
Preparation of cylindrical multi-compartment micelles by the hierarchical self-assembly of ABC triblock polymer in solution
Arzugul Muslimac,
Yi Shib,
Yechao Yanb,
Dongdong Yaob and
Abulikemu Abudu Rexit*ac
aSchool of Chemistry and Chemical Engineering, Xinjiang Normal University, Urumqi, China. E-mail: aarexit@mail.ustc.edu.cn; Fax: +86 0991 4332683
bLaboratory of Polymer Physics and Chemistry, Institute of Chemistry, The Chinese Academy of Science, Beijing, China
cSchool of Chemistry and Material Science, North West University, Xi'an, China. E-mail: arzu_hma@yahoo.com
First published on 24th September 2015
Abstract
Amphiphilic linear ABC triblock copolymer PnBA28-b-PS37-b-P2VP73 was prepared by the RAFT method and its two-step hierarchical self-assembly in selected solvents was explored. The triblock copolymer was refluxed in methanol and the micelle product was further dialyzed against acidic water. The effects of the concentration of copolymer on the morphology of the aggregates formed in methanol in the first step were investigated. The structures and morphologies of the micelles formed by the self-assembly were characterized with SEC, 1H NMR, TEM and DLS. Spherical patchy micelles were obtained in the poor solvent, methanol, for PS and PnBA blocks during the first step assembly. The patchy micelles were dialyzed against water with a pH value of ∼3.0 and aggregated to form multi-compartment micelles (MCMs). The MCMs further aggregated when the concentration of patchy micelles was increased. Cylindrical MCMs were formed with 2 mg mL−1 patchy micelles.
Introduction
Block copolymers can be self-assembled to form various controllable nanoscale ordered structures and morphologies,1–3 which has attracted considerable attention in nanotechnology research field.4 ABC triblock copolymers can be self-assembled in the appropriate selected solvent, according to the assembling conditions for each block, to form nanoscale aggregates with various structures and morphologies. In the selected solvent for one block where the other two blocks are insoluble, the insoluble two blocks form a micelle core. These two blocks can separate into two phases in a tens of nanometer space, resulting in multi-compartment micelles (MCMs).1,5–10 Large number of micro-segments are formed in the cores and shells of MCMs, leading to a complex micellar morphology. MCMs incorporate different payloads in individual compartments and can be used to load particles with different chemical properties. Therefore, it is potentially useful in drug delivery and controllable release as well as the preparation of special nanostructured materials.2Generally, MCMs are prepared by the self-assembly of diblock copolymer blends,11,12 self-assembly of triblock copolymers with various topologies2,13–32 and multiblock33–35 copolymers in selective solvents, annealing of the self-assembled copolymer aggregates, changing the self-assembly behavior of the copolymers in the selected solvent by the fluorination of one block in the copolymers, self-assembly between copolymers and assembly between copolymer and small molecules. The most widely used method for the preparation of MCMs with ABC triblock copolymers is to select a good solvent for one block that is soluble to the solvent to form the corona of MCMs. The other insoluble two blocks form the core domain of compartmentalized core of the MCMs.2,16,17,19–21,36–40 Müller et al. proposed a two-step hierarchical self-assembly approach for the preparation of MCMs.2,17,18,22,36,41,42 Primary micelles, with one block as corona and the other blocks as compartmentalized core were prepared in the selected solvent for the corona block. Secondary aggregation of the micelles was then induced by dialysis to form multi-compartment micelles. This is a striking success in the preparation of MCMs with controllable aggregation between MCMs.8
The self-assembly behavior of ABC triblock copolymers in solution especially to form MCMs, is not well understood as that of diblock copolymers.5 There is no efficient simple synthesis method for the preparation of MCMs with triblock copolymers. Strong phase separation ability is required for the triblock copolymers. Generally, fluorine-containing polymers are selected, which is a challenge for polymer synthesis and limits the performances and applications of resultant MCMs. The morphology of the triblock MCMs aggregates can be tuned by the optimizing assembly conditions, such as the block sequence, ratio of the composite blocks, composite of solvent, concentration of the copolymer, temperature, time and so on.1,9,42,43 The initial concentration of the copolymer can affect the stretch of the compartmentalized core, the interfacial tension between the micellar core and solvent, interactions between coronas, and so on,42 which further affects the self-assembly of the triblock copolymers in solution. In the present work, cylindrical MCMs were prepared from ABC triblock copolymer by tuning the concentration of the copolymer and solvents.
The micro-segments formed in compartmentalized core or shell are the prerequisite for the formation of the cylindrical MCMs. Thus, the structural design of the ABC triblock copolymer is critical. In the present work, triblock copolymer, PnBA-b-PS-b-P2VP, was designed due to the different solubility of the three blocks in methanol and acidic water and its easy synthesis. It is well known that P2VP is quaternized under pH stimuli and can form hydrogen bonds with metal salt complexes and phenolic hydroxyl groups. PnBA is in rubbery state at room temperature and has a strong interfacial tension with water. PS has a high glass transition temperature (Tg) of 100 °C and is in glassy state at room temperature. It can stabilize the morphology of the MCMs during the self-assembly process.44 The properties of each block provide the multiple functions of the prepared MCMs and stabilize the MCMs solution. Linear triblock copolymer, PnBA28-b-PS37-b-P2VP73, with narrow molecular weight range was successfully synthesized by the reversible addition–fragmentation chain transfer (RAFT) radical polymerization. The ABC triblock copolymer was refluxed in methanol. The PnBA and PS blocks formed the core with phase-separated domains of the resultant primary MCMs and soluble P2VP formed the shell of the micelles. The primary MCMs were dialyzed against acidic water to increase their phase separation ability. The high level cylindrical MCMs were obtained by adjusting the initial concentration of the triblock copolymer. The mechanism of this process is shown in Scheme 1.
 | ||
| Scheme 1 Mechanism of hierarchical self-assembly process of PnBA28-b-PS37-b-P2VP73 in solution. | ||
Experimental
Materials and reagents
Azobisisobutyronitrile (AIBN) was recrystallized in ethanol. N-butyl acrylate (nBA), styrene (St) and 2-vinylpyridine (2VP) were purchased from Alfa Aesar (Ward Hill, MA), dehydrated on CaH2 and distilled under reduced pressure. N,N-Dimethylformamide (DMF) was dried over anhydrous magnesium sulfate and distilled under reduced pressure. Other reagents were of analytical grade and used as received.Instruments and methods
Size Exclusive Chromatography (SEC) measurements were performed on Styragel HT2, HT4 and HT5 gel columns at 50 °C. The analytes were eluted with DMF driven by Waters 515 HPLC pumps and at 1.0 mL min−1. PS was used as a standard and the analytes were monitored with a Waters 2414 parallax refractive index detector. Nuclear magnetic resonance spectra (1H-NMR) were recorded on a Bruker DMX400 spectrometer at room temperature with CDCl3 as solvent and TMS as internal standard. Transmission electron microscopy (TEM) images were taken with on a TECNAI G220 TEM operated at an accelerating voltage of 200 kV. The machine was equipped with a digital camera. The micellar solution was dispersed on a copper lined pure carbon film and dried at room temperature. The primary patchy micelles were stained with I2 and the secondary micellar products were stained with RuO4. DLS measurements were performed at a scattering angle of 30° on an ALV DLS/SLS-SP 5022F equipment consisting of an ALV-SP 125 laser goniometer, an ALV 5000/E correlator and a He–Ne laser operating at a wavelength of λ = 632.8 nm. Test temperature was 25 °C and the test time was 1 h. Organic sample were filtered with 0.45 μm Millipore nylon filters and aqueous samples were filtered with 0.45 μm Millipore PVDF filters.Synthesis of PnBA28-b-PS37-b-P2VP73 copolymer
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The supernatant was removed and the solvent residue was removed by rotary evaporation. The precipitation was repeated three times and the final product PnBA28 was vacuum-dried at room temperature for 12 h.:1) and leached. The precipitation was repeated three times and the final product PnBA28-b-PS37 was vacuum-dried at room temperature for 12 h.
:1). The supernatant was removed and the solvent residue was removed by rotary evaporation. The precipitation was repeated three times and the final product PnBA28 was vacuum-dried at room temperature for 12 h.:1) and leached. The precipitation was repeated three times and the final product PnBA28-b-PS37 was vacuum-dried at room temperature for 12 h.Results and discussion
Synthesis of triblock copolymer by RAFT method
PnBA28, PnBA28-b-PS37 and PnBA28-b-PS37-b-P2VP73 were synthesized by RAFT method as shown in Scheme 2. Their SEC chromatograms and 1H-NMR spectra are shown in Fig. 1 and 2, respectively. | ||
| Scheme 2 Synthetic route of block copolymers. | ||
 | ||
| Fig. 1 SEC curves of block copolymers in DMF. | ||
 | ||
| Fig. 2 1H NMR spectrum of block copolymers. | ||
All PnBA28, PnBA28-b-PS37 and PnBA28-b-PS37-b-P2VP73 polymers showed a single symmetrical peak and the peaks of diblock and triblock copolymers shifted to higher molecular weight region, indicating that the copolymerization was effectively controlled. In addition, the molecular weight distributions of the resultant polymers are in a narrow range (PDI≤1.32) (Table 1), indicating the formation of the products in a narrow molecular range. The 1H NMR spectra of PnBA-b-PS-b-P2VP indicate the desired product was produced. The degree of polymerization (DP) of PnBA was calculated with the area ratio of the peak at δ = 2.3 ppm that was assigned to the hydrogen of the methine group in nBA repeating unit to the peak at δ = 3.8 ppm that was assigned to the hydrogen of methylene in EDMAT (Table 1). Similarly, the DPs of PS and P2VP were calculated with the area ratios of the hydrogen peak of methine in nBA repeating units at δ = 2.3 ppm to the hydrogen peak of benzene ring in PS units at δ = 6–7.2 ppm and to the hydrogen peak of –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– of pyridine ring in P2VP units at δ = 7.8–8.6 ppm, respectively (Table 1). The polymer molecular weights obtained with SEC are the relative molecular weights to PS, and thus they are different from those obtained with 1H NMR (Table 1).
CH– of pyridine ring in P2VP units at δ = 7.8–8.6 ppm, respectively (Table 1). The polymer molecular weights obtained with SEC are the relative molecular weights to PS, and thus they are different from those obtained with 1H NMR (Table 1).
Self-assembly behaviors of PnBA28-b-PS37-b-P2VP73 solution
Linear triblock copolymer PnBA28-b-PS37-b-P2VP73 was hierarchically self-assembled with a two-step procedure. At the primary step as shown in Fig. 3, PnBA28-b-PS37-b-P2VP73 copolymer samples with amounts of 2 mg, 5 mg, 10 mg, 20 mg and 40 mg were, respectively, refluxed in 20 mL methanol at 80 °C overnight, cooled to room temperature and stained with I2. The secondary micelle product was prepared by dialyzing the primary assembled micelles at the first step against the water with a pH value of ∼3. Fig. 4 shows the TEM images of the dispersed secondary micellar particles stained with RuO4. Fig. 5 is the DLS curves for the micelles obtained before and after dialysis, the relative data are shown in Table 2. | ||
| Fig. 3 TEM images of primary self-assembled micelles of PnBA28-b-PS37-b-P2VP73. (a)–(e) were the self-assembled aggregates in different concentration in methanol: (a) 0.1 mg mL−1, (b) 0.25 mg mL−1, (c) 0.5 mg mL−1, (d) 1.0 mg mL−1, (e) 2.0 mg mL−1, staining with I2. | ||
 | ||
| Fig. 4 TEM images of secondary self-assembled micelles of PnBA28-b-PS37-b-P2VP73 prepared by dialyzing the primary assembled micelles at the first step against the water with a pH value of ∼3, the first self-assembly concentration of samples were as following: (a) 0.1 mg mL−1, (b) 0.25 mg mL−1, (c) 0.5 mg mL−1, (d) 1.0 mg mL−1, (e) 2.0 mg mL−1, staining with RuO4. (f) is the image of (e) taken in 7 months later. | ||
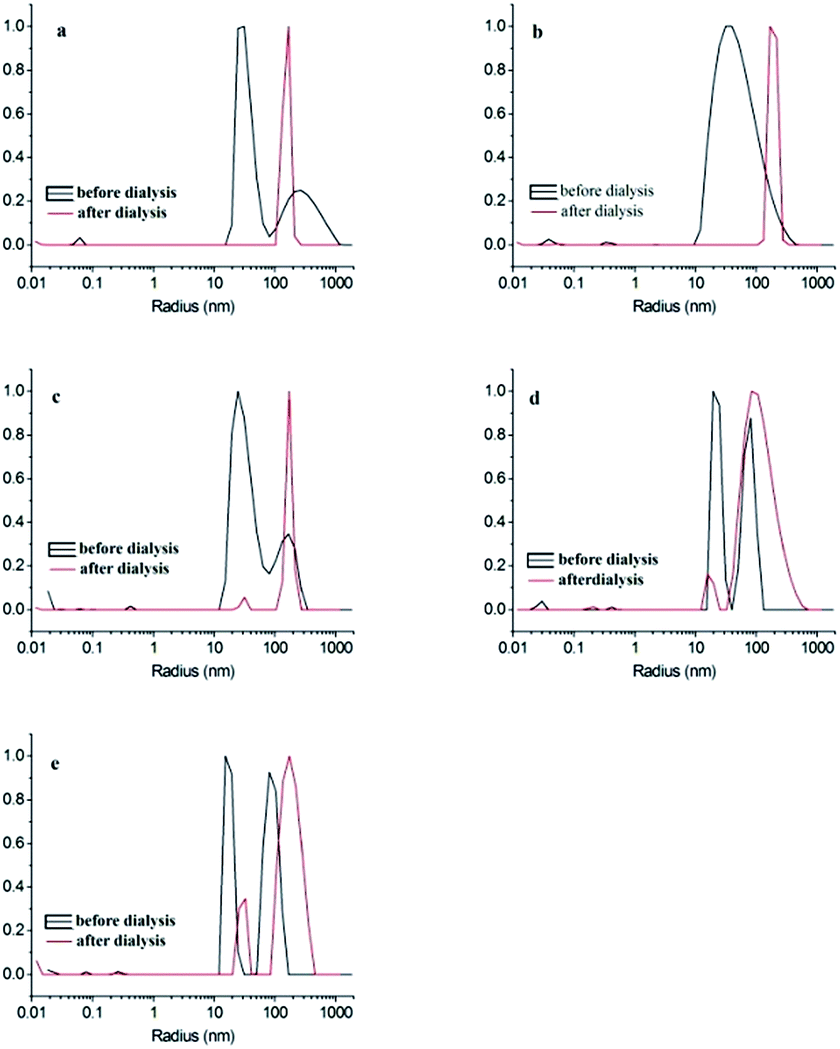 | ||
| Fig. 5 DLS curves of PnBA28-b-PS37-b-P2VP73 at different initial concentration: (a) 0.1 mg mL−1, (b) 0.25 mg mL−1, (c) 0.5 mg mL−1, (d) 1.0 mg mL−1, (e) 2.0 mg mL−1. | ||
| Concentration of samples | Self-assembly process | Radius (nm) |
|---|---|---|
| 0.1 mg mL−1 | Before dialysis | 67.9 |
| After dialysis | 154.7 | |
| 0.25 mg mL−1 | Before dialysis | 46.3 |
| After dialysis | 190 | |
| 0.5 mg mL−1 | Before dialysis | 44.9 |
| After dialysis | 30.6 | |
| 172.5 | ||
| 1.0 mg mL−1 | Before dialysis | 22.3 |
| 75.4 | ||
| After dialysis | 17.4 | |
| 111.4 | ||
| 2.0 mg mL−1 | Before dialysis | 17.4 |
The TEM images of the primary self-assembled products formed on this step show that they are uniform 27 ± 5 nm spherical micelles (Fig. 3). It is worth noting that 2–5 black spots with sizes of several nanometers were observed on each micellar particle, indicating the phase separation in the micelle particles. I2 stains P2VP microphase that is visualized as the dark patchy area on the TEM image. PS and PnBA microphase cannot be visualized with I2 staining and cannot be identified on the TEM images. Therefore, the gray round area in the TEM images is corresponding to the PS and PnBA blocks. Methanol is a good solvent for P2VP block and poor solvent for PS and PnBA blocks. The polarity of PnBA is slightly higher than PS. Therefore, PS block was tangled together to form the core of the micelle during the first-step assembly. PnBA block collapsed on the PS surface to form island-like microphases, which, in combination with PS core, formed the multi-compartmentalized core of MCMs. P2VP extended on the core surface to form the shell of the micelle. In methanol that is the selected solvent for P2VP block, the microphases are in thermodynamic metastability due to their interactions with P2VP block.44 Therefore, refluxing in methanol can activate the micelles and promote their self-assembly. P2VP block could also collapse on the PS surface due to solvent evaporation when the sample was dried for TEM imaging. Generally, the stained P2VP in the simple core–shell micelles exhibits anuniform continuous annular morphology. The island-like spots of the P2VP block in the MCMs prepared here indicate the phase separation between P2VP and PnBA, which leads to the morphologies as shown in Fig. 3a–e. Lodge et al.37,38 demonstrated that the morphology of triblock copolymer micelle was decided by the competition between interface where the blocks with weak interactions tends to reach and the interface between covalent-bonded blocks. P2VP and PnBA are the blocks with weak interactions in their poor solvent methanol and PnBA/PS blocks and P2VP/PS blocks are covalent-bonded. P2VP block tends to expend on the surface of the micelle and PnBA block collapses on the core surface, leading to the formation patchy micelles. DLS results further confirm the assembly mechanism as described above and showed in Scheme 1. The assembled micelles from the block copolymer solutions with all concentrations except 0.25 mg mL−1 showed two narrow distribution peaks. Most particles have a radius of 20 nm (a diameter of 40 nm), which are patchy micelles. The number of the large micelles was increased with the increase of the concentration of block copolymers, indicating the possible aggregation between the patchy micelles. The scattered light of large particles is stronger than that of smaller particle. The amount of particles with large sizes is actually very low even the intensity of its DLS peak is high.
As for the secondary micelle products shown in Fig. 4, RuO4 stains PS block, which is appeared as the dark area on the TEM images of the secondary micelle products. Uniform spherical particles with a diameter of 41 ± 10 nm were observed in Fig. 4a–c. It is worth noting that gray lines were formed between these particles, indicating the possible formation of oligo-aggregates. According to the DLS images of the secondary micelle products, the Rh of these particles reached 150 nm (i.e., 300 nm in diameter), which is much larger than the theoretical size of regular spherical micelles. These results indicate the formation of aggregate in the micellar solution. Further increase of the concentration of copolymers to 1 mg mL−1 led to short bead chain-like aggregates (Fig. 4d). The short 200 nm long cylindrical aggregates consisting of ∼100 nm spherical particles were formed in the micelle solution prepared with 2 mg mL−1 triblock copolymers (Fig. 4e). Based on the structural feature and sizes of these aggregates, the particles with clear boundaries between black and gray regions are not formed in methanol during the first step of the self-assembly. The black area is the image of PS microphase and the gray particles are the PnBA phase, which form the cylindrical aggregated particles in an alternating order. The bigger spherical particles are also composed of black and gray areas. The acidic solution is a good solvent for P2VP. Thus, P2VP can extend on the surface of the phase separated PS-PnBA compartmentalized core during TEM imaging. PS is in a frozen state in methanol at room temperature due to its high Tg. The size reassembly of the block copolymer in the patchy micelles could not occur when the solvent was changed to high polar water. The PnBA microphases on the surfaces of different PS microphases can adhere together to form the secondary self-assembled aggregates(Scheme 1). Lodge et al.38 prepared wormlike segmented MCMs by the self-assembly of star shaped ABC tri-block copolymer μ-EOF. Although they believed that the aggregation between MCMs could be achieved through sharing their micelle coronas, but they also demonstrated micelle coronas have steric repulsions between each other. Therefore, P2VP cannot stabilize the hydrophobic core due to the environmental change, leading to the secondary self-assembly. Meanwhile, the frozen PS microphase hinders the molecular level self-assembly to form a more stable structure, leading to the merging of PnBA patches to reduce the free energy. The relationship between the secondary self-assembly and the concentration of polymer concentration further confirms this hypothesis.
The shape diversity of the secondary micelles is related to the number of patches in the primary micelles. As shown in Fig. 3, most of primary micellar particles contain two patches, which form segmented cylindrical micelles during secondary self-assembly.
These also promote the aggregation of MCMs assembled from primary patchy micelles to lower the free energy of the system. Therefore, chain-like aggregates of the MCMs and patchy micelles were observed in the secondary assembled samples (Fig. 4a–e). The length of the aggregate chain was increased with the increase of the concentration of the triblock copolymer and complete segmented cylindrical MCMs with separated phases were obtained with 2.0 mg mL−1 PnBA28-b-PS37-b-P2VP73 solution (Fig. 4e). The dark area on the TEM image of the MCMs is the PS microphase and the lighter area is the PnBA microphase. P2VP block cannot be visualized with the RuO4 staining. The PS blocks (dark area in Fig. 4e) are segregated by the bigger PnBA blocks (light area in Fig. 4e) in the cylindrical MCMs. The high concentration and enhanced interfacial tension between PnBA and water leads the aggregation of patchy micelles through PnBA blocks, resulting in cylindrical MCMs (Scheme 1). Linear MCMs were formed through the adhesion between the micelles with two patches. Micelles with more than 2 patches functioned as the joint of the bifurcation in cylindrical MCMs. A considerate amount of bifurcated cylindrical MCMs were first observed in MCMs formed with 2 mg mL−1 triblock copolymers (Fig. 4e). Zhu et al.45 found that wormlike PS-b-P2VP-b-PEO self-assembled segmented MCMs remained stable under operations as strong mechanic stirring and ultrasonic vibrating. They believed that the micelle chains strongly interacted with each other, leading to the wormlike morphology. Fig. 4f shows the TEM image stained with RuO4 of segmented cylindrical MCMs taken in 7 months later. As can be seen, the segmented cylindrical MCMs obtained in the present work were slightly broken. The number of cylindrical micelles was slightly decreased. However, the spherical micelles tend to form cylindrical micelles. Compared with the micelles in Fig. 4e, the compartmentalized cores which are at the middle part of MCMs significantly were swelled. This might be attributed to the evaporation of the water during the storage. Therefore, the broken morphology of segmented cylindrical MCMs can be attributed to the change in their interaction with the solvent. In addition, Lodge et al.38 reported that μ-EOF self-assembled segmented wormlike MCMs were in thermodynamic metastability due to the slow or inhibited chain exchange between spherical MCMs. The results obtained in the present work may also be caused by the similar reasons.
The DLS analysis shows that the average radius of the secondary assembled MCMs is longer than that of the primary assembled MCMs, indicating severe PnBA collapse on the core surface, bigger multi-compartmentalized core and the accelerated aggregation between MCMs during the secondary self-assembly (Fig. 5). The size distribution of secondary MCMs, prepared with copolymer solutions with concentrations ≤0.5 mg mL−1, is narrow and the intensities of their peaks are identical, indicating the MCMs or the high level aggregates are similar under these conditions (Fig. 5a–d). The MCMs became bigger when the concentration of the copolymer was increased to 2.0 mg mL−1, indicating the formation of large amount of patchy micelles or the involvement of MCMs in higher level aggregation (Fig. 5e). P2VP shell swells during dialysis, which segregates the patchy micelles or MCMs prepared with low concentration copolymer solutions during the dialysis. More patchy micelles or MCMs are formed with high concentration of copolymers and they are closer or even contact directly with each other, leading to “effective collision” and aggregation between the insoluble core blocks to decrease the interfacial tension.41 Generally, the size distribution of the MCMs formed by the dialysis of the micelle with concentrations lower than its critical aggregation concentration (CAC) is narrow. In contrast, the dialysis of the micelle with concentration higher than its CAC or containing more primary aggregated micelles leads to “effective collision”. Thus, the CAC of the secondary assembly is slightly lower than 1.0 mg mL−1.
Conclusions
In the present work, PnBA28-b-PS37-b-P2VP73 triblock copolymer was prepared and its two-step hierarchical self-assembly was investigated. The effects of the concentration of copolymer used during the primary self-assembly on the morphology of MCMs were explored. Patchy micelles with PS and PnBA as multi-compartmentalized core and P2VP as shell were obtained in the poor solvent for PS and PnBA during the primary self-assembly process. The secondary self-assembly was conducted by dialyzing the patchy micellar solutions with various concentrations against the water with pH value of ∼3.0 to form MCMs. When the concentration was increased to 2 mg mL−1, the concentration of PnBA28-b-PS37-b-P2VP73 in the dialysis solution is higher than its CAC. “Effective collision” occurred between the aggregated patchy micelles and MCMs or between the closely contacted patchy micelles and MCMs, resulting in cylindrical MCMs with separated phases. In full, cylindrical MCMs with multi-phase can be obtained simply by tuning selective solvent and the concentration of the copolymer.Acknowledgements
Financial support by National Science Foundation of China for Youth Scholars (21104063) is greatly acknowledged.Notes and references
- W. Ian and G. J. Liu, Polymer, 2013, 54, 1950 CrossRef PubMed.
- F. Schacher, A. Walther and A. H. E. Müller, Langmuir, 2009, 25, 10962 CrossRef CAS PubMed.
- J. W. Hu, G. J. Liu and G. Nijkang, J. Am. Chem. Soc., 2008, 130, 3236 CrossRef CAS PubMed.
- C. M. Park, J. S. Yoon and E. L. Thomas, Polymer, 2003, 44, 6725 CrossRef CAS PubMed.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis, S. Pispas and A. A. Vgeropoulos, Prog. Polym. Sci., 2005, 30, 725 CrossRef CAS PubMed.
- H. J. Dou, G. J. Liu, J. Dupont and L. Z. Hong, Soft Matter, 2010, 6, 4214 RSC.
- C. A. Fustin, V. Abetz and J. F. Gohy, Eur. Phys. J. E, 2005, 16, 291 CrossRef CAS PubMed.
- A. O. Moughton, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2012, 45, 2 CrossRef CAS.
- T. Jiang, L. Q. Wang, S. L. Lin, J. P. Lin and Y. L. Li, Langmuir, 2011, 27, 6440 CrossRef CAS PubMed.
- A. Hanisch, A. H. Gröschel, M. Förtsch, D. Markus, J. Hiroshi, T. M. Ruhland, F. H. Schache and A. H. E. Müller, ACS Nano, 2013, 7, 4030 CrossRef CAS PubMed.
- J. T. Zhu and R. C. Hayward, Macromolecules, 2008, 41, 7794 CrossRef CAS.
- E. W. Price, Y. Y. Guo, C. W. Wang and M. G. Moffitt, Langmuir, 2009, 25, 6398 CrossRef CAS PubMed.
- G. Yu and A. Eisenberg, Macromolecules, 1998, 31, 5546 CrossRef CAS.
- J. F. Gohy, N. Willet, S. Varshney, J. X. Zhang and R. Jérôme, Angew. Chem., Int. Ed., 2001, 40, 3214 CrossRef CAS.
- X. Z. Jiang, G. Y. Zhang, N. Ravin and S. Y. Liu, Langmuir, 2009, 25, 2046 CrossRef CAS PubMed.
- K. Skrabania, A. Laschewsky, H. Berlepsch and C. Böttcher, Langmuir, 2009, 25, 7594 CrossRef CAS PubMed.
- F. Schacher, A. Walther, M. Ruppel, M. Drechsler and A. H. E. Müller, Macromolecules, 2009, 42, 3540 CrossRef CAS.
- A. Wolf, A. Walther and A. H. E. Mülle, Macromolecules, 2011, 44, 9221 CrossRef CAS.
- S. Kubowicz, J. F. Baussard, J. F. Lutz, A. F. Thünemann, H. Berlepsch and A. Laschewsky, Angew. Chem., Int. Ed., 2005, 44, 5262 CrossRef CAS PubMed.
- H. Berlepsch, C. Böttcher, K. Skrabania and A. Laschewsky, Chem. Commun., 2009, 17, 2290 RSC.
- K. Skrabania, H. Berlepsch, C. Böttcher and A. Laschewsky, Macromolecules, 2010, 43, 271 CrossRef CAS.
- A. Walther, C. Barner-Kowollik and A. H. E. Müller, Langmuir, 2010, 26, 12237 CrossRef CAS PubMed.
- Z. Y. Chen, H. G. Cui, K. Hales, Z. B. Li, K. Qi, D. J. Pochan and K. L. Wooley, J. Am. Chem. Soc., 2005, 127, 8592 CrossRef CAS PubMed.
- Z. B. Li, Z. Y. Chen, H. G. Cui, K. Hales, K. Qi, K. L. Wooley and D. J. Pochan, Langmuir, 2005, 21, 7533 CrossRef CAS PubMed.
- H. G. Cui, Z. Y. Chen, K. L. Wooley and D. J. Pochan, Macromolecules, 2006, 39, 6599 CrossRef CAS.
- M. Uchman, M. Štěpánek and K. Procházka, Macromolecules, 2009, 42, 5605 CrossRef CAS.
- B. Fang, A. Walther, A. Wolf, Y. Y. Xu, J. Y. Yuan and A. H. E. Müller, Angew. Chem., Int. Ed., 2009, 48, 2877 CrossRef CAS PubMed.
- A. Kotzev, A. Laschewsky and R. H. Rakotoaly, Macromol. Chem. Phys., 2001, 202, 3257 CrossRef CAS.
- S. Ritzenthaler, F. Court, L. David, E. Girard-Reydet, L. Leibler and J. P. Pascault, Macromolecules, 2002, 35, 6245 CrossRef CAS.
- S. Ritzenthaler, F. Court, L. David, L. Leibler and J. P. Pascault, Macromolecules, 2003, 36, 118 CrossRef CAS.
- Z. W. Ma, H. Z. Yu and W. Jiang, J. Phys. Chem., 2009, 113, 3333 CrossRef CAS PubMed.
- J. F. Gohy, E. Khousakoun, N. Willet, S. K. Varshney and R. Jérôme, Macromol. Rapid Commun., 2004, 25, 1536 CrossRef CAS PubMed.
- A. K. Brannan and F. S. Bates, Macromolecules, 2004, 37, 8816 CrossRef CAS.
- A. F. Thünemann, S. Kubowicz, H. Berlepsch and H. Möhwald, Langmuir, 2006, 22, 2506 CrossRef PubMed.
- R. Hoogenboom, F. Wiesbrock, M. A. M. Leenen, H. M. L. Thijs, H. Huang, C. Fustin, P. Guillet, J. F. Gohy and U. S. Schubert, Macromolecules, 2007, 40, 2837 CrossRef CAS.
- J. F. Lutz and A. Laschewsky, Macromol. Chem. Phys., 2001, 206, 813 CrossRef PubMed.
- Z. B. Li, M. A. Hillmyer and T. P. Lodge, Macromolecules, 2006, 39, 765 CrossRef CAS.
- Z. B. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98 CrossRef CAS PubMed.
- S. Zhong, H. G. Cui, Z. Y. Chen, K. L. Wooley and D. J. Pochan, Soft Matter, 2008, 4, 90 RSC.
- Z. L. Zhou, Z. B. Li, Y. Ren, M. A. Hillmyer and T. P. Lodge, J. Am. Chem. Soc., 2003, 125, 10182 CrossRef CAS PubMed.
- F. Schacher, E. Betthausen, A. Walther, H. Schmalz, D. V. Pergushov and A. H. E. Müller, ACS Nano, 2009, 3, 2095 CrossRef CAS PubMed.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. E. Müller, Nat. Commun., 2012, 3, 710 CrossRef PubMed.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Loweand and C. L. McCormick, Macromolecules, 2006, 39, 1724 CrossRef CAS.
- J. L. Qin, Y. M. Chen, D. D. Yan and F. Xi, Macromolecules, 2010, 43, 10652 CrossRef CAS.
- J. T. Zhu and W. Jiang, Macromolecules, 2005, 38, 9315 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |