2O2 (1.13 g, 0.010 mol), MeCN (5 mL) and NEt3 (0.05 g, 0.0005 mol) at 80 °C; entry no. 2 of Table S3† for benzyl alcohol and entry no. 4 of Table S4† for cumic alcohol] along with the conversion of alcohols.
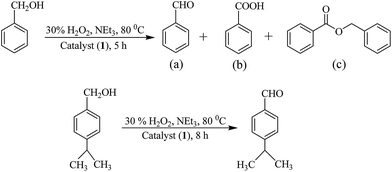 |
| | Scheme 5 Oxidation of benzyl alcohol to (a) benzaldehyde, (b) benzoic acid, (c) benzylbenzoate, and oxidation of cumic alcohol to cuminaldehyde. | |
Under the optimized reaction conditions other catalysts have also been tested for the oxidation of benzyl alcohol (for the selectivity of different reaction products see Table S5†) and cumic alcohol and results are presented in Table 10. In the presence of additive (NEt3), conversions of these substrates are 61–74% and 53–68%, respectively while in the absence of additive, the conversions are 54–68% and 48–61%, respectively. Though catalytic potential of these complexes towards the oxidation of both substrates are not very high, the time required to achieve the equilibrium in the absence and in the presence of additive (i.e. 24 h vs. 5–8 h), suggests that additive plays an important role in improving the catalytic efficiency of complexes.
Table 10 Oxidation of different aromatic alcohols and TOF's using different molybdenum(VI) complexes as catalyst precursor
| Substrates |
Catalysts (conc. in mmol) |
With additivea |
Without additive |
| Conv. [%] |
TOFb [h−1] |
Conv. [%] |
TOFc [h−1] |
| NEt3 = 0.05 mmol. TOF values calculated at 5 h and 8 h of reaction time for benzyl alcohol and cumic alcohol, respectively. TOF values calculated at 24 h of reaction time. |
| Benzyl alcohol (0.54 g, 5 mmol) |
1 (3.8 × 10−3) |
66 |
173 |
59 |
32 |
| 2 (3.8 × 10−3) |
61 |
160 |
54 |
29 |
| 3 (3.8 × 10−3) |
74 |
194 |
68 |
37 |
| 4 (3.8 × 10−3) |
69 |
181 |
63 |
34 |
| Cumic alcohol (0.75 g, 5 mmol) |
1 (3.8 × 10−3) |
59 |
97 |
55 |
30 |
| 2 (3.8 × 10−3) |
53 |
87 |
48 |
26 |
| 3 (3.8 × 10−3) |
68 |
111 |
61 |
34 |
| 4 (3.8 × 10−3) |
60 |
99 |
57 |
29 |
Oxidation of cyclohexanol. The oxidation of cyclohexanol catalyzed by complexes 1, 2, 3 and 4 using H2O2 as oxidant in the presence of NEt3 gave cyclohexanone selectively (Scheme 6). Using complex 1, the best suited reaction conditions, after various trials (Table 11, Fig. S5†), concluded for the maximum oxidation of 0.005 mol (0.5 g) of cyclohexanol (entry no. 2 of Table 11) are: [MoO2(L1)(MeOH)] 1 (0.002 g, 3.8 × 10−6 mol), 30% aqueous H2O2 (1.69 g, 0.015 mol), MeCN (5 mL), NEt3 (0.05 g, 0.0005 mol) at 80 °C. About 4 h was required to attain the equilibrium with 53% conversion and TOF value of 174 h−1. Complex 2, 3 and 4 gave 50% (TOF = 164 h−1), 61% (TOF = 201 h−1) and 57% (TOF = 187 h−1) conversion, respectively in 4 h of reaction time. Without additive, as observed above also, it requires 24 h and the obtained conversions are 47% (TOF = 26 h−1), 42% (TOF = 23 h−1), 55% (TOF = 30 h−1) and 51% (TOF = 28 h−1) for complexes 1, 2, 3 and 4, respectively (Table 12). In the absence of catalyst, conversion obtained was only 10%.
 |
| | Scheme 6 Oxidation of cyclohexanol to cyclohexanone. | |
Table 11 Conversion of cyclohexanol (0.5 g, 0.005 mol) using [MoVIO2(L1)(MeOH)] 1 as catalyst precursor in the presence of NEt3 (0.05 g, 0.0005 mol) in 4 h of reaction time under different reaction conditions
| Entry no. |
Catalyst [g (mmol)] |
H2O2 [g (mmol)] |
MeCN [mL] |
Temp. [°C] |
Conv. [%] |
| 1 |
0.001 (1.8 × 10−3) |
1.69 (15) |
5 |
80 |
45 |
| 2 |
0.002 (3.8 × 10−3) |
1.69 (15) |
5 |
80 |
53 |
| 3 |
0.003 (5.6 × 10−3) |
1.69 (15) |
5 |
80 |
51 |
| 4 |
0.002 (3.8 × 10−3) |
1.13 (10) |
5 |
80 |
44 |
| 5 |
0.002 (3.8 × 10−3) |
2.26 (20) |
5 |
80 |
56 |
| 6 |
0.002 (3.8 × 10−3) |
1.69 (15) |
7 |
80 |
50 |
| 7 |
0.002 (3.8 × 10−3) |
1.69 (15) |
9 |
80 |
47 |
| 8 |
0.002 (3.8 × 10−3) |
1.69 (15) |
5 |
70 |
49 |
| 9 |
0.002 (3.8 × 10−3) |
1.69 (15) |
5 |
60 |
51 |
Reactivity of dioxidomolybdenum(VI) complexes with H2O2 and possible reaction intermediate
The [MoVIO(O2)]2+ complexes have been isolated by reacting in situ generated oxidoperoxidomolybdenum(VI) species with ligands I and II (see experimental). The generation of such species has also been established in methanol by reacting [MoVIO2(L1)(MeOH)] 1 and [MoVIO2(L2)(MeOH)] 2 with H2O2 and monitoring progress of the reaction by electronic absorption spectroscopy. In a typical reaction, 20 mL of 4.3 × 10−5 M solution of 1 was treated with one drop portion of 30% aqueous H2O2 (0.053 g, 0.47 mmol) dissolved in 10 mL of methanol and the resultant spectroscopic changes are presented in Fig. 9. Thus, the band at 210 nm shows considerable increase in intensity and slightly shifts to 220 nm. Simultaneously, the intensity of bands at 245 and 298 nm increases along with a generation of isosbestic point at 313 nm; Fig. 9(a). In the presence of excess of H2O2 the 245 band slowly disappears; Fig. 9(a). Considering higher concentration of 1 (7.8 × 10−5 M) where 412 nm band is visible and adding drop wise portions of 6.1 × 10−2 M H2O2 causes slow decrease of 412 nm band along with the generation of an isosbestic point at 313 nm; Fig. 9(b). This band finally disappears up on the addition of excess of H2O2 solution. Complex 2 presents very similar spectral changes upon treating with a solution of H2O2 in methanol. The final spectral patterns {see Fig. 9(c) and (d)} are similar to the one recorded for the peroxido complexes 3 and 4 indicating the formation of similar peroxido complexes in solution upon the addition of H2O2 to complexes 1 and 2, respectively.
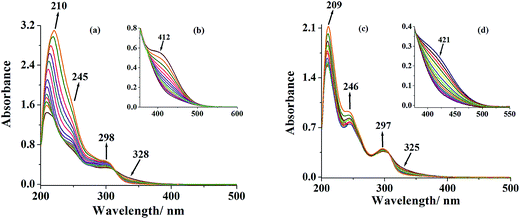 |
| | Fig. 9 (a) Spectral changes recorded after successive addition of one drop portion of 30% H2O2 (0.053 g, 0.47 mmol) dissolved in 10 mL of MeOH (final concentration of H2O2; 4.6 × 10−2 M) to 20 mL of (4.3 × 10−5 M) solution of [MoVIO2(L1)(MeOH)] 1. (b) Inset: spectral changes with 30% H2O2 (0.064 g, 0.56 mmol) dissolved in 10 mL of methanol (final concentration of H2O2; 6.1 × 10−2 M) to 20 mL of methanolic solution (7.8 × 10−5 M) of complex 1. (c) Spectral changes with 30% H2O2 (0.062 g, 0.55 mmol) dissolved in 10 mL of methanol (final concentration of H2O2; 5.4 × 10−2 M) to 20 mL of methanolic solution (6.0 × 10−5 M) of [MoVIO2(L2)(MeOH)] 2. (d) Inset: spectral changes with 30% H2O2 (0.069 g, 0.61 mmol) dissolved in 10 mL of methanol (final concentration of H2O2; 5.6 × 10−2 M) to 20 mL of methanolic solution (8.1 × 10−5 M) of complex 2. | |
Table 12 Oxidation of cyclic alcohol and TOF's using different molybdenum(VI) complexes as catalyst precursor
| Substrates |
Catalysts (conc. in mmol) |
With additivea |
Without additive |
| Conv. [%] |
TOFb [h−1] |
Conv. [%] |
TOFc [h−1] |
| NEt3 = 0.05 mmol. TOF values calculated at 4 h of reaction time. TOF values calculated at 24 h of reaction time. |
| Cyclohexanol (0.50 g, 5 mmol) |
1 (3.8 × 10−3) |
53 |
174 |
47 |
26 |
| 2 (3.8 × 10−3) |
50 |
164 |
42 |
23 |
| 3 (3.8 × 10−3) |
61 |
201 |
55 |
30 |
| 4 (3.8 × 10−3) |
57 |
187 |
51 |
28 |
Conclusions
The [MoVIO2]2+ complexes, [MoVIO2(L1)(MeOH)] 1 and [MoVIO2(L2)(MeOH)] 2, and their corresponding [MoVIO(O2)]2+ complexes, [MoVIO(O2)(L1)(MeOH)] 3 and [MoVIO(O2)(L2)(MeOH)] 4 have been prepared from potential tridentate ligands, 4-[3,5-bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]benzoic acid (H2L1, I) and 3,5-bis(2-hydroxyphenyl)-1-phenyl-1,2,4-triazole (H2L2, II) and characterized. The single crystal X-ray study of 1a (i.e. DMSO coordinated in place of methanol) and 2 confirms the dibasic tridentate behavior of ligands. These complexes emerge out as efficient and selective homogenous catalysts for the oxidation of bicyclic, cyclic and aromatic alcohols in MeCN in the presence of N-based additive (NEt3) where about 4–8 h was required to achieve the equilibrium. However, in the absence of additive reactions take about 24 h to reaching equilibrium. The N-based additive, NEt3, abstracts hydrogen from H2O2 due to its strong basic nature which in turns accelerates the formation of peroxido intermediate complex. This way it reduces the time period of the catalytic reaction with equally good conversion.30 Thus, the additive plays an important role in improving the catalytic efficiency of complexes and reduces the time of oxidation considerably (24 h to 4–8 h). Amongst the two type of complexes studied, the catalytic potentials of [MoVIO(O2)]2+ complexes are slightly better than [MoVIO2]2+ complexes. Reactivity of the [MoVIO2]2+ complexes with H2O2 provides evidence for the possible peroxido intermediate species formation during the catalytic action.
References
- A. Kamimura, Y. Nozaki, M. Nishiyama and M. Nakayama, RSC Adv., 2013, 3, 468–472 RSC
 .
. - X. T. Zhou, H. Bing and S. G. Liu, Tetrahedron Lett., 2013, 54, 3882–3885 CrossRef CAS .
-
(a) C. Aellig, U. Neuenschwander and I. Hermans, ChemCatChem, 2012, 4, 525–529 CrossRef CAS ;
(b) W. Feng, G. J. Wu, L. D. Li and N. J. Guan, Green Chem., 2011, 13, 3265–3272 RSC ;
(c) Y. Xie, Z. F. Zhang, S. Q. Hu, J. L. Song, W. J. Li and B. X. Han, Green Chem., 2008, 10, 278–282 RSC ;
(d) B. Z. Zhan, M. A. White, T. K. Sham, J. A. Pincock, R. J. Doucet, K. V. R. Rao, K. N. Robertson and T. S. Cameron, J. Am. Chem. Soc., 2003, 125, 2195–2199 CrossRef CAS PubMed .
- D. Sahu, C. Sarmah and P. Das, Tetrahedron Lett., 2014, 55, 3422–3425 CrossRef CAS .
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS .
- K. C. Gupta, A. K. Sutar and C. C. Lin, Coord. Chem. Rev., 2009, 253, 1926–1946 CrossRef CAS .
-
(a) J. A. L. da Silva, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2011, 255, 2232–2248 CrossRef CAS ;
(b) M. Sutradhar, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2015, 301–302, 200–239 CrossRef CAS .
- M. R. Maurya, J. Chem. Sci., 2011, 123, 215–228 CrossRef CAS .
- A. V. Biradar, M. K. Dongare and S. B. Umbarkar, Tetrahedron Lett., 2009, 50, 2885–2888 CrossRef CAS .
- M. T. Raisanen, A. A. Hunaiti, E. Atosuo, M. Kemell, M. Leskela and T. Repo, Catal. Sci. Technol., 2014, 4, 2564–2573 CAS .
- J. Taghavimoghaddam, G. P. Knowles and A. L. Chaffee, J. Mol. Catal. A: Chem., 2012, 35, 879–888 Search PubMed .
- G. Chatela, C. Monniera, N. Kardosa, C. Voironc, B. Andriolettib and M. Drayea, Appl. Catal., A, 2014, 478, 157–164 CrossRef .
- M. M. D. Anna, M. Malia, P. Mastrorillia, P. Cotugno and A. Monopoli, J. Mol. Catal. A: Chem., 2014, 386, 114–119 CrossRef .
-
(a) M. R. Maurya, N. Kumar and F. Avecilla, J. Mol. Catal. A: Chem., 2014, 392, 50–60 CrossRef CAS ;
(b) M. R. Maurya, S. Dhaka and F. Avecilla, Polyhedron, 2014, 67, 145–159 CrossRef CAS ;
(c) M. R. Maurya, S. Dhaka and F. Avecilla, Polyhedron, 2014, 81, 154–167 CrossRef CAS ;
(d) M. R. Maurya, S. Dhaka and F. Avecilla, Polyhedron, 2015, 96, 79–87 CrossRef CAS ;
(e) M. R. Maurya, S. Dhaka and F. Avecilla, New J. Chem., 2015, 39, 2130–2139 RSC ;
(f) M. R. Maurya and N. Kumar, J. Mol. Catal. A: Chem., 2015, 406, 204–212 CrossRef CAS ;
(g) M. R. Maurya, L. Rana and F. Avecilla, Inorg. Chim. Acta, 2015, 429, 138–147 CrossRef CAS ;
(h) M. R. Maurya, N. Saini and F. Avecilla, Polyhedron, 2015, 90, 221–232 CrossRef CAS ;
(i) M. R. Maurya, N. Saini and F. Avecilla, Inorg. Chim. Acta, 2015, 438, 168–178 CrossRef CAS .
- G. J. J. Chen, J. W. McDonald and W. E. Newton, Inorg. Chem., 1976, 15, 2612–2615 CrossRef CAS .
- S. Steinhauser, U. Heinz, M. Bartholoma, T. Weyhermuller, H. Nick and K. Hegetschweiler, Eur. J. Inorg. Chem., 2004, 21, 4177–4192 CrossRef .
- G. M. Sheldrick, SADABS, version 2.10, University of Göttingen, Germany, 2004 Search PubMed .
- G. M. Sheldrick, SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed .
- A. Rezaeifard, I. Sheikhshoaie, N. Monadi and H. Stoeckli-Evans, Eur. J. Inorg. Chem., 2010, 799–806 CrossRef CAS .
- A. Syamal and M. R. Maurya, Coord. Chem. Rev., 1989, 95, 183–238 CrossRef CAS .
-
(a) D. Wang, M. Ebel, C. Schulzke, C. Grüning, S. K. S. Hazari and D. Rehder, Eur. J. Inorg. Chem., 2001, 935–942 CrossRef CAS ;
(b) J. C. Pessoa, M. J. Calhorda, I. Cavaco, I. Correia, M. T. Duarte, V. Felix, R. T. Henriques, M. F. M. Piedade and I. Tomaz, Dalton Trans., 2002, 4407–4415 RSC .
- A. D. Keramidas, A. B. Papaioannou, A. Vlahos, T. A. Kabanos, G. Bonas, A. Makriyannis, C. P. Rapropoulou and A. Terzis, Inorg. Chem., 1996, 35, 357–367 CrossRef CAS PubMed .
- S. Pasayat, S. P. Dash, S. Roy, R. Dinda, S. Dhaka, M. R. Maurya, W. Kaminsky, Y. P. Patil and M. Nethaji, Polyhedron, 2014, 67, 1–10 CrossRef CAS .
- S. Pasayat, S. P. Dash, Saswati, P. K. Majhi, Y. P. Patil, M. Nethaji, H. R. Dash, S. Das and R. Dinda, Polyhedron, 2012, 38, 198–204 CrossRef CAS .
- R. Dinda, P. Sengupta, S. Ghosh, H. M. Figge and W. S. Sheldrick, Dalton Trans., 2002, 23, 4434–4439 RSC .
- R. Dinda, P. Sengupta, S. Ghosh and W. S. Sheldrick, Eur. J. Inorg. Chem., 2003, 363–369 CrossRef CAS .
- J. Topich, Inorg. Chem., 1981, 20, 3704–3707 CrossRef CAS .
- M. Chaudhury, Dalton Trans., 1984, 2, 115–120 RSC .
- N. K. Ngan, K. M. Lo and C. S. R. Wong, Polyhedron, 2011, 30, 2922–2932 CrossRef CAS .
- R. R. Fernandes, J. Lasri, M. F. C. Guedes da Silva, J. A. L. da Silva, J. J. R. Frausto da Silva and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2011, 351, 100–111 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 (detail of thermogravimetric analysis of complexes). Tables S2–S4 (data on the oxidation of fenchyl alcohol, benzyl alcohol and cumic alcohol under various reaction conditions, respectively). Fig. S1 (electronic spectra of ligands, H2L1 I and H2L2 II). Fig. S2–S5 (catalytic plots of fenchyl alcohol, benzyl alcohol, cumic alcohol and cyclohexanol, respectively). CCDC 1433879 for 1a and 1414244 for 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra16490g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.