
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalysis of water oxidation in acetonitrile by iridium oxide nanoparticles†
Jonnathan C.
Hidalgo-Acosta
a,
Manuel A.
Méndez
a,
Micheál D.
Scanlon
a,
Heron
Vrubel
a,
Véronique
Amstutz
a,
Wojciech
Adamiak
b,
Marcin
Opallo
b and
Hubert H.
Girault
*a
aLaboratoire d'Electrochimie Physique et Analytique, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: Hubert.Girault@epfl.ch
bInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprazaka 44/52, 01-224 Warsaw, Poland. E-mail: mopallo@ichf.edu.pl
First published on 27th October 2014
Abstract
Water oxidation catalysed by iridium oxide nanoparticles (IrO2 NPs) in water–acetonitrile mixtures using [RuIII(bpy)3]3+ as oxidant was studied as a function of the water content, the acidity of the reaction media and the catalyst concentration. It was observed that under acidic conditions (HClO4) and at high water contents (80% (v/v)) the reaction is slow, but its rate increases as the water content decreases, reaching a maximum at approximately equimolar proportions (≈25% H2O (v/v)). The results can be rationalized based on the structure of water in water–acetonitrile mixtures. At high water fractions, water is present in highly hydrogen-bonded arrangements and is less reactive. As the water content decreases, water clustering gives rise to the formation of water-rich micro-domains, and the number of bonded water molecules decreases monotonically. The results presented herein indicate that non-bonded water present in the water micro-domains is considerably more reactive towards oxygen production. Finally, long term electrolysis of water–acetonitrile mixtures containing [RuII(bpy)3]2+ and IrO2 NPs in solution show that the amount of oxygen produced is constant with time demonstrating that the redox mediator is stable under these experimental conditions.
Introduction
Society's ever-increasing demand for energy is still primarily satisfied by the combustion of fossil fuels. The environmental degradation associated with their extraction from the earth, their finite availability and the deleterious effects of global warming due to the release of vast quantities of carbon dioxide (CO2) on combustion make the development of a clean energy supply one of the main challenges facing our generation. Nature meets its energy requirements through the process of photosynthesis, where the energy necessary to convert CO2 into carbohydrates is obtained from sunlight and water. Inspired by this process, much effort is currently being devoted to the production of environmentally friendly solar fuels (such as molecular hydrogen (H2)) via water splitting, ideally solely using sunlight as the energy source and water as the sacrificial electron donor.1,2 The water splitting reaction is best viewed as the sum of two half-reactions, the hydrogen evolution reaction (HER) and the water oxidation reaction (WOR). In practice, the HER is a much less energy-demanding process than the WOR, with the latter being considered the bottleneck of water splitting. The complexity of the WOR arises from the requirement to transfer four electrons and four protons for the total conversion of water into molecular oxygen (O2), a process that involves multiple intermediaries separated in many cases by significant energy barriers.3 Therefore, the use of a catalyst is required in order to produce O2 at reasonable rates and/or overpotentials.The catalytic WOR in aqueous media is well documented in the literature.4–21 However, relatively few studies have been devoted to the influence of primarily non-aqueous conditions, such as trace water present in organic solvents or ionic liquids, on this key reaction.22–25 Such a bias for aqueous conditions is surprising given that (i) nature carries out the WOR under hydrophobic conditions in the presence of trace water as the O2-evolving complex of photosystem (II) is embedded in the lipid environment of thylakoid membranes,26,27 (ii) the stabilities of molecular water oxidation catalysts (WOCs) are known to improve in non-aqueous environments28 and (iii) changes in the solvation environment of water are known to drastically modify its physicochemical properties29 and reactivity.22,30 In this study, we focus on the latter point regarding the changes in reactivity of water molecules dissolved in an organic solvent. Previously, for the HER, we have shown that the reactivity of dissolved protons, and in effect their susceptibility to reduction by electron donors, is dependent on their surrounding medium.31 In a series of articles, we highlighted that organic solubilized protons may be reduced using lipophilic weak electron donors, such as decamethylferrocene (DMFc), that are thermodynamically incapable of reducing aqueous protons. Utilizing ion-transfer voltammetry, we proved that the reduction potential of these transferred protons in 1,2-dichloroethane was shifted positively over half a volt, allowing their reduction to H2 by DMFc. This biphasic HER reaction was easily catalyzed by Pt (ref. 32) and other more earth-abundant “floating catalytic rafts” such as molybdenum disulfide (MoS2) and molybdenum carbide (Mo2C) nanoparticles grown on carbon supports such as carbon nanotubes or reduced graphene oxide.33–35
Meyer and co-workers have studied the mechanism and kinetics of the electrocatalytic WOR in miscible water–propylene carbonate (PC) solutions at nanostructured indium tin oxide (ITO) electrodes modified with the molecular catalysts [Ru(Mebimpy)(bpy)(OH2)]2+ (Mebimpy = 2,6-bis(1-methylbenzimidazol-2-yl)pyridine; bpy = 2,2′-bipyridine)23 and [FeIII(dpaq)(H2O)]2+ (dpaq = 2-[bis(pyridine-2-ylmethyl)] amino-N-quinolin-8-yl-acetamido).25 Their choice of organic solvent was based on the relatively high miscibility of PC with water (up to 8% (v/v)), the solvents wide polarizable potential window with an oxidative limit of >2 V (vs. SHE) and its weak coordinating ability in comparison to water.23 Switching from an acidic aqueous solution to the primarily non-aqueous environment of a miscible water–PC solution lead to an increase in the rate of the WOR by a factor of 300 for [Ru(Mebimpy)(bpy)(OH2)]2+ and gave the first clear well-defined experimental evidence of water oxidation electrocatalysis for the molecular Fe complex, [FeIII(dpaq)(H2O)]2+ (albeit a factor of 10 times slower than the Ru polypyridyl oxidant). These positive results were attributed in both instances to the known destabilization of water in PC, i.e. the water is in effect more reactive and susceptible to oxidation in PC.36
In parallel, Bond and co-workers have identified ionic liquids as another viable non-aqueous environment to modulate the reactivity of dissolved water, allowing its ease of oxidation and facilitating its unexpected use as a sacrificial electron donor under conditions where it normally remains inert in wet organic solvents or neat water.22,30 The unique environment provided by the ionic liquid 1-n-butyl-3-methylimidazolium tetrafluoroborate (BMIMBF4) allowed water to act as a sacrificial electron donor in the presence of Ag+ and tetracyanoquinodimethane (TCNQ) to evolve O2 quantitatively and produce exceptionally long AgTCNQ wires.22 In a separate study, water effectively acted as a sacrificial four-electron donor to photochemically reduce polyoxometalate anions (POMs) in wet ionic liquids.24 This exceptional reactivity was attributed to the modification of the structure of molecular water in ionic liquids where it may exist in a unique “free” state that excludes self-aggregation via hydrogen-bonding. Consequently, the water molecules tend to interact more strongly with the anions of the ionic liquid and, along with other possible contributory features of modified water in ionic liquids listed by Bond and co-workers,22 the net effect is a significant decrease of the activation energy for the WOR in ionic liquids.
Clearly, the influence of a primarily non-aqueous environment profoundly affects the structure, reactivity and mechanistic pathways adopted by dissolved water molecules during the catalytic WOR. To date, the limited number of reports in non-aqueous environments have all focused on homogeneous molecular water oxidation catalysts.23,25 Herein, we present a systematic analysis of the influence of the constituents of the reaction media (catalyst concentration, buffer employed and especially the water content) on the catalytic WOR in water–acetonitrile (ACN) miscible solutions using well-established iridium oxide nanoparticles (IrO2 NPs) as heterogeneous WOCs and [RuIII(bpy)3]3+ as the sacrificial electron acceptor or redox shuttle.
Experimental
Monitoring the kinetics of water oxidation in water–ACN mixtures by UV/vis spectroscopy
Kinetic measurements of the WOR in water–ACN mixtures were obtained by monitoring the disappearance of the UV/vis absorbance peak corresponding to the oxidized sacrificial electron acceptor, [RuIII(bpy)3]3+ (λmax = 673 nm, prepared as the hydrophobic salt RuIII(bpy)3(PF6)3 as detailed in the ESI†), with time using an Ocean Optics USB 4000 fiber optic spectrophotometer. Details of the synthesis and characterization by electrochemical methods, UV/vis spectroscopy, elemental analysis and 1H NMR spectroscopy, of RuIII(bpy)3(PF6)3 and RuII(bpy)3(PF6)2 are provided in the ESI.† All kinetic measurements were performed using aqueous solutions and dry ACN solvent thoroughly de-gassed with nitrogen, under anaerobic conditions in a glovebox purged with nitrogen (O2 < 1 ppm, H2O < 1 ppm) and at an ambient temperature of 23 ± 2 °C.Two separate kinetic studies were performed. The first involved investigating the influence of “acidity regulators” and the concentration of IrO2 NPs on the rates of the WOR. For the synthesis as well as the characterization of IrO2 NPs by UV/vis spectroscopy and transmission electron microscopy (TEM) see ESI.† Perchloric acid (HClO4) and a buffer designed specifically for the WOR by Mallouk and co-workers,37–39 a bicarbonate–hexafluorosilicate (NaHCO3–Na2SiF6) mixture, were employed as the “acidity regulators”. The latter was prepared as previously described.38 The total water content in the water–ACN mixture was maintained constant at 10% (v/v) for these measurements. In a typical experiment 1.5 mL of 2.67 mM [RuIII(bpy)3]3+ dissolved in dry ACN was placed in a quartz cuvette. Next, 0.5 mL of a water–ACN mixture (0.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) containing the acidity regulator and the catalyst was injected under vigorous stirring and the absorbance values at 673 nm and 900 nm recorded as a function of time. The final mixture contained either 4.3 mM NaHCO3–Na2SiF6 or 20 mM HClO4 and increasing concentrations of IrO2 NPs from 0 to 31 μM. The preparation of each sample with increasing IrO2 NPs concentration is detailed explicitly in the ESI.† The pre-mixing of water–ACN prior to injection into the pure dry ACN solvent was essential to obtain accurate kinetic rate constants in the first seconds after injection. Direct injection of a pure aqueous solution to dry ACN leads to slow mixing and dramatic changes in the optical properties of the solution, thereby, affecting accurate determination of the kinetic rate constants. However, it is noteworthy that implementing the pre-mixing protocol restricts the appearance of absorbance peak spikes or instabilities to the first second after catalyst injection, see Fig. S10, ESI.† Thus, data acquired in the first second after injection was not used for the kinetic analysis. The absorbance value at 900 nm was used for background correction. This methodology enabled changes in the baseline to be corrected, in particular immediately after injection when the two miscible liquids of different refractive indices mix, see Fig. S10, ESI.† The second set of kinetic data was obtained to highlight the influence of varying the water content (from 5 to 85 % (v/v)) in the water–ACN mixture on the rate of the WOR in the presence of 20 mM HClO4 and 27 μM IrO2 NPs. Once more, the preparation of each sample with increasing water content is detailed explicitly in the ESI.† Again, on injection under vigorous stirring, the absorbance values at 673 nm and 900 nm were recorded as a function of time. Additionally, changes in the viscosity of the water–ACN mixtures with increasing water content were determined (see ESI†).
:1 v/v) containing the acidity regulator and the catalyst was injected under vigorous stirring and the absorbance values at 673 nm and 900 nm recorded as a function of time. The final mixture contained either 4.3 mM NaHCO3–Na2SiF6 or 20 mM HClO4 and increasing concentrations of IrO2 NPs from 0 to 31 μM. The preparation of each sample with increasing IrO2 NPs concentration is detailed explicitly in the ESI.† The pre-mixing of water–ACN prior to injection into the pure dry ACN solvent was essential to obtain accurate kinetic rate constants in the first seconds after injection. Direct injection of a pure aqueous solution to dry ACN leads to slow mixing and dramatic changes in the optical properties of the solution, thereby, affecting accurate determination of the kinetic rate constants. However, it is noteworthy that implementing the pre-mixing protocol restricts the appearance of absorbance peak spikes or instabilities to the first second after catalyst injection, see Fig. S10, ESI.† Thus, data acquired in the first second after injection was not used for the kinetic analysis. The absorbance value at 900 nm was used for background correction. This methodology enabled changes in the baseline to be corrected, in particular immediately after injection when the two miscible liquids of different refractive indices mix, see Fig. S10, ESI.† The second set of kinetic data was obtained to highlight the influence of varying the water content (from 5 to 85 % (v/v)) in the water–ACN mixture on the rate of the WOR in the presence of 20 mM HClO4 and 27 μM IrO2 NPs. Once more, the preparation of each sample with increasing water content is detailed explicitly in the ESI.† Again, on injection under vigorous stirring, the absorbance values at 673 nm and 900 nm were recorded as a function of time. Additionally, changes in the viscosity of the water–ACN mixtures with increasing water content were determined (see ESI†).
The analysis of the products for the mixtures containing 10% water was carried out under identical conditions to those previously described but using a septum sealed cell. At t = 0, a fixed amount of catalyst was injected to the cell. Once the time necessary for the completion of the reaction had elapsed (in excess of 5 min), the headspace of the vial was sampled using a syringe with a push–pull valve (SGE Analytical Sciences) and subsequently analyzed by gas chromatography using a Perkin-Elmer gas chromatograph (Clarus 500, equipped with 5 Å molecular sieves 80/100 mesh) with a thermal conductivity detector (TCD) and argon as the carrier gas. The liquid was analyzed by 1H NMR spectroscopy and UV/vis spectroscopy. In this set of experiments the final concentration of IrO2 NPs was 31 μM. Further details of the methodologies followed for the analyses by gas chromatography and 1H NMR spectroscopy are provided in the ESI.†
Electrochemical measurements in organic media
All electrochemical measurements in organic media were performed in a three-electrode configuration using a PGSTAT 30 potentiostat (Metrohm, CH). No iR compensation was applied to the cell. All voltammetry experiments were completed using organic media thoroughly de-gassed with nitrogen, under anaerobic conditions in a glovebox filled with nitrogen and at an ambient temperature of 23 ± 2 °C.Bulk electrolysis experiments were carried out in water–ACN mixtures initially containing 2 mM RuII(bpy)3(PF6)2, 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6), 31 μM IrO2 NPs and 4.3 mM NaHCO3–Na2SiF6. A classic bulk electrolysis cell configuration was employed using a Duocel® reticulated vitreous carbon working electrode (RVC, pores/inch = 30; relative density 3%, supplied by ERG Aerospace Corporation, U.S.A.), a Pt-mesh counter electrode and a Ag/Ag+ double-junction organic reference electrode. The anodic and cathodic compartments were separated by a glass wool plug to prevent the re-oxidation of O2 at the Pt-mesh counter electrode. An image of the full electrolysis experimental setup (Fig. S2†) is available in the ESI.† Prior to experiments, RVC electrodes were cleaned by immersion in ethanol for 20 min, followed by drying under a stream of nitrogen gas. RVC was chosen as the working electrode due to its large specific area, robust mechanical properties and high overpotential for the WOR compared to Pt under similar conditions. The gas products accumulated in the headspace of the electrolysis cell were sampled and analyzed by gas chromatography, as previously described.
Results and discussion
Designing a model system for water oxidation in a non-aqueous environment
Current state-of-the-art research to overcome the WOR “bottleneck” in water-splitting is dominated by the search for novel WOCs, either homogenous molecular or heterogeneous NP-based species and ideally consisting entirely of earth-abundant non-precious elements, that necessitate the lowest possible overpotential to achieve exceptionally fast catalytic rates. In this article, we deviate from this common approach and instead focus on improving the reactivity of the water molecules themselves, by changing the reaction conditions under which the WOR takes place, and not on improving the reactivity of the catalyst. Thus, in designing these experiments, we chose the well-known bench-mark catalyst and oxidant combination of IrO2 NPs and [RuIII(bpy)3]3+ to test the influence of the composition of the reaction media on the WOR catalytic rates.As noted in detail vide supra, the reactivity of water molecules varies considerably in non-aqueous environments. We chose a primarily non-aqueous environment based on water–ACN mixtures as a large body of physicochemical40,41 and spectroscopic data is available for this solvent system.42–48 Additionally, ACN is an abundant solvent of low toxicity, is miscible with water in any proportion, has a wide polarizable potential window with an oxidative limit of >2 V (vs. SHE)49 and is thus stable in the presence of the powerful oxidant [RuIII(bpy)3]3+.
[RuIII(bpy)3]3+ is typically produced photochemically in situ, in order to act as an oxidant for the WOR in the presence of an appropriate catalyst, such as IrO2 NPs.7,12,37 In the presence of light, [RuII(bpy)3]2+ is excited and the metal-to-ligand charge transfer (MLCT) excited state, [Ru(bpy)3]2+*, is formed. The latter is oxidatively quenched by a suitable sacrificial electron acceptor, such as persulfate (S2O82−), to produce [RuIII(bpy)3]3+. In the presence of IrO2 NPs, the [RuIII(bpy)3]3+ is reduced back to [RuII(bpy)3]2+ and O2 is produced. Detailed kinetic studies of the WOR in aqueous media mediated by [RuIII(bpy)3]3+ and catalyzed by IrO2 NPs were reported by Morris and coworkers in a NaHCO3–Na2SiF6 buffer (pH 5.7).39 The analysis of the kinetic data showed that the rate-determining step (RDS) in the process is electron transfer between [RuIII(bpy)3]3+ and the surface Ir catalytic centers.39 In the present work, we have chemically synthesized [RuIII(bpy)3]3+ as its hydrophobic PF6 salt for direct use as a sacrificial oxidant. The advantages of this strategy in comparison to the aqueous photochemical approach were threefold: (i) it enabled us to carry out homogeneous kinetic measurements in hydrophobic ACN-rich media, (ii) the stability of [RuIII(bpy)3]3+ in dry ACN is drastically improved compared to an aqueous solution (see Fig. S9, ESI†) and (iii) we avoided the complications associated with the use of S2O82−, namely its poor solubility in organic media and the presence of highly oxidizing intermediates (e.g., SO4˙−) that may themselves promote the WOR and obscure the accurate determination of kinetic rate constants.
Before embarking on a detailed kinetic study, a simple preliminary experiment was devised to quickly test if water molecules were indeed more reactive in the primarily non-aqueous environment of the present model water–ACN system. The first step involved forming an immiscible biphasic water–ACN system by dissolving potassium chloride in the aqueous phase at a concentration of 1 M before contacting it with an equal volume of ACN.50 Upon vigorous stirring, a stable biphasic system was obtained with the less dense ACN-rich phase on top and the aqueous-rich phase at the bottom. Addition of a known amount of solid RuIII(bpy)3(PF6)3 (5 to 10 mg) to the resulting biphasic system in the absence of catalyst revealed a faster reduction of [RuIII(bpy)3]3+ in the ACN-rich phase. This clear increase in reactivity was visible to the naked eye as the ACN-rich phase immediately turns orange whereas the aqueous-rich phase retained the green colour of unreacted [RuIII(bpy)3]3+ (see Fig. 1 and Movie S1, ESI†). The facile reduction of [RuIII(bpy)3]3+ in the ACN-rich phase may be attributed to the WOR and, perhaps, to an extent to the co-evolution of Cl2. Hence, this simple experiment provides a qualitative example of the increased reactivity of water molecules in an ACN-rich phase.
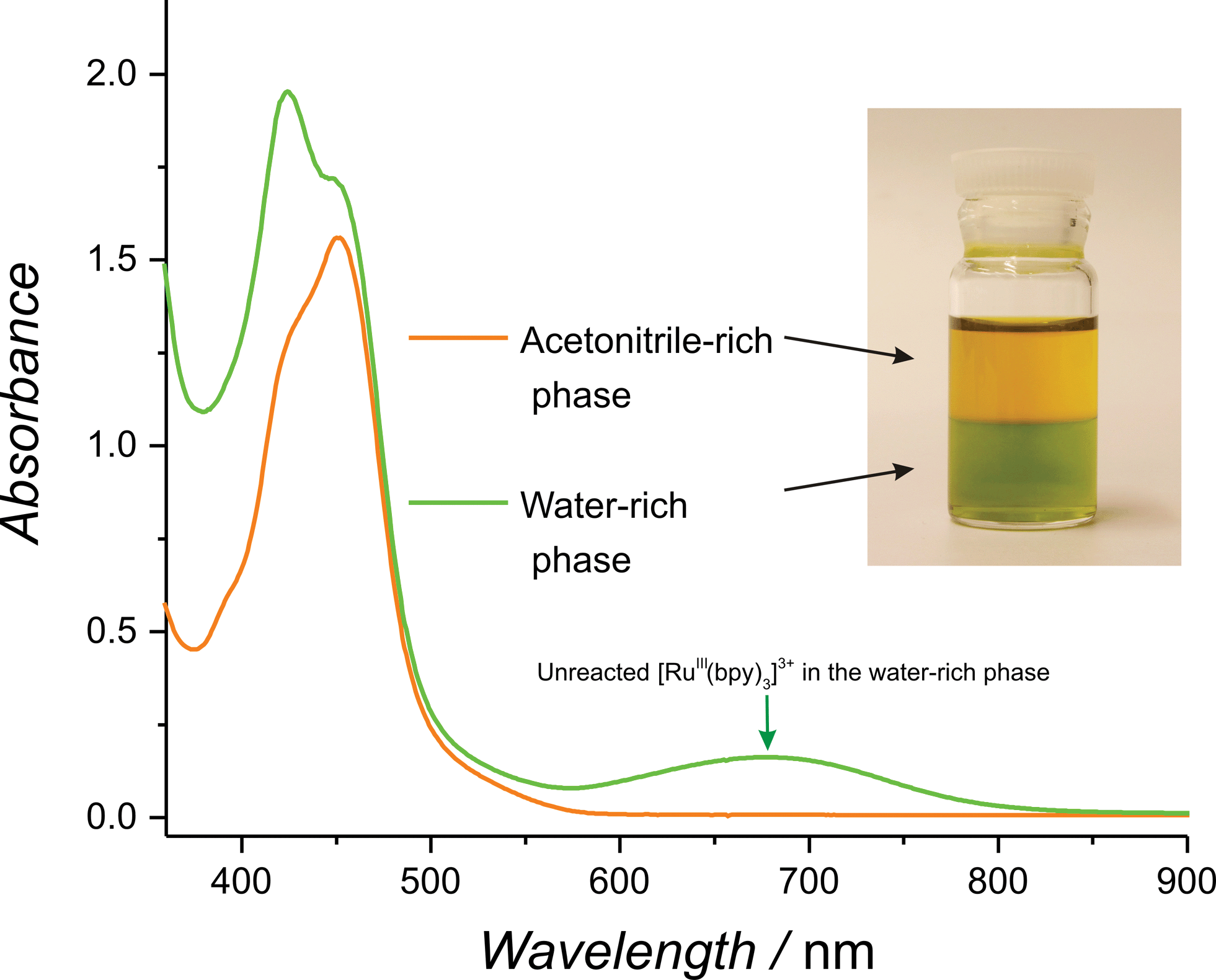 | ||
| Fig. 1 Highlighting the increased reactivity of water molecules in a non-aqueous environment. UV/vis spectra of both phases in a biphasic system composed of a water-rich phase (green in colour) and an organic-rich phase (orange in colour) after the addition of small amounts of RuIII(bpy)3(PF6)3 (for details see the text and Movie S1, ESI†). | ||
Consistent with these observations, UV/vis spectra of both phases taken after 1 min. of reaction (Fig. 1) revealed a significant characteristic absorption band for unreacted [RuIII(bpy)3]3+ at 673 nm in the water-rich (bottom) phase and its complete absence in the ACN-rich (top) phase. Even more strikingly, rapid and vigorous O2 evolution was observed to occur preferentially in the organic phase (Movie S1, ESI†).
Influence of “acidity regulators” and the IrO2 NPs concentration on the kinetics of the WOR
The preliminary experiment depicted in Fig. 1 and Movie S1 (ESI†), clearly shows that the WOR proceeds faster in the primarily non-aqueous water–ACN environment even in the absence of catalyst. Thus, initial kinetic experiments were designed to see if the reactivity of the water molecules with [RuIII(bpy)3]3+ as the electron acceptor in the water–ACN mixture could be optimally tuned by introducing catalytic IrO2 NPs and “acidity regulator” aqueous solutions of HClO4 or NaHCO3–Na2SiF6 (pH = 5.7). Although the pH is difficult to control given the obvious difficulties in the evaluation of the activity coefficient of the proton, the weak basicity of ACN ensures that it is not directly protonated to form CH3CNH+ in water–ACN mixtures. Thus, in the event of protons being introduced either by dissociation of a strong acid or released during the WOR, the solvation shell of the proton is practically in the form of (H2O)nH+·ACN.29 Therefore, the aqueous buffer will still act as an acidity regulator in this mixed environment. The use of buffer is crucial since changes of the proton activity during the WOR significantly influence the thermodynamic driving force of the reaction, making analysis of the data rather complex. A set of experiments highlighting the difficulty in obtaining meaningful kinetic data in the absence of buffer is available in the ESI† (Fig. S11).The corrected UV/vis absorbance profiles for the disappearance of the absorbance band at 673 nm representing the consumption of [RuIII(bpy)3]3+ during the WOR are presented in Fig. 2A (HClO4) and 2B (NaHCO3–Na2SiF6), both without catalyst and in the presence of increasing concentrations of IrO2 NPs. This real time absorbance data was re-plotted on a logarithmic scale in Fig. 2C (HClO4) and 2D (NaHCO3–Na2SiF6) to obtain first-order rate constants. Linear trends were observed for both acidity regulators and at all IrO2 NP concentrations investigated up to 5 s after injecting the water–ACN mixture (containing IrO2 NPs and the acidity regulator) to the dry ACN solvent (containing [RuIII(bpy)3]3+). Deviations from this linear behavior at longer time scales were attributed to significant changes in the acidity of the environment due to the release of considerable quantities of protons during the WOR, the ensuing larger thermodynamic driving force required to overcome the WOR overpotential under these conditions and the resultant slower rates of reaction in comparison to initial conditions.
 | ||
| Fig. 2 The influence of “acidity regulators”. Real time absorbance measurements for [RuIII(bpy)3]3+ solutions after the addition of a mixture containing water, acetonitrile, buffer, and IrO2 nanoparticles in different concentrations. The acidity regulators used were (A) 20 mM HClO4 and (B) 4.3 mM NaHCO3–Na2SiF6. In both cases the absorbances were plotted as the difference between the values at 673 nm and 900 nm. The final content of water was 10% (v/v). ln[absorbance] vs. time plots derived from the data in (A) for 20 mM HClO4 and (B) for 4.3 mM NaHCO3–Na2SiF6 are shown in (C) and (D), respectively. The inset in each case represents the plot of the first order constant determined as the slope of the curve vs. the concentration of catalyst. | ||
Considering that water is always in large excess with respect to the electron donor, the rate law can be the expressed as:
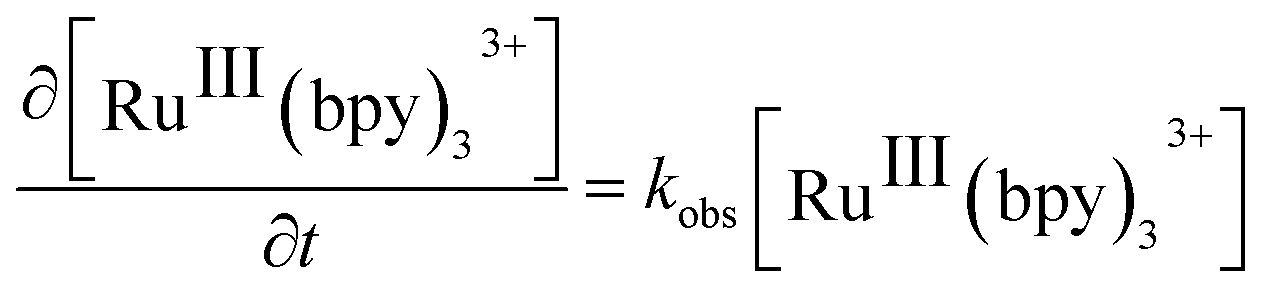 | (1) |
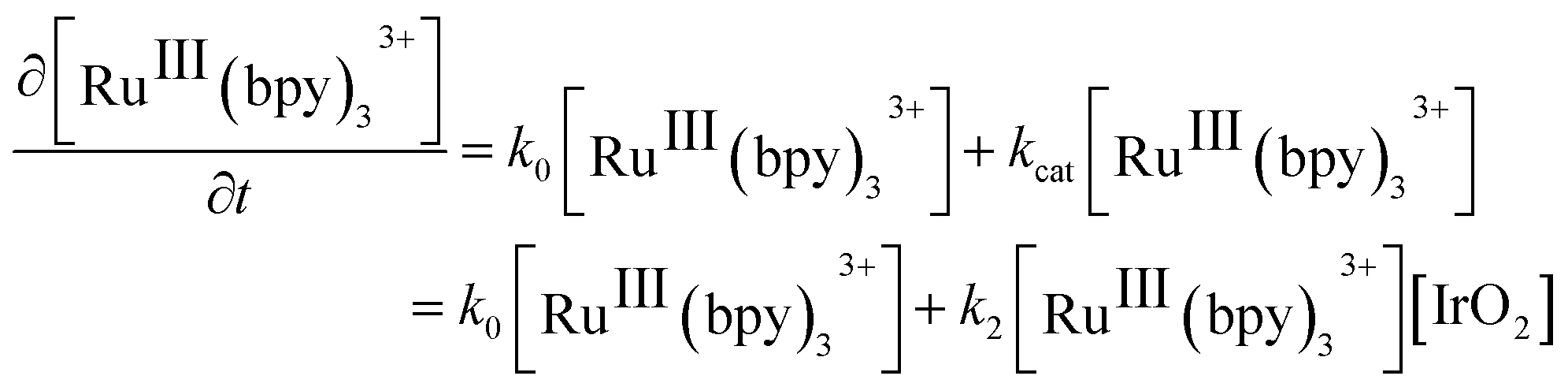 | (2) |
A number of clear trends emerge on inspection of the kinetic data extracted from Fig. 2 and summarized in Table 1. Firstly, higher rates of reaction were observed when NaHCO3–Na2SiF6 was used as the acidity regulator, both in the absence and presence of IrO2 NPs, in comparison to those obtained with HClO4. The former trend (without catalysis) is solely due to the higher thermodynamic driving force required to oxidize water in the presence of higher HClO4 concentrations. Meanwhile, the latter trend (with catalysis) is additionally influenced by the pH dependent redox properties of the IrO2 NP surface active sites.51−53 Indeed, although the conditions of the NaHCO3–Na2SiF6 buffered WOR studied by Morris et al. are different to those discussed herein, in terms of reaction-media composition and [RuIII(bpy)3]3+ concentration, the magnitude order of the rate constants between both studies were identical, giving credibility to our obtained values.39 Secondly, a linear relationship exists between the concentration of IrO2 NPs and the observed rates of catalysis with both buffering systems, supporting the bimolecular nature of the mechanism under the present conditions (eqn (2)).
| System | k 0 /10−4 s−1 | [IrO2]/μM | k cat/10−3 s−1 | k 2/M−1 s−1 |
|---|---|---|---|---|
| HClO4 | 7 | 6.2 | 4.0 | 209 |
| 12.4 | 5.5 | |||
| 18.6 | 5.9 | |||
| 24.8 | 7.9 | |||
| 31.0 | 9.3 | |||
| NaHCO3–Na2SiF6 | 154 | 6.2 | 55.1 | 14130 |
| 12.4 | 96.5 | |||
| 18.6 | 218.6 | |||
| 24.8 | 299.6 | |||
| 31.0 | 391.6 |
In addition to the rates of catalysis, another key attribute of a water oxidation system is the stability of the redox acceptor. Ghosh et al. have reported that in the absence of a catalyst the reduction of [RuIII(bpy)3]3+ in water yields negligible amounts of O2 or H2O2. This observation was attributed to the irreversible degradation of the [RuIII(bpy)3]3+ into a variety of products including several Ru complexes and CO2.54 Similar results were found herein, with no O2 detected by gas chromatography. Efforts to detect H2O2 by the well-known starch-iodine method were hampered by the poor solubility of the reagents in the ACN–water mixture. Thus, although very low amounts of H2O2 were detected, the results were irreproducible and not reliable.
However, unlike previous reports, irreversible degradation of [RuIII(bpy)3]3+ was not observed by 1H NMR spectroscopy (Fig. S12, ESI†). In the presence of catalyst (31 μM) and NaHCO3–Na2SiF6 buffer, the yield of O2 was 61% based on the initial amount of [RuIII(bpy)3]3+ present. Once more, in the presence of catalyst, no degradation of [RuIII(bpy)3]3+ was observed by 1H NMR spectroscopy (Fig. S13, ESI†). Tentatively, non-catalyzed reduction of [RuIII(bpy)3]3+ may take place with concomitant oxidation of the buffer species, thus explaining the non-stoichiometric evolution of O2 even in the presence of catalyst. The absence of degradation products both with and without catalyst is noteworthy since many of the practical difficulties associated with the use of [RuIII(bpy)3]3+ are related to its degradation over extended periods of time.
Influence of the water content on the kinetics of the WOR in a non-aqueous environment
A second series of kinetic studies were performed to elucidate the optimal water content in the water–ACN mixtures to achieve the fastest WOR kinetic rates in the presence of 27 μM IrO2 NPs and HClO4 as the acidity regulator. HClO4 was chosen in preference to NaHCO3–Na2SiF6 as (i) its slower base rate of catalysis was favored to experimentally observe clear changes in the kinetic rate constants, (ii) the experimental design necessitated the preparation of [RuIII(bpy)3]3+ in an aqueous solution, i.e., at large water contents of 70 to 85% (v/v), and HClO4 provided an acidic environment where [RuIII(bpy)3]3+ was stable for a few minutes (see ESI†).The influence of the water content in the water–ACN mixture on the normalized second order rate constant k2 (see eqn (2)) is shown in Fig. 3. The observed trend may be divided into three distinct regions of water mole fractions (XH2O): (i) a water-rich region (0.8 ≤ XH2O ≤ 0.95), (ii) a region consisting of water–ACN micro-heterogeneities or micro-domains (0.4 ≤ XH2O ≤ 0.8) and (iii) an ACN-rich region (0.1 ≤ XH2O ≤ 0.4). The term micro-heterogeneity refers to the situation where molecules of the solvent, in this case water and/or ACN, are preferentially surrounded by molecules of their own kind. Thus, in this region, reported by Takamuku et al. to extend over the range 0.4 ≤ XH2O ≤ 0.8,48 both water and ACN clusters co-exist in the mixture. The observed trend in Fig. 3 indicates that the rate of the WOR is slow in the water-rich region, progressively increases until XH2O ≈ 0.7 and then rapidly increases to a maximum in the micro-heterogeneity region. Finally, in the ACN-rich region the rate rapidly declines in a linear fashion from the peak rate reached at XH2O = 0.5.
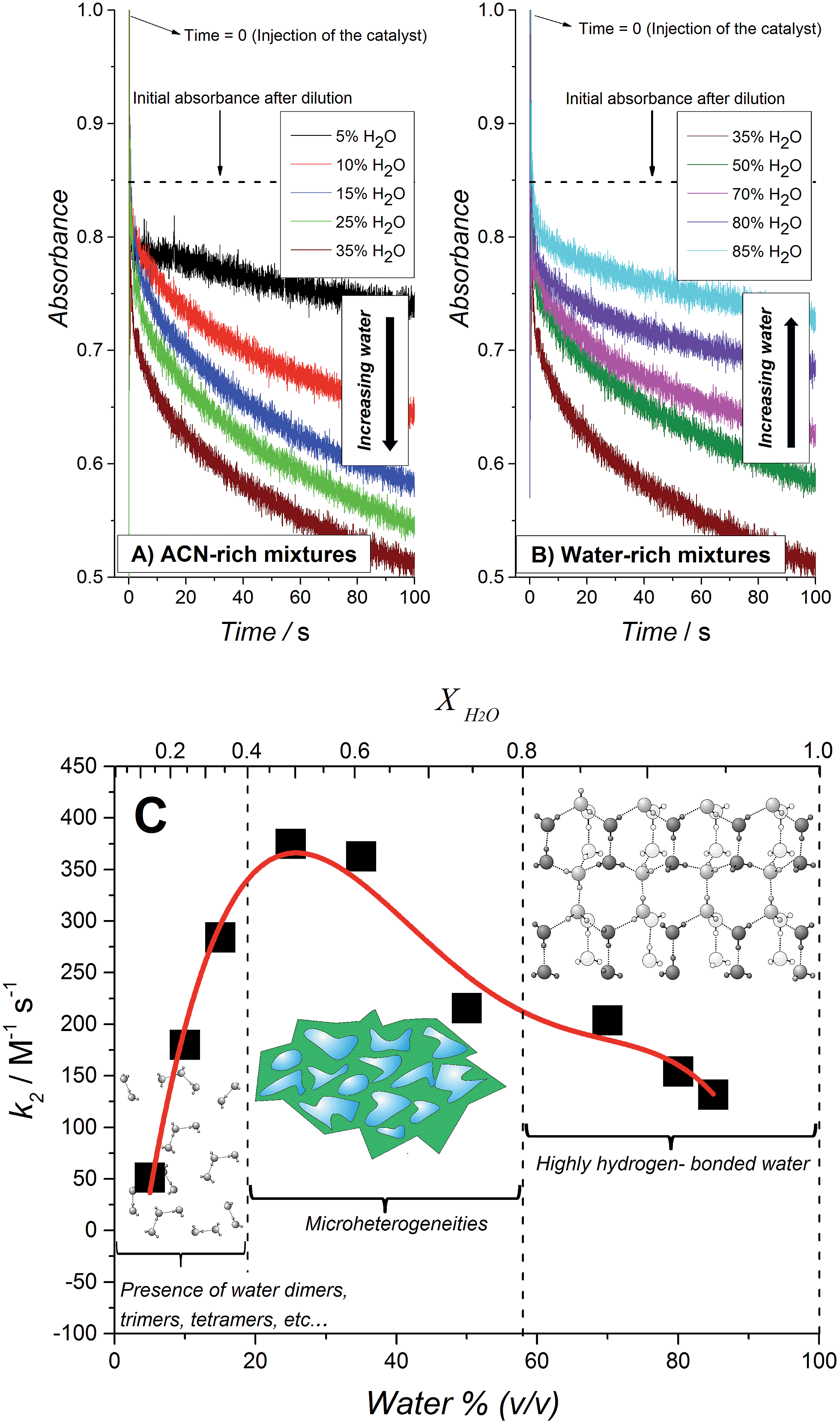 | ||
| Fig. 3 The influence of water content on the kinetics of the WOR. (A and B) Real time absorbance measurements for [RuIII(bpy)3]3+ solutions after the addition of a mixture containing water, acetonitrile, HClO4 and IrO2 nanoparticles. The final concentration of water was varied in each experiment and the concentrations of HClO4 and IrO2 were fixed at 20 mM and 27 μM, respectively. (C) Normalized second order constant (k2) for [RuIII(bpy)3]3+ reduction vs. the total content of water expressed both as water percentage (v/v) and water mole fraction (XH2O). | ||
The initial progressive increase in the rate of the WOR with decreasing XH2O is attributed to a gradual decrease in the strength of the hydrogen-bonding network with increasing ACN content, as previously postulated for water molecules dissolved in ionic liquids.22,24,30 X-ray diffraction and infra-red (IR) spectroscopy studies of water–ACN mixtures by Takamuku et al. revealed that the strong hydrogen-bond network begins to gradually degrade on the introduction of ACN, this process rapidly accelerates at XH2O ≈ 0.7 (mirroring the rapid increase in rate observed experimentally in Fig. 3) and the hydrogen-bond network essentially disappears and the water molecules are present in their “free” state, free of self-aggregation, in the range 0.2 ≤ XH2O ≤ 0.4. The removal of the water–water H-bonds diminishes the “protection” of the water molecules and increases the susceptibility of the O-atom in water to interact with the catalytically active iridium species at the surface of the IrO2 NPs. The subsequent rapid linear decrease in the rate of the WOR in the ACN-rich region (0.1 ≤ XH2O ≤ 0.4) may be initially surprising as one would expect that the “free” water molecules in this regime would be the most reactive.
Takamuku et al. reported that the electron donicity and acceptability properties of water–ACN mixtures, measured as the ability to solvate the VO(acac)2 complex48 (D coefficient), decreases monotonically from pure water to pure ACN showing two inflection points, one of them at low water concentrations (XH2O = 0.2). As water has a larger acceptor number (54.8) than ACN (19.3), the D coefficient value reflects the solvation ability by water rather than ACN molecules. In other words, the ability of water to scavenge protons at low molar fractions is decreased even though the O-atom is “more free” to participate in the oxygen transfer step needed to carry out water oxidation. This results in a bell-shaped dependence of the rate of water oxidation with varying XH2O, as observed in Fig. 3.
Control IR spectroscopy experiments (see Fig. S14 and S15, ESI†) were performed in an identical manner to those described by Takamuku et al. but incorporating the diverse additional species present in the water–ACN mixtures during kinetic experiments for the WOR, namely, sensitizer molecules (Ru(bpy)3(PF6)2), acid (HClO4) and heterogeneous catalytic species (IrO2 NPs), as outlined in Fig. 3. The trends observed by Takamuku et al. were not affected by the presence of the aforementioned additional species, thus validating our comparative analysis between the kinetic data shown in Fig. 3 and the spectroscopic data obtained by Takamuku et al.
Concerning the interaction of the IrO2 NPs with water in the mixed solvent media, we propose that water interacts preferentially with the IrO2 surface. We base our hypothesis on the experimental observation that at water contents below 10% and [IrO2] > 60 μM, the catalytic NPs aggregate and precipitate. This behavior is indicative of reduced NP stability at low water content. It also indicates that water, besides being the reagent for the WOR, suppresses aggregation of IrO2 NPs due to its preferential interaction with the NP surface. Further qualitative evidence of the hydrophilic nature of the IrO2 surface is the observation of the preferential partition of IrO2 NPs in immiscible biphasic systems to the water-rich phase rather than the ACN-rich phase, as clearly shown in Fig. S8, ESI.†
As a result, reactive weakly hydrogen-bonded water micro-domains are brought in direct contact with IrO2 NPs and stabilize the catalytic NPs (Scheme 1). Upon addition of [RuIII(bpy)3]3+ dissolved in ACN, it will partition across the interface formed between the water-rich domain at the NP surface and the ACN-rich bulk phase. Following its partition, the electron acceptor will diffuse into the water-rich overlayer, reach the surface of the catalytic NP and undergo electron-transfer to produce [RuII(bpy)3]2+ and oxidized Ir centers. Concomitant water oxidation occurs at the IrO2 NP producing O2 and protons. O2, being neutral and more soluble in ACN will rapidly partition to the organic-rich phase. The importance of proton acceptor species (B in Scheme 1) is also a crucial factor in the WOR kinetics, as B will interact with protons, probably in a concerted way with the electron transfer process, to form HB. Partition of ionic species (e.g., [RuIII(bpy)3]3+) across the interface formed between the two liquid phases can in turn lead to the polarization of this liquid–liquid interface. The effect of the polarization herein proposed requires further study, and may provide another parameter to drive the reaction.
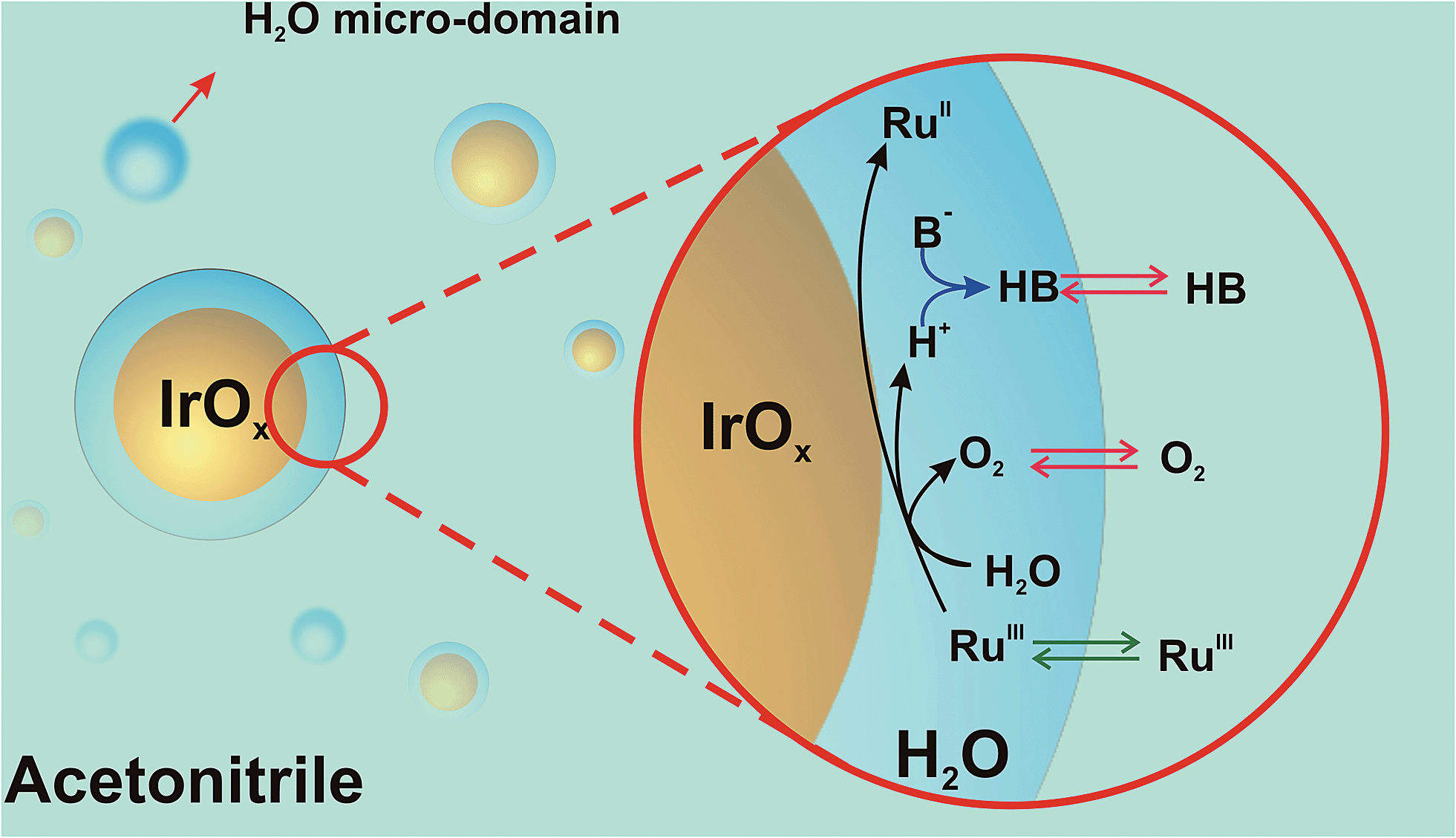 | ||
| Scheme 1 Proposed formation of reactive water overlayers on IrO2 nanoparticles. RuIII = [RuIII(bpy)3]3+, RuII = [RuII(bpy)3]2+ and B− = proton acceptor species (in the absence of any additional basic species B = H2O). | ||
Continuous electrocatalytic O2 evolution in water–ACN mixtures with [RuII(bpy)3]2+ as a redox shuttle
The robust nature of the electrocatalytic WOR in non-aqueous conditions was probed by bulk electrolysis experiments whereby the WOR was catalyzed by colloidal IrO2 NPs floating in solution and [RuII(bpy)3]2+ acted as a recyclable redox shuttle transferring electrons from the surface of the electrode to the IrO2 NPs, whereupon they were used to drive the WOR (see Fig. 4 for the reaction scheme).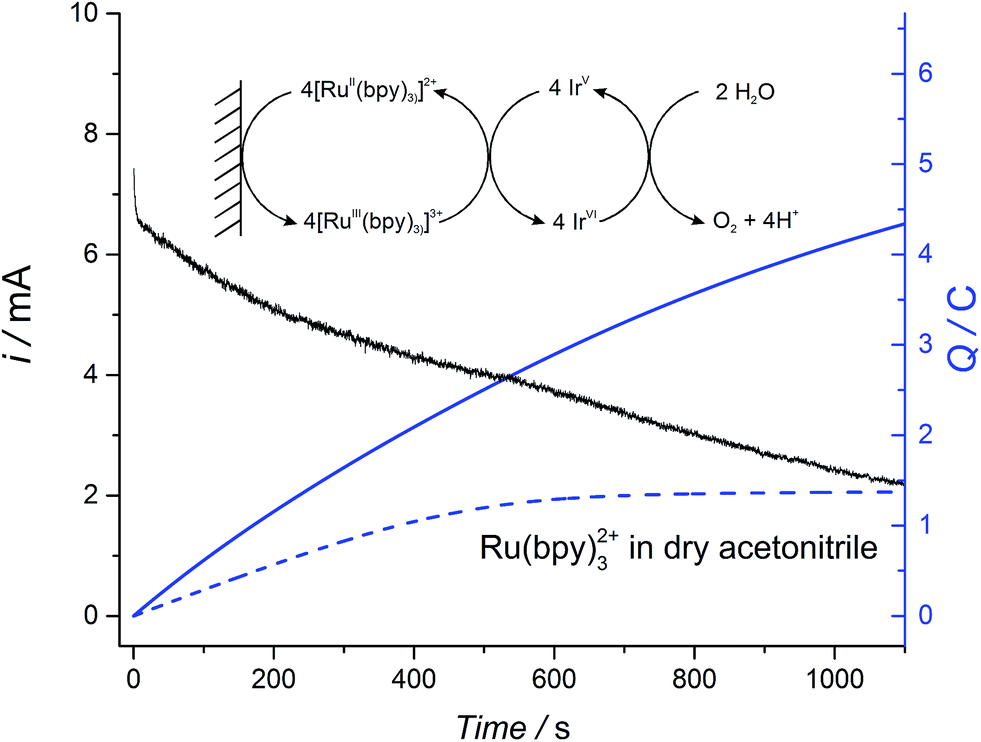 | ||
| Fig. 4 Plots of the current (black line) and charge (blue line) during bulk electrolysis of a solution containing 2 mM Ru(bpy)3(PF6)2, 31 μM IrO2 NPs and 4.3 mM buffer NaHCO3–Na2SiF6, in a water–ACN mixture (2.5:7.5 (v/v)). For comparison, the plot of charge during bulk electrolysis of 2 mM Ru(bpy)3(PF6)2 (blue dotted line) in dry acetonitrile was included. In each case the supporting electrolyte was 0.1 M TBAPF6. The electrode was RVC (for details see Experimental section). | ||
The optimal potential at which to carry out bulk electrolysis was determined from cyclic voltammograms of 2 mM [RuII(bpy)3]2+ at two different water contents (10% and 25% (v/v)) and in the presence of IrO2 NPs (Fig. S16 ESI†). A quasi-reversible voltammetric wave corresponding to the [RuIII(bpy)3]3+/[RuII(bpy)3]2+ redox couple was observed at ∼0.9 V (vs. Fc+/Fc) in both cases. The considerable iR drop present due to the electrolysis cell design leads to the observation of a broad wave rather than a sharp peak for the anodic response. Thus, all bulk electrolysis experiments were conducted at a potential 400 mV more positive than the less-distorted cathodic peak of the [RuIII(bpy)3]3+/[RuII(bpy)3]2+ redox couple.
Representative current-time and charge-time bulk electrolysis profiles are shown in Fig. 4. A progressive decrease in the current was observed due to the change in the pH-dependent thermodynamic driving force with the continuous release of protons during the WOR (shown for the catalyzed reaction with 25% (v/v) water as the black trace in Fig. 4; data for 10% (v/v) water not shown). In the presence of IrO2 NPs and 10% or 25% (v/v) water, a non-negligible current at long time scales (>1000 s) was observed. This behavior is characteristic of a catalytic reaction, in which the redox shuttle is continuously recycled at the electrode surface (see inset Fig. 4), leading to a sustained increase in the amount of charge passed. By comparison, however, a plateau of charge passed is reached once all of the [RuII(bpy)3]2+ is oxidized at the electrode surface in the absence of a recycling mechanism when the experiment was performed in dry ACN and in the absence of catalyst (blue dotted line, Fig. 4). A further control experiment was performed to ensure that the extent of the direct WOR at the electrode surface during bulk electrolysis in the absence of both the [RuIII(bpy)]3+/[RuII(bpy)]2+ redox shuttle and catalytic IrO2 NPs is minimal (Fig. S17 ESI†).
Analysis of the effect of water content on the kinetics of the WOR measured by chronoamperometry is rather complex due to the large changes in viscosity as the water–ACN ratio increases (viscosity values at different water contents are shown in Fig. S18 ESI†). However, the values of O2 measured under recycling conditions were found to be dependent on the water content. Table 2 shows the Faradaic efficiency, determined as a function of water content, as well as the O2 yield for the chemical reduction of [RuIII(bpy)3]3+ under the same experimental conditions. With 10% (v/v) water present, the yields of O2 evolved obtained by both chemical reduction and electrochemical reduction are essentially the same. Nonetheless, as the water content is increased to 25% (v/v), the yields of O2 evolved via chemical reduction and bulk electrolysis deviated by 10%. This difference is attributed to faster degradation of the redox shuttle upon electrochemical cycling as the water content increases.
| Water content/% (v/v) | Yield chemical oxidation/% | Bulk electrolysis | |
|---|---|---|---|
| Charge/C | Faradic efficiency/% | ||
| 10 | 61 | 4.05 | 63 |
| 25 | 64 | 4.34 | 55 |
Conclusions
The present results clearly demonstrate that IrO2-catalyzed water oxidation is dependent on the water content for water–acetonitrile mixtures. Based on kinetic measurements, the maximum activity for the catalyst in acidic media was exhibited when the biphasic media was composed of approximately equimolar proportions of water and acetonitrile. Under such experimental conditions hydrogen bonding is weakened and isolated reactive pools of water are generated. Formation of reactive water overlayers on the surface of IrO2 NPs is also proposed. All in all, the present strategy represents a better approach to characterize the catalytic activities of water oxidation catalysts in organic media that could be applied to a wide range of catalysts and organic solvents. On the other hand, the use of water–acetonitrile mixtures appears a viable alternative to enhance the stability of redox shuttles, a fundamental problem in photocatalytic water oxidation systems in general.1Acknowledgements
We acknowledge financial support from the project Polswiss PSPB-35/2010.Notes and references
- J. Barber, Chem. Soc. Rev., 2009, 38, 185 RSC.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946 CrossRef CAS PubMed.
- W. Rüttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1 CrossRef PubMed.
- H. Yamazaki, A. Shouji, M. Kajita and M. Yagi, Coord. Chem. Rev., 2010, 254, 2483 CrossRef CAS PubMed.
- M. Barth, M. Lapkowski, W. Urek, J. Muszynski and S. Lefrant, Synth. Met., 1997, 84, 111 CrossRef CAS.
- A. Sartorel, M. Carraro, F. M. Toma, M. Prato and M. Bonchio, Energy Environ. Sci., 2012, 5, 5592 CAS.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795 RSC.
- A. Harriman, M.-C. Richoux, P. A. Christensen, S. Mosseri and P. Neta, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 3001 RSC.
- P. K. Dutta and S. K. Das, J. Am. Chem. Soc., 1997, 119, 4311 CrossRef CAS.
- F. Locatelli, B. Didillon, D. Uzio, G. Niccolai, J. P. Candy and J. M. Basset, J. Catal., 2000, 193, 154 CrossRef CAS.
- M. Hara, J. T. Lean and T. E. Mallouk, Chem. Mater., 2001, 13, 4668 CrossRef CAS.
- P. G. Hoertz, Y.-I. Kim, W. J. Youngblood and T. E. Mallouk, J. Phys. Chem. B, 2007, 111, 6845 CrossRef CAS PubMed.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia and G. F. Swiegers, Acc. Chem. Res., 2009, 42, 1935 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2010, 132, 11467 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS.
- C. Sens, I. Romero, M. Rodríguez, A. Llobet, T. Parella and J. Benet-Buchholz, J. Am. Chem. Soc., 2004, 126, 7798 CrossRef CAS PubMed.
- T. Wada, K. Tsuge and K. Tanaka, Angew. Chem., Int. Ed., 2000, 39, 1479 CrossRef CAS.
- T. Wada, K. Tsuge and K. Tanaka, Inorg. Chem., 2000, 40, 329 CrossRef PubMed.
- R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802 CrossRef CAS PubMed.
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462 CrossRef CAS.
- Y. Xu, A. Fischer, L. Duan, L. Tong, E. Gabrielsson, B. Åkermark and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 8934 CrossRef CAS PubMed.
- C. Zhao and A. M. Bond, J. Am. Chem. Soc., 2009, 131, 4279 CrossRef CAS PubMed.
- Z. Chen, J. J. Concepcion, H. Luo, J. F. Hull, A. Paul and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 17670 CrossRef CAS PubMed.
- G. Bernardini, C. Zhao, A. G. Wedd and A. M. Bond, Inorg. Chem., 2011, 50, 5899 CrossRef CAS PubMed.
- M. K. Coggins, M.-T. Zhang, A. K. Vannucci, C. J. Dares and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 5531 CrossRef CAS PubMed.
- A. R. Holzwarth, M. G. Müller, M. Reus, M. Nowaczyk, J. Sander and M. Rögner, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6895 CrossRef CAS PubMed.
- G. Renger and T. Renger, Photosynth. Res., 2008, 98, 53 CrossRef CAS PubMed.
- Y. Liu, S.-X. Guo, A. M. Bond, J. Zhang and S. Du, Electrochim. Acta, 2013, 101, 201 CrossRef CAS PubMed.
- N. B.-M. Kalish, E. Shandalov, V. Kharlanov, D. Pines and E. Pines, J. Phys. Chem. A, 2011, 115, 4063 CrossRef CAS PubMed.
- G. Bernardini, A. G. Wedd, C. Zhao and A. M. Bond, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11552 CrossRef CAS PubMed.
- I. Hatay, B. Su, F. Li, R. Partovi-Nia, H. Vrubel, X. Hu, M. Ersoz and H. H. Girault, Angew. Chem., Int. Ed., 2009, 48, 5139 CrossRef CAS PubMed.
- J. J. Nieminen, I. Hatay, P. Ge, M. A. Mendez, L. Murtomaki and H. H. Girault, Chem. Commun., 2011, 47, 5548 RSC.
- P. Ge, M. D. Scanlon, P. Peljo, X. Bian, H. Vubrel, A. O'Neill, J. N. Coleman, M. Cantoni, X. Hu, K. Kontturi, B. Liu and H. H. Girault, Chem. Commun., 2012, 48, 6484 RSC.
- X. Bian, M. D. Scanlon, S. Wang, L. Liao, Y. Tang, B. Liu and H. H. Girault, Chem. Sci., 2013, 4, 3432 RSC.
- M. D. Scanlon, X. Bian, H. Vrubel, V. Amstutz, K. Schenk, X. Hu, B. Liu and H. H. Girault, Phys. Chem. Chem. Phys., 2013, 15, 2847 RSC.
- J. Muzikar, T. van de Goor, B. Gaš and E. Kenndler, Anal. Chem., 2002, 74, 428 CrossRef CAS.
- M. Hara, C. C. Waraksa, J. T. Lean, B. A. Lewis and T. E. Mallouk, J. Phys. Chem. A, 2000, 104, 5275 CrossRef CAS.
- N. D. Morris and T. E. Mallouk, J. Am. Chem. Soc., 2002, 124, 11114 CrossRef CAS PubMed.
- N. D. Morris, M. Suzuki and T. E. Mallouk, J. Phys. Chem. A, 2004, 108, 9115 CrossRef CAS.
- C. Moreau and G. Douhéret, Thermochim. Acta, 1975, 13, 385 CrossRef CAS.
- M. A. Villamanan and H. C. Van Ness, J. Chem. Eng. Data, 1985, 30, 445 CrossRef CAS.
- E. Von Goldammer and H. G. Hertz, J. Phys. Chem., 1970, 74, 3734 CrossRef CAS.
- H. Kovacs and A. Laaksonen, J. Am. Chem. Soc., 1991, 113, 5596 CrossRef CAS.
- D. Jamroz, J. Stangret and J. Lindgren, J. Am. Chem. Soc., 1993, 115, 6165 CrossRef CAS.
- J. E. Bertie and Z. Lan, J. Phys. Chem. B, 1997, 101, 4111 CrossRef CAS.
- A. Wakisaka, Y. Shimizu, N. Nishi, K. Tokumaru and H. Sakuragi, J. Chem. Soc., Faraday Trans., 1992, 88, 1129 RSC.
- A. Wakisaka, S. Takahashi and N. Nishi, J. Chem. Soc., Faraday Trans., 1995, 91, 4063 RSC.
- T. Takamuku, M. Tabata, A. Yamaguchi, J. Nishimoto, M. Kumamoto, H. Wakita and T. Yamaguchi, J. Phys. Chem. B, 1998, 102, 8880 CrossRef CAS.
- T. R. O'Toole, J. N. Younathan, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1989, 28, 3923 CrossRef.
- J. D. Watkins, F. Amemiya, M. Atobe, P. C. Bulman-Page and F. Marken, Electrochim. Acta, 2010, 55, 8808 CrossRef CAS PubMed.
- A. A. Gambardella, N. S. Bjorge, V. K. Alspaugh and R. W. Murray, J. Phys. Chem. C, 2011, 115, 21659 CAS.
- K. E. Michaux and R. W. Murray, Langmuir, 2013, 29, 12254 CrossRef CAS PubMed.
- T. Nakagawa, C. A. Beasley and R. W. Murray, J. Phys. Chem. C, 2009, 113, 12958 CAS.
- P. K. Ghosh, B. S. Brunschwig, M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1984, 106, 4772 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, syntheses, characterization, details concerning the kinetic experiments. See DOI: 10.1039/c4sc02196g |
| This journal is © The Royal Society of Chemistry 2015 |