
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein ubiquitination and formation of polyubiquitin chains without ATP, E1 and E2 enzymes†
Sungjin
Park
,
David T.
Krist
and
Alexander V.
Statsyuk
*
Department of Chemistry, Center for Molecular Innovation and Drug Discovery, Chemistry of Life Processes Institute, Northwestern University, Silverman Hall, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: a-statsyuk@northwestern.edu
First published on 26th November 2014
Abstract
Studying protein ubiquitination is difficult due to the complexity of the E1–E2–E3 ubiquitination cascade. Here we report the discovery that C-terminal ubiquitin thioesters can undergo direct transthiolation with the catalytic cysteine of the model HECT E3 ubiquitin ligase Rsp5 to form a catalytically active Rsp5∼ubiquitin thioester (Rsp5∼Ub). The resulting Rsp5∼Ub undergoes efficient autoubiquitination, ubiquitinates protein substrates, and synthesizes polyubiquitin chains with native Ub isopeptide linkage specificity. Since the developed chemical system bypasses the need for ATP, E1 and E2 enzymes while maintaining the native HECT E3 mechanism, we named it “Bypassing System” (ByS). Importantly, ByS provides direct evidence that E2 enzymes are dispensable for K63 specific isopeptide bond formation between ubiquitin molecules by Rsp5 in vitro. Additionally, six other E3 enzymes including Nedd4-1, Nedd4-2, Itch, and Wwp1 HECT ligases, along with Parkin and HHARI RBR ligases processed Ub thioesters under ByS reaction conditions. These findings provide general mechanistic insights on protein ubiquitination, and offer new strategies for assay development to discover pharmacological modulators of E3 enzymes.
Introduction
Protein ubiquitination is a highly conserved post-translational modification that regulates fundamental cellular processes.1–3 Ubiquitin conjugation is controlled by the sequential action of three enzymes: ubiquitin-activating enzymes (E1, ∼2 known), ubiquitin-conjugating enzymes (E2, ∼37 known), and ubiquitin ligases (E3, ∼600 known).3 Among these, E3 ligases stand out due to the astonishing complexity and diversity of biochemical reactions they catalyze. E3 ligases control polyubiquitin chain linkages and polyubiquitin chain length, select specific substrates and specific residues to be ubiquitinated, as well as select and activate specific E2∼Ub thioesters for subsequent ubiquitin transfer events.4 Such complexity makes it difficult to study the biochemical properties of E3 ligases, and to design assays to discover and to characterize pharmacological modulators of E3s. Typical biochemical assays to study E3 enzymes require at least three enzymes E1/E2/E3, ubiquitin, and ATP. The situation is more complex in the case of multi-subunit E3s such as cullin–RING ligases and the APC/C, where 3–15 protein subunits are required to assemble the functional E3 ligase.5–7As a part of our long-term research program aimed at deciphering physiological roles of protein ubiquitination, we faced the need to address this challenge and simplify the highly complex E1 → E2 → E3 enzymatic cascade. We envisioned that removal of ATP, E1, and E2 enzymes from the enzymatic reaction mixture could serve the desired purpose.
Initially, we focused our efforts on Homologous to E6-AP Carboxyl Terminus (HECT) E3 ubiquitin ligases that have a catalytic cysteine and form a mandatory HECT E3∼Ub thioester conjugate during the E1–E2–HECT E3 enzymatic cascade.8–10 Since HECT E3 ubiquitin ligases are frequently misregulated in cancers and neurodegenerative diseases, tools to study the biochemistry and physiological functions of these enzymes are of significant importance.11–13 In addition, HECT E3 ubiquitin ligases frequently cross-talk with disease relevant kinase signaling pathways, suggesting an emerging therapeutic importance of HECT E3s.14
In the HECT E3 ubiquitination cascade, E1 enzymes activate the C-terminus of ubiquitin by forming a high energy E1∼Ub thioester adduct, while E2 enzymes transfer ubiquitin from E1 enzymes to the catalytic cysteine of HECT E3 ubiquitin ligases (Fig. 1A).9 Thus, for ubiquitin to travel from the E1 enzyme to E3 enzyme, two transthiolation reactions are needed. Since E1 enzymes activate the C-terminus of ubiquitin, we hypothesized that C-terminal ubiquitin thioesters, such as Ub–MES (mercaptoethanesulfonate),15 will mimic E1∼Ub thioesters, thereby allowing us to circumvent the need for E1 enzyme and ATP.
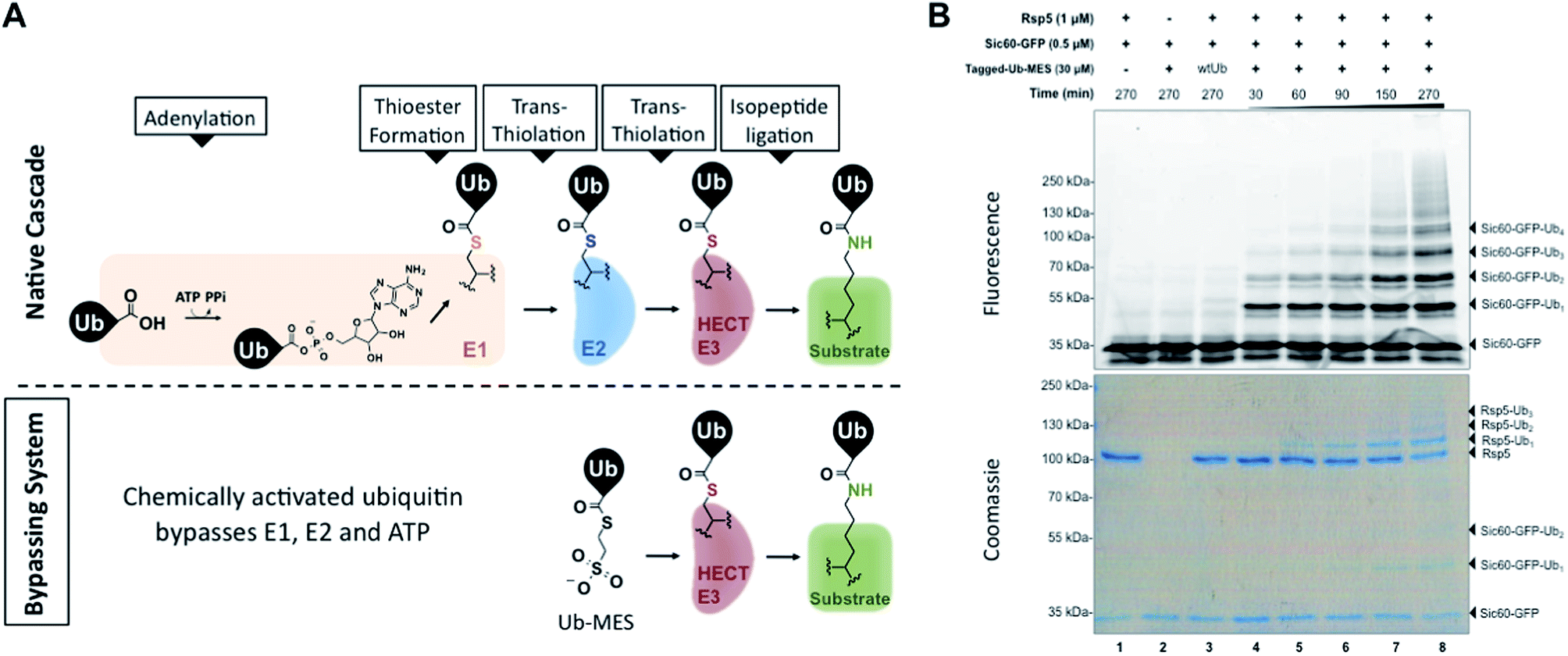 | ||
| Fig. 1 Bypassing System (ByS). (A) C-terminal ubiquitin thioester Ub–MES can form a catalytically active HECT E3∼Ub thioester adduct and conjugate ubiquitin to protein substrates, bypassing ATP, E1, and E2. (B) A time course of Sic60-GFP ubiquitination by Rsp5 and Tagged-Ub–MES. Reaction mixtures were incubated at room temperature for indicated times, quenched with Laemmli buffer, resolved by SDS-PAGE and imaged with in-gel fluorescence scanning and coomassie staining. | ||
We also envisioned that if C-terminal ubiquitin thioesters can undergo a transthiolation reaction with the catalytic cysteine of HECT E3, such ubiquitin thioester can also chemically mimic and circumvent the need for E2∼Ub thioester in the native HECT ubiquitination cascade. If all these assumptions are correct, C-terminal ubiquitin thioesters, such as Ub–MES, should mono- and polyubiquitinate protein substrates in the presence of HECT E3 enzyme, without a requirement for E1 enzyme, E2 enzyme, or ATP (Fig. 1A). Importantly, the proposed Ub–MES probe is fundamentally different from previously introduced activity-based ubiquitin probes for HECT E3 enzymes and deubiquitinating enzymes, which are based on C-terminal ubiquitin electrophiles such as Ub–VME.16,17 While Ub–VME acts as a suicide inhibitor of these enzymes, the proposed Ub–MES forms catalytically active HECT E3∼Ub covalent complex.
One application of such a system is to investigate the mechanism of polyubiquitin chain formation. Models by which HECT E3s catalyze polyubiquitin chain formation have been the focus of long-standing debate: (1) the sequential addition model (currently favored), and (2) other models, including the indexation and seesaw models.18 The first model implies that HECT E3s catalyze the formation of isopeptide linkages between ubiquitin molecules and are the primary determinants of polyubiquitin chain linkage specificity. In this case, HECT E3s assemble polyubiquitin chains via the sequential transfer of ubiquitin from their catalytic cysteine to lysine of acceptor ubiquitin at the end of a growing polyubiquitin chain. Other models imply that E2 and E3 enzymes pre-assemble polyubiquitin chains with specific linkages on their catalytic cysteines prior to en bloc transfer of the preassembled chains from the E3 enzyme to protein substrate. To this end, we believe that the bypassing system provides a very direct and simple experimental design to address whether E2 enzymes are needed for polyubiquitination.
Additionally, the bypassing system has potential to facilitate the development of simple assays to screen for small molecule modulators of HECT E3 enzymes. The advantages of these assays include a lower cost from the removal of E1 and E2 enzymes, and a lower number of false positives associated with the off-target inhibition of E1 and E2s.
This paper describes the discovery of a novel two-component enzymatic reaction in which the C-terminal ubiquitin thioester Ub–MES directly reacts with the model HECT E3 Rsp5 to produce Rsp5∼Ub thioester, which then ubiquitinates protein substrates, autoubiquitinates, and synthesizes polyubiquitin chains with specific isopeptide linkages. The developed reaction provides direct evidence that E2 enzymes are dispensable for the formation of K63-specific Ub–Ub isopeptide linkages formed by HECT E3s in vitro. Also, the reaction appears to be generally applicable to E3s that bear catalytic cysteines since the HECT E3s Nedd4-1, Nedd4-2, Itch, Wwp1 and the RING-in-between-RING (RBR) E3s Parkin and HHARI are active under these reaction conditions.
Results
Rsp5 can ubiquitinate its artificial substrate Sic60-GFP in the presence of Ub–MES
To test our initial hypothesis, we prepared N-terminal 3×FLAG–6×His-tagged ubiquitin (1–76) mercaptoethanesulfonate thioester (Tagged-Ub–MES, MW 12.5 kDa) (Fig. S1†).15 For a model HECT E3 ubiquitin ligase, we used Rsp5, a yeast homolog of the human HECT E3 Nedd4-1, and a fluorescent substrate Sic60-GFP, as described previously (Fig. S2†).19,20 Intriguingly, treatment of Rsp5 and Sic60-GFP with Tagged-Ub–MES, caused the formation of higher molecular weight fluorescent bands that corresponded to the combined molecular weights of Sic60-GFP (35 kDa) and increments of Tagged-ubiquitin (12.5 kDa) (Fig. 1B).The reaction was time- and concentration dependent with respect to Tagged-Ub–MES (Fig. 1B and S3†), and the ubiquitinated Sic60-GFP could be visualized by coomassie staining. Similar to native ubiquitination, we also observed the autoubiquitination of Rsp5, as judged by coomassie staining. Remarkably, Sic60-GFP was not labeled by Tagged-Ub–MES in the absence of Rsp5 despite the presence of 27 nucleophilic lysines in Sic60-GFP (Fig. 1B, lane 2), indicating high chemical specificity of the reaction. Since the discovered system bypasses the need for ATP, E1 and E2 to carry out ubiquitination, we refer to it as the ‘Bypassing System’ (ByS).
Protein ubiquitination via Rsp5/ByS depends on the catalytic cysteine of Rsp5, and enzyme/substrate recognition
Since Rsp5 could ubiquitinate Sic60-GFP in the presence of Tagged-Ub–MES, we hypothesized that Tagged-Ub–MES undergoes a transthiolation reaction with the catalytic Cys777 of Rsp5 to produce catalytically active Rsp5∼Ub thioester. We therefore tested if ubiquitination of Sic60-GFP by Rsp5 in the Bypassing System (Rsp5/ByS) depends on the catalytic or other solvent exposed surface cysteines of Rsp5.21To do so, we prepared the following Rsp5 mutants: (1) Rsp5 C777A in which the catalytic Cys777 is mutated to alanine, (2) Rsp5Δ3C in which three non-catalytic cysteines are removed (C455A, C517S, and C721A mutations), and (3) Rsp5Δ4C in which all cysteines including the catalytic cysteine are removed (C777A, C455A, C517S, and C721A mutations). As anticipated, only native Rsp5 and Rsp5Δ3C could ubiquitinate Sic60-GFP under ByS conditions (Fig. 2A). Notably, Rsp5 C777A bearing the three non-catalytic cysteines Cys455, Cys517, and Cys721 could not ubiquitinate Sic60-GFP or itself (Fig. 2A, lane 5), indicating that protein ubiquitination via Rsp5/ByS strictly depends on the catalytic cysteine of Rsp5.
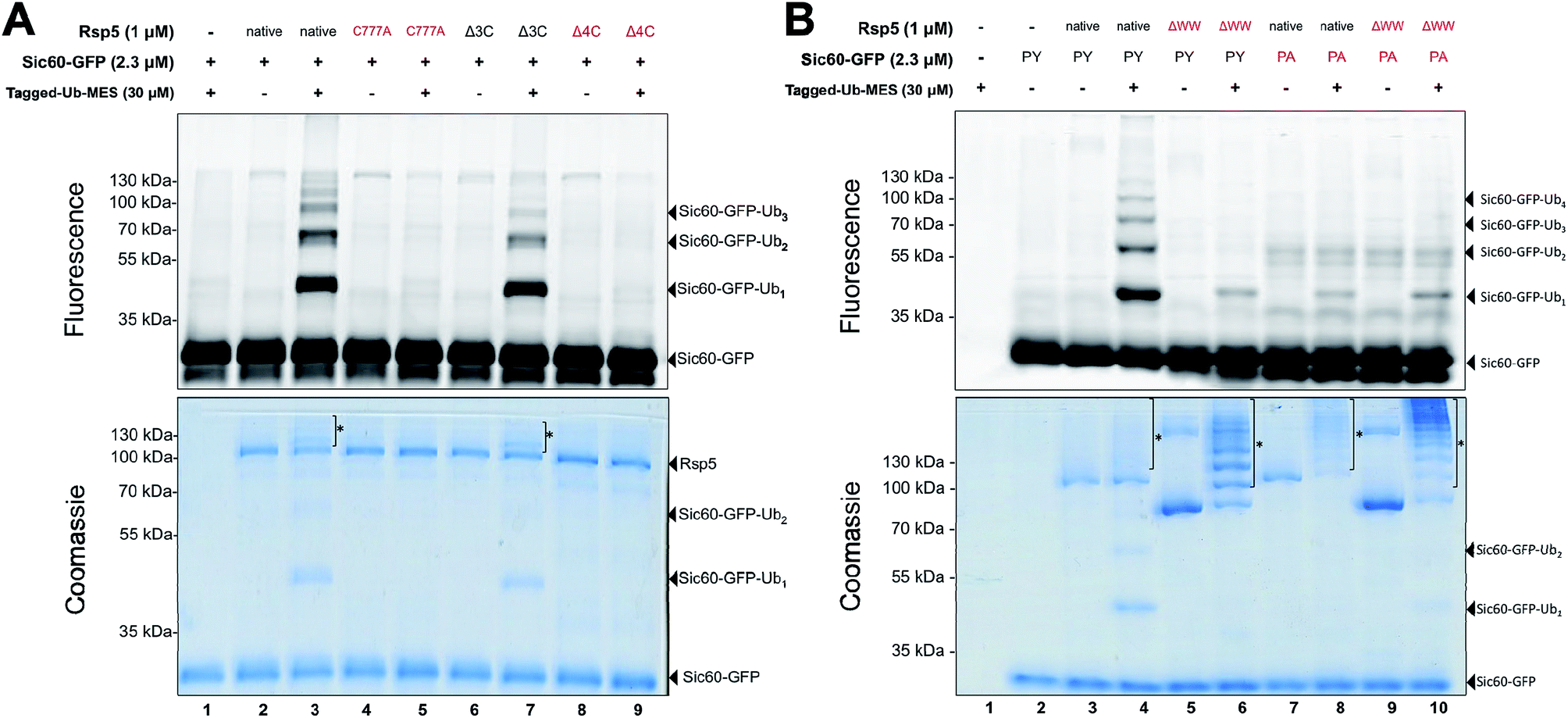 | ||
| Fig. 2 Ubiquitination via Rsp5/ByS depends on the catalytic cysteine of Rsp5 and enzyme/substrate recognition. (A) Rsp5/ByS depends on the presence of the Rsp5 catalytic cysteine. Ubiquitination of Sic60-GFP by Rsp5 mutants via ByS was analyzed by in-gel fluorescence scanning. Rsp5 variants without the catalytic cysteine (Cys777) are colored red. Autoubiquitinated Rsp5 is marked with *. Δ3C:Rsp5 lacks three non-catalytic cysteines. Δ4C:Rsp5 lacks all four cysteines. (B) Ubiquitination of Sic60-GFP via Rsp5/ByS depends on enzyme–substrate recognition. Ubiquitination of Sic60-GFP or Sic60PA-GFP in the presence of Rsp5 or Rsp5ΔWW was analyzed by in-gel fluorescence scanning. Rsp5ΔWW and Sic60PA-GFP are colored in red. Autoubiquitinated Rsp5 and Rsp5ΔWW are marked with *. ΔWW:Rsp5 lacks its WW domains, which recognize PY motifs on the substrate. PY: Sic60-GFP has its PY motif, while Sic60PA-GFP has a PY → PA mutation. | ||
We further investigated if the ubiquitination of Sic60-GFP via Rsp5/ByS depends on the specific binding between Rsp5 and Sic60-GFP. To test this, we prepared mutant Rsp5 and Sic60-GFP that lack key interacting elements: (1) Rsp5ΔWW in which the three WW domains of Rsp5 are deleted, and (2) Sic60PA-GFP in which the PY motif was mutated (PPPY → PPPA).22,23 Ablation of these interaction motifs led to the decreased ubiquitination of Sic60-GFP in the native protein ubiquitination cascade due to disruption of the enzyme–substrate interaction.19 Similarly, the ubiquitination of Sic60-GFP via Rsp5/ByS was decreased when Rsp5ΔWW or Sic60PA-GFP was used (Lanes 5–10 of Fig. 2B). The decrease in Sic60-GFP ubiquitination was not due to a lack of enzymatic activity since both native Rsp5 and Rsp5ΔWW were autoubiquitinated (Lanes 4, 6, 8, and 10 in Fig. 2B coomassie staining). Taken together, these results suggest that the ubiquitination of Sic60-GFP via Rsp5/ByS depends on enzyme–substrate recognition.
Rsp5-mediated ubiquitination under ByS conditions exploits the native mechanism
One possibility accounting for the observed ubiquitination via Rsp5/ByS could be a simple proximity-based transfer reaction, in which the Rsp5∼Tagged-Ub intermediate adopts a catalytic architecture that is distinct from that of the native system.24 In this scenario, ubiquitin charged to the catalytic cysteine of Rsp5 randomly collides with and is transferred to nearby Sic60-GFP lysine residues due to the innate conformational flexibility of Rsp5.25–27 Such concern is further supported by our recent work with chemical cross-linkers that indicated the catalytic cysteine of Rsp5 and the target lysine of Sic60-GFP become proximal even in the absence of E1, E2, ATP and loaded ubiquitin.19 If so, the observed protein ubiquitination via Rsp5/ByS does not reflect the native enzymatic reaction, but is rather a proximity driven ubiquitin transfer.To address this question, we prepared the mutant Rsp5806stop, in which the last four C-terminal residues of Rsp5 are deleted. In the native ubiquitination reaction, Rsp5806stop cannot transfer ubiquitin onto protein substrates, although it can still receive ubiquitin from E2∼Ub and form Rsp5806stop∼Ub thioester.28 Recently, Kamadurai et al. demonstrated that Phe806, located on the Rsp5 C-lobe, is crucial for the stabilization of a C-lobe/N-lobe bilobal composite catalytic site on Rsp5 to ligate ubiquitin onto substrate.24 As expected, the ubiquitination of Sic60-GFP via Rsp5806stop/ByS was severely impaired similar to Rsp5806stop in the native ubiquitination cascade (Fig. 3A, lane 4). In addition, similar to the native ubiquitination reaction, Rsp5806stop did form Rsp5806stop∼Ub thioester adducts (Fig. 3B, lane 4), as judged by the sensitivity of the Rsp5806stop∼Ub band to hydroxylamine (Fig. 3B, lane 6). Taken together our results suggest that Rsp5/ByS ubiquitinates Sic60-GFP via the intrinsic catalytic mechanism of Rsp5 utilized in the native ubiquitination cascade.
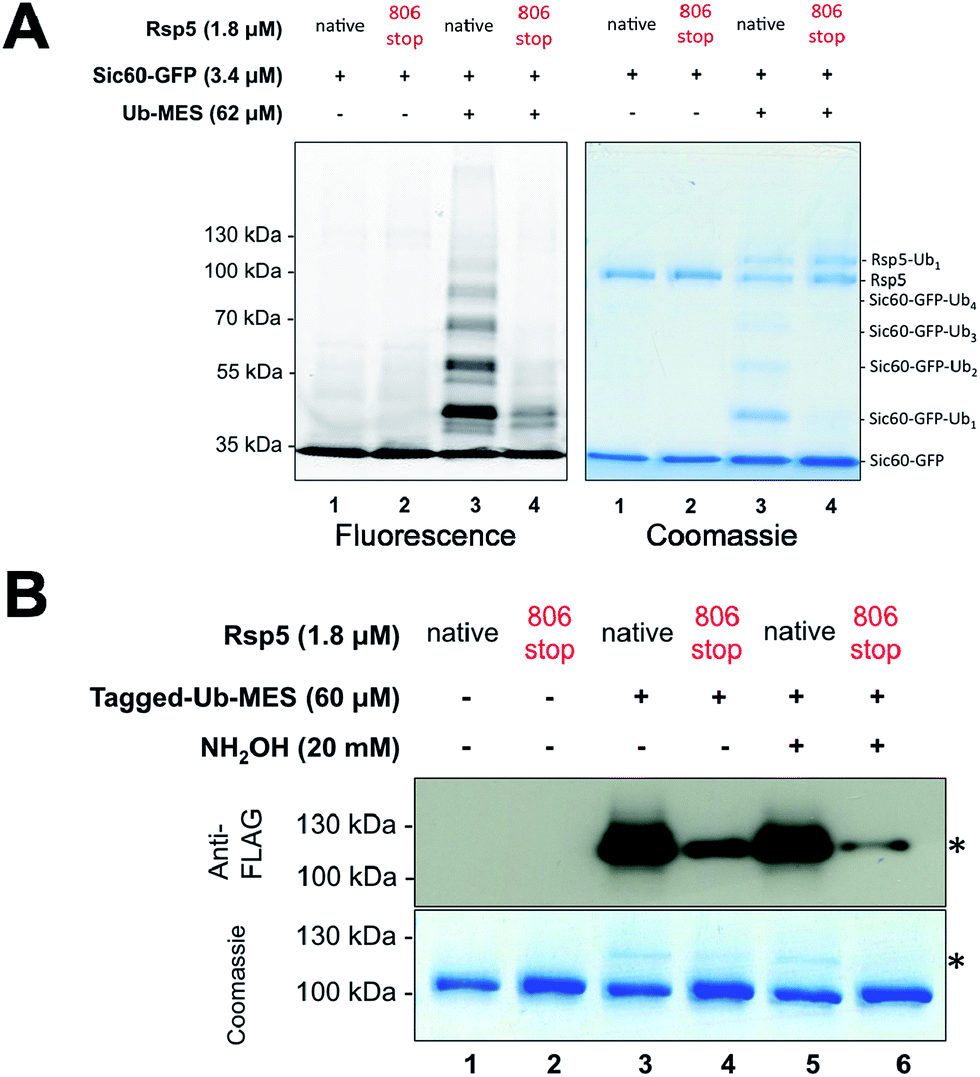 | ||
| Fig. 3 Ubiquitination via Rsp5/ByS recapitulates the instrinsic mechanism of Rsp5. (A) Ubiquitination of Sic60-GFP via Rsp5/ByS depends on the last four C-terminal amino acids of Rsp5. The ubiquitination of Sic60-GFP via Rsp5806stop/ByS is analyzed by in-gel fluorescence scanning after 4 hours of reaction time. (B) Rsp5806stop forms an inactive thioester adduct with Tagged-Ub-MES (Rsp5806stop∼Tagged-Ub). The formation of Rsp5806stop∼Tagged-Ub in the Rsp5806stop/ByS reaction was confirmed by Western-blotting with anti-FLAG antibody and coomassie staining. Reaction mixtures were incubated for 45 minutes at room temperature and quenched by either non-reducing Laemmli buffer or reducing Laemmli buffer containing NH2OH (20 mM, final concentration). The band corresponding to Rsp5806stop∼Tagged-Ub adduct is marked with *. | ||
Rsp5 can synthesize K63-linked polyubiquitin chains without E2 enzymes
Since our previous results showed that Rsp5/ByS ubiquitinates Sic60-GFP via the intrinsic catalytic mechanism, we asked if Rsp5 can form K63-linked polyubiquitin chains under ByS reaction conditions as in the native ubiquitination cascade.29–31 To test this, Rsp5ΔWW was treated with Tagless-Ub–MES (i.e. Ub–MES containing native ubiquitin) variants to monitor the formation of polyubiquitin chains. We used the following Tagless-Ub–MES variants: (1) Tagless-Ub(wt)–MES, (2) Tagless-Ub(K48R)–MES and (3) Tagless-Ub(K63R)–MES. The formation of K63-linked polyubiquitin chains at different time points was then evaluated using K63-ubiquitin linkage specific antibodies (Fig. 4A) and MALDI-TOF analysis (Fig. 4B–D). Gratifyingly, we detected K63-linked polyubiquitin chains under ByS reaction conditions when Rsp5 was treated with Tagless-Ub(wt)–MES or Tagless-Ub(K48R)–MES (Fig. 4A, lane 2–7). Treatment of Rsp5ΔWW with Tagless-Ub(K63R)–MES, however, did not produce K63-linked polyubiquitin chains (Fig. 4A, lane 8–10).To further validate our findings, we excised the gel region of 100–250 kDa, which contained most polyubiquitin chains as judged by coomassie (Fig. 4A, lane 4, 7 and 10), performed in-gel trypsin digestion, and analyzed the resulting peptides using MALDI-TOF. Similar to our western blotting results, we observed K63-linkage signal for the reaction mixtures of Rsp5ΔWW treated with Tagless-Ub(wt)–MES or Tagless-Ub(K48R)–MES (Fig. 4B and C, respectively), but not with the Tagless-Ub(K63R)–MES (Fig. 4D, Table S2–6†). Moreover, MALDI-TOF analysis of reactions with Tagless-Ub(K63R)–MES showed peptides from K48-linked chains, which are also known catalytic products of Rsp5 in the native cascade (Fig. 4D).31,32
Besides K63 and K48 isopeptide linkages, we did not detect any other major linkage signals of polyubiquitin chains in our MALDI-TOF experiments, although we could detect a weak signal of the K11-polyubiquitin linkage for Tagless-Ub(K63R)–MES. The ability of Rsp5 to utilize Lys11 of ubiquitin for polyubiquitin chain synthesis has also been previously documented, thus further supporting the overall similarities between ByS and the native ubiquitination reaction.32 Similar results were observed when polyubiquitin chains were prepared using the native ubiquitination cascade with Rsp5ΔWW and wtUb, Ub(K48R) and Ub(K63R) (Fig. S4†). We also confirmed that Rsp5/ByS assembles K63-linked polyubiquitin chains on the Sic60-GFP protein substrate (Fig. S5†).
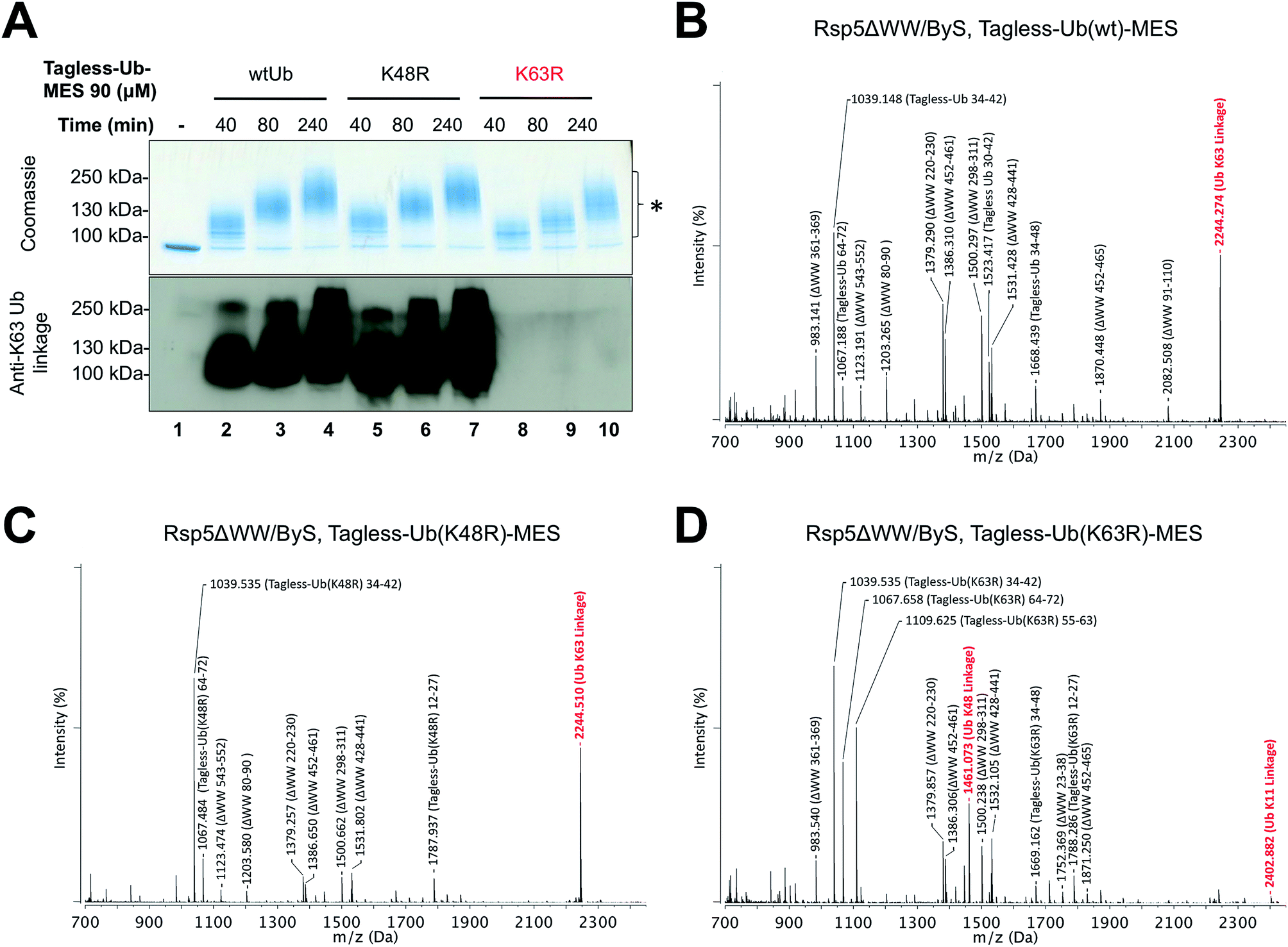 | ||
| Fig. 4 Rsp5/ByS forms K63-linked polyubiquitin chains. (A) K63-linked polyubiquitin chains formed by Rsp5ΔWW and Tagless-Ub–MES were visualized by western-blotting with K63-ubiquitin linkage specific antibodies. A mixture of Rsp5ΔWW (1.8 μM) and each Ub–MES mutant was incubated for indicated times and analyzed by coomassie staining and western-blotting. MALDI-TOF analysis was performed for the higher MW bands produced in the reaction with Rsp5ΔWW and (B) Tagless-Ub(wt)–MES (lane 4), (C) Tagless-Ub(K48R)–MES (lane 7), and (D) Tagless-Ub(K63R)–MES (lane 10). The gel region of 100–250 kDa from lane 4, 7, and 10 was excised for MALDI-TOF analysis. Any peak corresponding to the calculated polyubiquitin linkage signal (Table S2–S4†) is marked in red. Autoubiquitinated Rsp5ΔWW is marked with *. | ||
Rsp5ΔWW was used for these experiments because it was more active in autoubiquitination assays compared to full length Rsp5 and formed polyubiquitin chains more efficiently. However, we have made similar observations for full length Rsp5/ByS, suggesting that the WW domains of Rsp5 are not critical for the assembly of linkage specific polyubiquitin chains (Fig. S6†).
During our experiments, we noticed, however, that one of the Rsp5ΔWW tryptic peptides (220QYSSFEDQYGR230, m/z = 1379.402, Fig. 4B, Table S1C†) and a peptide from the K6-specific ubiquitin linkage (1MQIFVK6(GG)TLTGK,11m/z = 1379.677, Table S2B†) have very similar m/z ratios, such that it was difficult to distinguish these peptides in our MALDI-TOF experiments. To resolve this issue, the tryptic peptides of Rsp5ΔWW and polyubiquitin chains generated with Tagless-Ub(wt)–MES (Fig. 4A, lane 4) were acetylated with acetic anhydride and analyzed by MALDI-TOF (Fig. S7, Table S5†). The tryptic peptide of Rsp5ΔWW has only one free amine at the N-terminus, whereas that of the K6-linked polyubiquitin chain has two: its N-terminal amine and a C-terminal lysine. MALDI-TOF analysis of acetylated tryptic peptides of both Rsp5ΔWW (Fig. S7C†) and polyubiquitin chains formed by Rsp5/ByS with Tagless-Ub(wt)–MES (Fig. S7D†) showed the signal that corresponds to the singly acetylated peptide of Rsp5ΔWW (Ac-220QYSSFEDQYGR230, calculated average m/z = 1421.424), but not the signal for the doubly acetylated K6-linkage Ub peptide (Ac-MQIFVK6(GG)TLTGK(Ac),11 calculated average m/z = 1464.464).
Furthermore, signal from the 220QYSSFEDQYGR230 peptide is always accompanied by signal from the Rsp5 peptide 452EYVELYTQWR461 (calculated average m/z = 1386.6688). We noticed that the signal ratio for these peptides is similar for both Rsp5 and Rsp5ΔWW in all MALDI experiments, even without ubiquitination (Fig. S6B and S7A†). This further suggests that both of these peptides originate from the same protein sample, i.e. Rsp5 or Rsp5ΔWW. These observations coupled with our peptide acetylation experiments suggest that most likely the observed peptide signal with m/z = 1379.290 (Fig. 4B) is a tryptic peptide derived from Rsp5ΔWW, and not from a K6-linked polyubiquitin chain.
Taken together, our experiments suggest that Rsp5/ByS can synthesize K63-specific polyubiquitin chains in vitro in the absence of E1, E2 and ATP. Moreover, our experiments provide direct evidence that E2 enzymes are dispensable for the Rsp5-catalyzed Ub–Ub isopeptide bond formation with K63 specificity in vitro.
N-terminal tags in Ub–MES inhibit polyubiquitin chain synthesis by Rsp5 in both ByS and native ubiquitination reactions
While conducting our experiments, we discovered that the N-terminally modified Ub–MES (Tagged-Ub–MES, Table S1†) interfered with the formation of K63-linked polyubiquitin chains by Rsp5 both under ByS and the native cascade conditions (Fig. 5). To test the effect of N-terminal tagging on polyubiquitin chain synthesis, we prepared Tagged- and Tagless-Ub by basic hydrolysis of the corresponding Ub–MES thioesters as previously described.33 Subsequently, the Tagged-Ub and Tagless-Ub constructs were purified and used in autoubiquitination reactions under native ubiquitination conditions with E1, E2, Rsp5ΔWW and ATP. The incubation time of each reaction was optimized to produce similar amounts of higher molecular weight bands, as judged by coomassie staining. We then examined the formation of K63-linked polyubiquitin chains with K63-linkage specific antibodies. Our western blotting experiments demonstrated that Tagged-Ub and Tagged-Ub–MES are unable to participate in the formation of K63-linked polyubiquitin chains under both the native ubiquitination cascade and ByS reaction conditions, respectively (Fig. 5A). Tagless-Ub, on the other hand, could be used to form K63-linked polyubiquitin chains, indicating that the reaction conditions for hydrolyzing Tagged-Ub–MES thioester are not contributing to the inactivity of Tagged-Ub in forming polyubiquitin chains.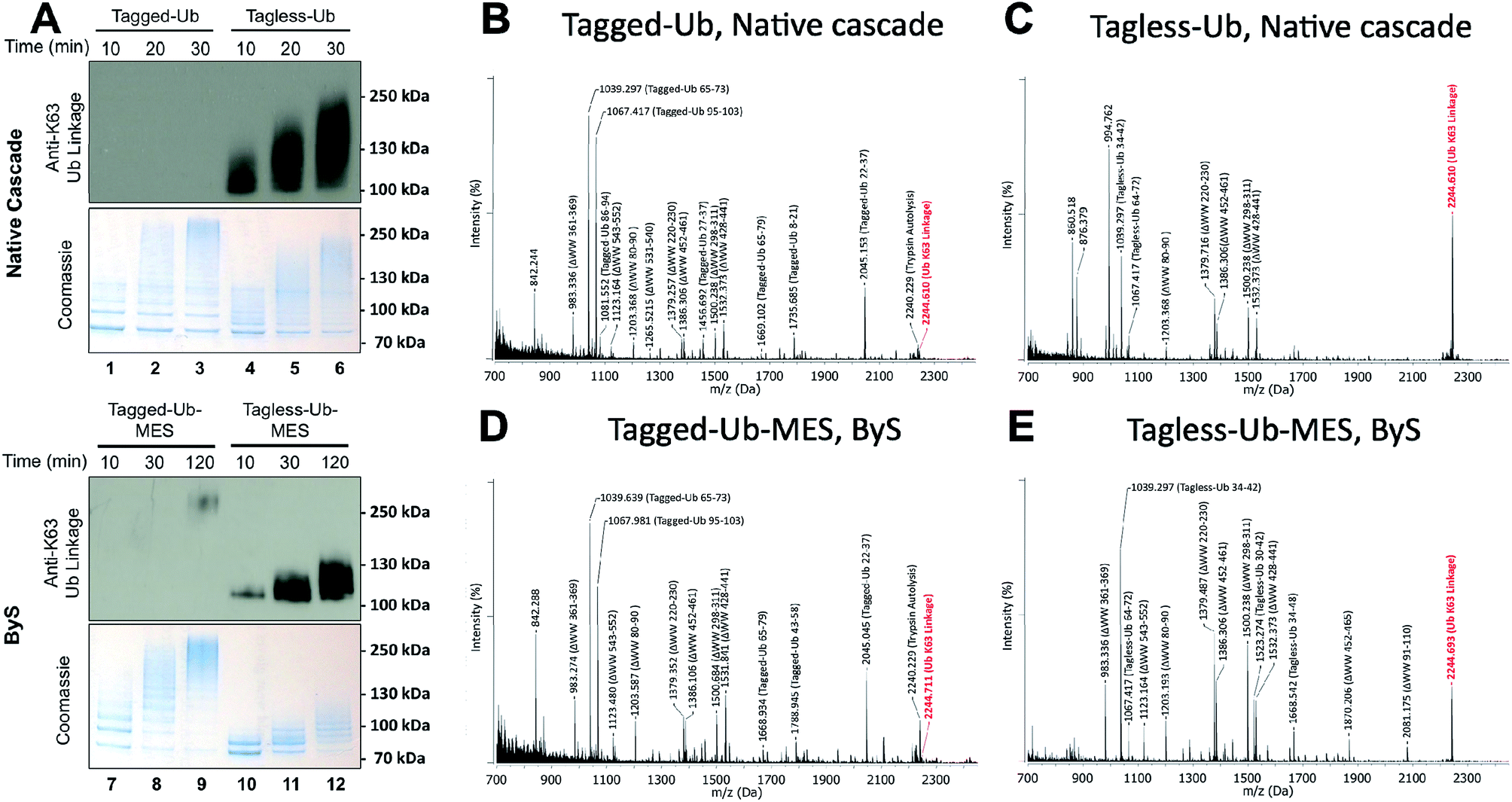 | ||
| Fig. 5 N-terminal tag on ubiquitin (Tagged-Ub) interferes with the formation of K63-polyubiquitin chains by Rsp5ΔWW under native and ByS reaction conditions. (A) The presence of K63-linkages in polyubiquitin chains formed by Rsp5 with Tagged- and Tagless-ubiquitin under both native cascade and ByS conditions was detected with western-blotting using anti-K63-linkage antibody. Native ubiquitination reactions contained UBE1 (0.09 μM), UbcH5a (1.0 μM), hydrolyzed Tagged- or Tagless-ubiquitin (90 μM), Rsp5ΔWW (2.0 μM) and ATP (4 mM). For ByS reactions, Rsp5ΔWW (2.0 μM) was treated with Tagless-Ub–MES (50 μM) or Tagged-Ub–MES (40 μM) and incubated at room temperature for the indicated times. MALDI-TOF analysis was performed for polyubiquitin chains formed by Rsp5ΔWW with (B) Tagged-Ub via native cascade (lane 3), (C) Tagless-Ub via native cascade (lane 6), (D) Tagged-Ub–MES via ByS (lane 9) and (E) Tagless-Ub–MES via ByS (lane 12). The gel region from 100–250 kDa in lanes 3, 6, 9 and 12 from a coomassie-stained gel was excised, digested by trypsin and analyzed by MALDI-TOF. Any peak corresponding to calculated polyubiquitin linkage signals (ESI Tables S2 and S5†) is marked in red. | ||
To further validate our observations, the gel region between 100 kDa to 250 kDa (Fig. 5A, lane 3, 6, 9 and 12) from the native and ByS ubiquitination reactions was excised, digested by trypsin and analyzed by MALDI-TOF (Fig. 5B–E). As expected, the m/z signal corresponding to K63-linkage was significantly diminished when Tagged-Ub or Tagged-Ub–MES was used in native or ByS reaction conditions. Intriguingly, we could not detect any dominant Ub-linkages in the case of Tagged-Ub or Tagged-Ub–MES with MALDI-TOF analysis, indicating that the N-terminal modification on Ub may stimulate non-specific multi-ubiquitination (Fig. 5A, higher molecular weight bands in lanes 1-3 and 7-9). Other than this polyubiquitin chain synthesis defect, Tagged-Ub–MES displayed biochemical properties identical to those of Tagless-Ub–MES in ByS conditions (Fig. S8†).
Our original Tagged-Ub–MES construct is composed of a 3×FLAG–6×His tag that is immediately followed by Glu2 of ubiquitin (Table S1†), such that Met1 of ubiquitin is replaced with a histidine residue. Structurally, Met1 of ubiquitin is proximal to Lys63 (∼7 Å), which in turn is close to the negatively charged Glu64 residue.34 In our Tagged-Ub–MES constructs, the hydrophobic Met1 side chain is replaced with a polar histidine side chain, which may lead to the inhibition of isopeptide bond ligation via Lys63. Other possible reasons for the inhibition of K63-linked polyubiquitin chain formation may include physical obstruction between the ubiquitin N-terminal tag and ubiquitin binding sites on the C- or N- lobe of HECT E3.
To test if the lack of Met1 in Tagged-Ub–MES is responsible for the inhibition of polyubiquitin chain formation, we prepared a Tagged-Ub(Met1)-MES that has Met1 inserted after the N-terminal tag and discovered that the absence of Met1 is not a major contributor to the observed inhibition of polyubiquitin chain synthesis. Even after the insertion of Met1, N-terminal tag on ubiquitin causes significant inhibition of K63-linked polyubiquitin chains in both ByS and the native cascade (Fig. S9†). Taken together our experiments suggest that the N-terminal 3×FLAG–6×His tag on ubiquitin inhibits the formation of K-63 linked polyubiquitin chains in bypassing and in native ubiquitination systems. The mechanism for how it interferes with the polyubiquitination reaction requires further investigation, which may provide clues on how E3 enzyme processivity is regulated, and how to design small molecule inhibitors of E3 enzyme processivity.
Overall, we suggest that caution must be exercised when conducting cell-based transfection experiments with N-terminally modified ubiquitin. Based on our in vitro experiments, N-terminal ubiquitin modifications may interfere with the formation of polyubiquitin chains and lead to non-specific multi-ubiquitination of intracellular proteins, possibly causing aberrant cellular phenotypes. Although the inhibition of protein turnover both in vivo and in vitro by N-terminally tagged ubiquitin was described before,35 to our best knowledge, this is the first report to unambiguously demonstrate that N-terminal modification on ubiquitin can inhibit K63-specific chain formation by HECT E3s in vitro.
Comparison of ByS with the native ubiquitination system
Having developed ByS, we next decided to compare the ubiquitination efficiency between the native system and ByS. Since E2∼Ub thioester in ByS is replaced with Ub∼MES thioester, we expected that Ub∼MES will have much lower binding affinity to Rsp5, and perhaps slower transthiolation kinetics, to render a lower ubiquitination efficiency for ByS. To conduct a direct comparison, we used pulse-chase experiments,27 and monitored the consumption of UbcH5a∼Ub and Tagless-Ub–MES thioesters by Rsp5ΔWW (Fig. S10†). Our observations suggest that a native cascade is much more efficient at autoubiquitination than ByS. We observed complete consumption of UbcH5a∼Ub thioester after 10 s of the reaction time, while Tagless-Ub–MES was not consumed after 90 min. Similarly, we observed the formation of monoubiquitinated Rsp5 after 10 seconds in the native system, yet in the bypassing system, monoubiquitinated Rsp5 was not observed at 1 minute, as judged by coomassie staining. Since we found the N-terminal ubiquitin tag to inhibit the formation of K63-linked polyubiquitin chains, we asked if the same N-terminal tag affects the activity of Tagged- and Taggless-Ub–MES probes in autoubiquitination assays with Rsp5ΔWW (Fig. S11†). To analyze the ubiquitination efficiency, we monitored the consumption of Tagless- and Tagged-Ub–MES by Rsp5ΔWW in ByS. Both thioesters were similarly consumed after 60 minutes, as judged by coomassie. Interestingly, in the case of Tagged-Ub–MES a major band of monoubiquitinated Rsp5ΔWW formed, while in the case of Taggless-Ub–MES multi- and poly-ubiquitinated forms of Rsp5 were observed. This is perhaps related to the fact that the N-terminal tag in Tagged-Ub–MES inhibits polyubiquitin chain synthesis.Kinetic characterization of the bypassing system
To quantitatively assess the enzymatic activity of HECT E3s in ByS, we developed a robust protocol to quantify the remaining amount of Ub–MES using fluorescein labeled cysteine (FCys, Scheme S1†). We envisioned that FCys would undergo a native chemical ligation reaction with the residual Ub–MES such that fluorescently labelled ubiquitin will correlate with residual Ub–MES in the reaction mixture (Fig. 6A).36,37 To quench a bypassing system reaction, FCys (6 mM) and urea (6 M) are added prior to 16 hour incubation at 37 °C. Tagless-Ub–FCys is then visualized by fluorescence scanning after the conjugate is resolved on a polyacrylamide gel. We confirmed that under these reaction conditions, the conjugation reaction yields Tagless-Ub–FCys by MS analysis (Fig. S12A†). We also confirmed that the fluorescence intensity of Tagless-Ub–FCys displayed linear correlation to the loading amount of Tagless-Ub–MES over a wide range (50 ng–2.0 μg, Fig. S12B†).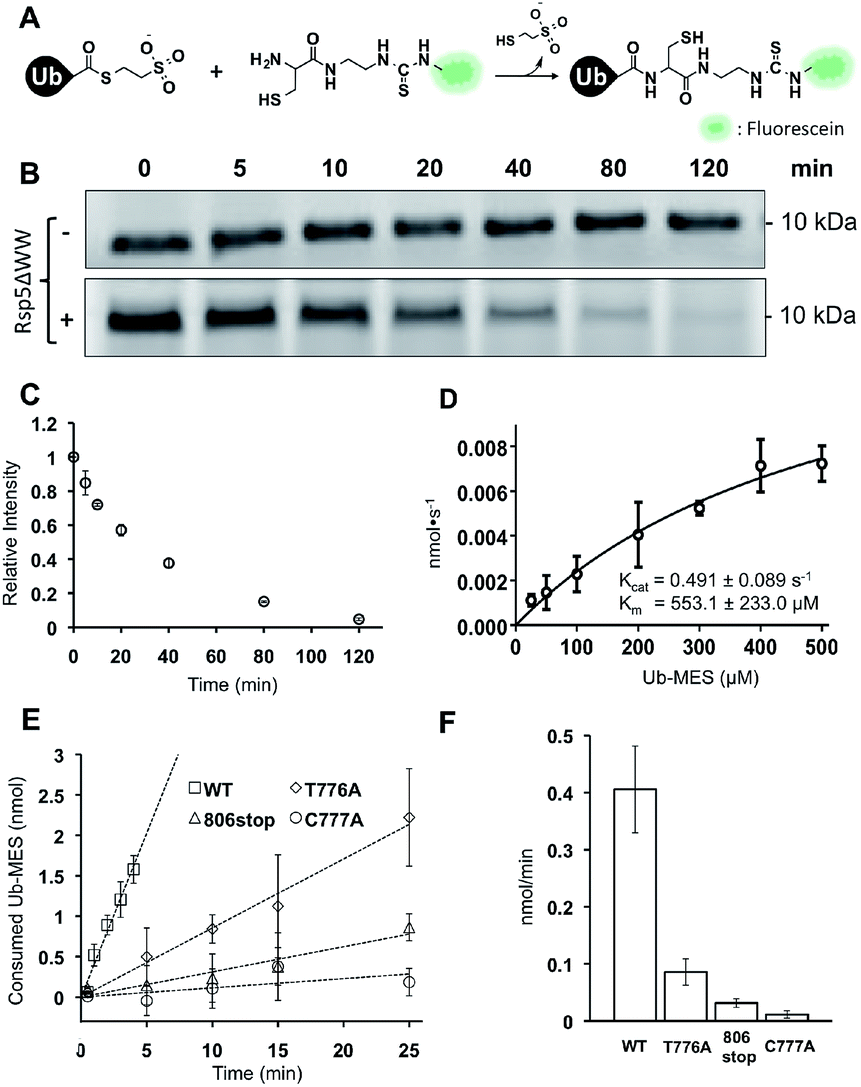 | ||
| Fig. 6 Kinetic characterization of Rsp5/ByS. (A) Native chemical ligation between Tagless-Ub–MES and FCys. (B) Reaction mixtures were quenched at different time points and the remaining Tagless-Ub–MES was labeled with FCys. Tagless-Ub–MES (250 μM) was incubated for indicated times without Rsp5ΔWW (upper panel) or with Rsp5ΔWW (lower panel, 1.5 μM). (C) Consumption of Tagless-Ub–MES (250 μM) by Rsp5ΔWW (1.5 μM). (D) The initial reaction rates at different Ub–MES concentrations were plotted, and kcat and Km values estimated. (E) Tagless-Ub–MES (250 μM) was incubated with the indicated mutant of Rsp5ΔWW (5 μM) and quenched with FCys at five time points starting at 30 seconds. After linear fitting, the y-intercept of each line was adjusted to equal zero. (F) Comparison of gross consumption rates of Tagless-Ub–MES by Rsp5ΔWW mutants. | ||
In the absence of Rsp5ΔWW, the amount of Tagless-Ub–MES did not change over 120 min (Fig. 6B, top). In the presence of Rsp5ΔWW, however, the amount of Tagless-Ub–MES decreased over time (Fig. 6B and C). Also, we observed that the initial consumption rates (0–5 min) of Tagless-Ub–MES by Rsp5 depend on the concentration of Tagless-Ub–MES and Rsp5 (kcat = 0.491 ± 0.089 s−1 and Km = 553.1 ± 223 μM; Fig. 6D and S13†). The Km and kcat are estimated based on the data points obtained with the highest concentration of Ub–MES achievable (500 μM). Rsp5ΔWW could not be saturated at this Ub–MES concentration. Although FCys can react with the Rsp5∼Ub thioester intermediate in addition to remaining Ub–MES, the large excess of Tagless-Ub–MES over Rsp5 along with the short lifetime of Rsp5∼Ub thioester,21 lead us to expect the contribution from this species to be negligible.
It is important to note that these kinetic parameters are obtained using Tagless-Ub–MES as a surrogate substrate, and are not estimates of Rsp5 kcat and Km in the native ubiquitination cascade. Also, the developed assay measures gross consumption of Ub–MES over time by Rsp5. In this reaction setup, enzymatic turnover will generate multiple enzymatically active autoubiquitinated forms of Rsp5. Therefore, the obtained kcat and Km are derived from an ensemble of different Rsp5–Ubx variants.
Nevertheless, the developed FCys protocol is particularly useful for quantitative assessment of the ligation efficiency of HECT E3s. As an example, we prepared three Rsp5ΔWW mutants with distinct mechanistic defects in the native ubiquitination cascade: Rsp5ΔWWC777A that lacks the catalytic cysteine, Rsp5ΔWWT776A which has a diminished ability to undergo transthiolation with E2∼Ub thioester, and Rsp5ΔWW806stop which is deficient at the ubiquitin isopeptide ligation step.24 As expected, these Rsp5 mutants displayed decreased initial rates of Tagless-Ub–MES consumption compared to wtRsp5ΔWW: wtRsp5ΔWW (0.406 nmol min−1), Rsp5ΔWWT776A (0.085 nmol min−1), Rsp5ΔWW806stop (0.031 nmol min−1), and Rsp5ΔWWC777A (0.011 nmol min−1) (Fig. 6E and F and S14†). This result demonstrates that the ByS/FCys protocol can be used to measure the inhibitory effect of biochemical mutations on the activity of HECT E3s. Thus, it is reasonable to predict that small molecule inhibition or activation of HECT E3s could be detected and measured under ByS conditions using the FCys protocol.
Other E3 ligases are active under ByS reaction conditions
We questioned if other E3 ligases that harbour catalytic cysteines are active under ByS reaction conditions and synthesize polyubiquitin chains via ByS. Thus, we tested the generality of the reaction on a panel of E3 ligases (1) Nedd4 family HECT E3s including Nedd4-1, Nedd4-2, Wwp1 and Itch, and (2) RBR E3 ligases including Parkin and HHARI (Fig. S15†). Nedd4-1, a founding member of the Nedd4 family and a human homologue of Rsp5, along with eight other members of the Nedd4 HECT E3 family share a similar protein structure and catalytic mechanism.38 It has been shown that Nedd4 family ligases produce predominantly K63-linked polyubiquitin chains as reaction products.39 RBR E3 ligases, on the other hand, contain RING domains, and harbour a catalytic cysteine that forms an obligatory RBR E3∼Ub thioester intermediate, and are thus considered RING–HECT hybrids.40 Recently, a thorough biochemical analysis showed that Parkin forms K6, K11, K48, and K63 chain linkages both in vivo and in vitro.41 To our knowledge, the ubiquitin chain specificity for HHARI has not been investigated. To test if these enzymes are active under ByS reaction conditions, we incubated 2 μM of each E3 enzyme with 200 μM of Tagless-Ub(wt)–MES, Ub(K48R)–MES or Ub(K63R)–MES and then analyzed the formation of polyubiquitin chains by HECT E3s or RBR E3s using K63- or K48-ubiquitin linkage specific antibodies, respectively (Fig. 7A and B). As expected, HECT and RBR E3s used Tagless-Ub–MES as a substrate to catalyze the formation of polyubiquitin chains with K63- or K48-linkages, respectively. While the formation of other linkages cannot be excluded, our data show that these enzymes are active under ByS reaction conditions and can catalyze the formation of polyubiquitin chains. Taken together, our data suggest that ByS can be generally used to study the biochemistry of transthiolation and isopeptide ligation steps of HECT and RBR E3s, which can be conveniently decoupled from E1 and E2 enzymes.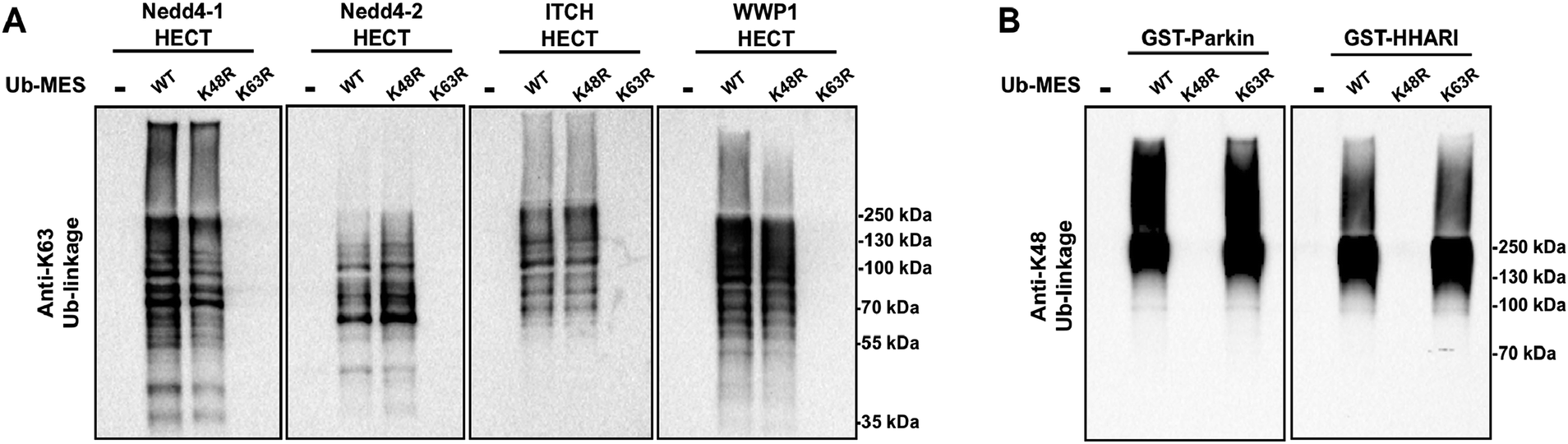 | ||
| Fig. 7 Activity of HECT E3 ligases and RBR E3 ligases under ByS reaction conditions. (A) The HECT domains of Nedd4-1, Nedd4-2, Itch and Wwp1 (2 μM) were incubated with either Ub(wt)–MES, Ub(K48R)–MES or Ub(K48R)–MES (200 μM) for 90 minutes at room temperature. The reaction mixtures were then analyzed using K63-ubiquitin linkage specific antibody. (B) GST-tagged RBR domains of Parkin (rat) and HHARI (human) (2 μM) were treated with Ub–MES as in (A) and were analyzed using K48-ubiquitin linkage antibody. HHARI and Parkin constructs lack auto-inhibitory domains. | ||
Conclusion
Using Rsp5 HECT E3 as a model ubiquitin ligase, we discovered an unusual phenomenon in which protein ubiquitination and polyubiquitin chain synthesis occur in the absence of ATP, E1 and E2 enzymes in vitro, requiring only the E3 enzyme and chemically activated ubiquitin. This stands in sharp contrast to the dogmatic auto- and substrate ubiquitination that require ATP, E1, E2, E3 enzymes, and ubiquitin. Interestingly, we have established that the discovered bypassing system recapitulates the mechanism and the isopeptide linkage specificity of the native ubiquitination reaction in vitro. To our knowledge, the discovered two-component ubiquitination reaction represents in its essence a new, previously unknown and substantially simplified form of enzymatic mono- and polyubiquitination. Since the developed reaction bypasses the need for ATP, E1 and E2 enzymes we call it “Bypassing System” or “ByS”. Importantly, this system requires only two components to ubiquitinate protein substrates, thereby significantly simplifying biochemical studies in vitro.Our initial preliminary mechanistic studies demonstrate that Ub–MES undergoes a transthiolation reaction with the catalytic cysteine of Rsp5, and forms an active Rsp5∼Ub enzymatic intermediate. Rsp5∼Ub thioester formed under these reaction conditions ubiquitinates protein substrates, autoubiquitinates, and synthesizes polyubiquitin chains with specific isopeptide linkages in the absence of E1, E2 enzymes and ATP. Based on the reaction mechanism, the developed two-component ubiquitination reaction could be generally applied to other E3 ligases that have catalytic cysteines. In an initial proof-of-concept study, we found that Nedd4 family HECT E3s including Nedd4-1, Nedd4-2, Wwp1 and Itch, and RING-in-between-RING (RBR) E3 ligases including Parkin and HHARI, process Ub–MES efficiently to produce K63- and K48-linked polyubiquitin chains. Thus, we believe that C-terminal Ub-thioester probes can serve as general and useful tools to study biochemical properties of HECT E3s and RBR E3s.
Moreover, using the Ub–MES probe, we provide direct experimental evidence that E2 enzymes are dispensable for the formation of linkage specific polyubiquitin chains by Rsp5 as well as by Nedd4 family HECT E3s including Nedd4-1, Nedd4-2, Itch, Wwp1 and RBR E3s Parkin and HHARI in vitro. This further supports the sequential addition model of polyubiquitin chain synthesis by Nedd4 family HECT E3s. Important assumptions of the sequential addition model are that Nedd4 family HECT E3s are solely responsible for the (1) K63-chain type specificity as well as (2) catalysis of isopeptide bond formation between ubiquitin molecules. Although previous work demonstrates that HECT domain alone can encode the chain type specificity,32,39,42 we present direct experimental evidence that E2s are dispensable for Nedd4-catalyzed Ub–Ub isopeptide bond formation. Our results cannot exclude, however, that in vivo both sequential addition and en bloc transfer mechanisms may exist. Similarly, our results suggest that RBR E3s are capable of assembling polyubiquitin chains by forming isopeptide linkages in the absence of E1 and E2 enzymes.
We envision that the developed ByS can serve as a useful platform to study other biochemical properties of HECT E3∼Ub and RBR E3∼Ub thioesters by decoupling ubiquitin ligation from the preceding steps mediated by E1 and E2 enzymes. For example, point mutation experiments to study the role of ubiquitin surface residues during ligation by HECT ligases have been difficult to implement because the mutated ubiquitin has to be compatible with E1 and E2 enzymes.43 As a consequence, mutagenesis has largely been restricted to studying the role of HECT E3 surface residues during ubiquitin ligation, but not those of ubiquitin. Alternatively, the Ub–MES surface can be engineered to selectively bind specific E3 enzymes preceding substrate ubiquitination.44 Such ubiquitin probes could be used to identify protein substrates of HECT E3s and other E3s harboring catalytic cysteines.
Finally, the developed ByS can be used to design simple assays to discover and to characterize HECT E3 and RBR E3 enzyme inhibitors or activators. Current assays require E1 and E2 enzymes present in the reaction mixture along with ATP, which often lead to false positive results due to off-target inhibition of E1 and E2 enzymes. The developed ByS provides a simple and elegant solution to this problem, by obviating the need to use E1, E2 enzymes and ATP. To this end, the developed native chemical ligation protocol for quantitative assessment of the enzymatic activity of HECT E3s may serve as a prelude to conceptually novel assays to discover and characterize inhibitors or activators of HECT E3 and RBR E3 enzymes. Further developments of homogeneous fluorescent assays for HECT E3s and RBR E3s based on ByS, mechanistic investigations, and applications of ByS will be reported in the near future.
Acknowledgements
Funding from Northwestern University is greatly acknowledged. A.V.S. is a Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trusts. We would like to thank Simona Polo (Fondazione Istituto FIRC di Oncologia Molecolare, Milan, Italy) for providing Nedd4-1 constructs. We also acknowledge the Northwestern IMSERC facility where we used the Bruker Autoflex III MALDI and the AVANCE III 500 MHz NMR. Fluorescence scanning was done on the GE Healthcare Typhoon 9400 at the Northwestern Keck Biophysics Facility.Notes and references
- A. Varshavsky, Annu. Rev. Biochem., 2012, 81, 167 CrossRef CAS PubMed.
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1998, 67, 425 CrossRef CAS PubMed.
- C. M. Pickart, Annu. Rev. Biochem., 2001, 70, 503 CrossRef CAS PubMed.
- R. J. Deshaies and C. A. Joazeiro, Annu. Rev. Biochem., 2009, 78, 399 CrossRef CAS PubMed.
- J. R. Lydeard, B. A. Schulman and J. W. Harper, EMBO Rep., 2013, 14(12), 1050–1061 CrossRef CAS PubMed.
- J. Pines, Nat. Rev. Mol. Cell Biol., 2011, 12, 427 CrossRef CAS PubMed.
- Z. Zhang, J. Yang, E. H. Kong, W. C. Chao, E. P. Morris, P. C. da Fonseca and D. Barford, Biochem. J., 2013, 449, 365 CrossRef CAS PubMed.
- J. M. Huibregtse, M. Scheffner, S. Beaudenon and P. M. Howley, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2563 CrossRef CAS.
- M. Scheffner, U. Nuber and J. M. Huibregtse, Nature, 1995, 373, 81 CrossRef CAS PubMed.
- S. E. Schwarz, J. L. Rosa and M. Scheffner, J. Biol. Chem., 1998, 273, 12148 CrossRef CAS PubMed.
- M. Scheffner and S. Kumar, Biochim. Biophys. Acta, 2014, 1843, 61 CrossRef CAS PubMed.
- F. Bernassola, M. Karin, A. Ciechanover and G. Melino, Cancer Cell, 2008, 14, 10 CrossRef CAS PubMed.
- D. Rotin and S. Kumar, Nat. Rev. Mol. Cell Biol., 2009, 10, 398 CrossRef CAS PubMed.
- H. An, D. T. Krist and A. V. Statsyuk, Mol. BioSyst., 2014, 10, 1643–1657 RSC.
- K. D. Wilkinson, T. Gan-Erdene and N. Kolli, Methods Enzymol., 2005, 399, 37 CAS.
- A. Borodovsky, H. Ovaa, N. Kolli, T. Gan-Erdene, K. D. Wilkinson, H. L. Ploegh and B. M. Kessler, Chem. Biol., 2002, 9, 1149–1159 CrossRef CAS.
- K. R. Love, R. K. Pandya, E. Spooner and H. L. Ploegh, ACS Chem. Biol., 2009, 4, 275 CrossRef PubMed.
- M. Hochstrasser, Cell, 2006, 124, 27 CrossRef PubMed.
- S. Park, I. Ntai, P. Thomas, E. Konishcheva, N. L. Kelleher and A. V. Statsuk, Biochemistry, 2012, 51, 8327 CrossRef CAS PubMed.
- Y. Saeki, E. Isono and E. A. Toh, Methods Enzymol., 2005, 399, 215 CAS.
- G. Wang, J. Yang and J. M. Huibregtse, Mol. Cell. Biol., 1999, 19, 342 CAS.
- B. Andre and J. Y. Springael, Biochem. Biophys. Res. Commun., 1994, 205, 1201 CrossRef CAS PubMed.
- O. Staub and D. Rotin, Structure, 1996, 4, 495 CrossRef CAS.
- H. B. Kamadurai, Y. Qiu, A. Deng, J. S. Harrison, C. Macdonald, M. Actis, P. Rodrigues, D. J. Miller, J. Souphron, S. M. Lewis, I. Kurinov, N. Fujii, M. Hammel, R. Piper, B. Kuhlman and B. A. Schulman, eLife, 2013, 2, e00828 Search PubMed.
- S. Lorenz, A. J. Cantor, M. Rape and J. Kuriyan, BMC Biol., 2013, 11, 65 CrossRef PubMed.
- M. A. Verdecia, C. A. Joazeiro, N. J. Wells, J. L. Ferrer, M. E. Bowman, T. Hunter and J. P. Noel, Mol. Cell, 2003, 11, 249 CrossRef CAS.
- H. B. Kamadurai, J. Souphron, D. C. Scott, D. M. Duda, D. J. Miller, D. Stringer, R. C. Piper and B. A. Schulman, Mol. Cell, 2009, 36, 1095 CrossRef CAS PubMed.
- C. Salvat, G. Wang, A. Dastur, N. Lyon and J. M. Huibregtse, J. Biol. Chem., 2004, 279, 18935 CrossRef CAS PubMed.
- Y. Kee, N. Lyon and J. M. Huibregtse, EMBO J., 2005, 24, 2414 CrossRef CAS PubMed.
- Y. Kee, W. Munoz, N. Lyon and J. M. Huibregtse, J. Biol. Chem., 2006, 281, 36724 CrossRef CAS PubMed.
- Y. Saeki, T. Kudo, T. Sone, Y. Kikuchi, H. Yokosawa, A. Toh-e and K. Tanaka, EMBO J., 2009, 28, 359 CrossRef CAS PubMed.
- H. C. Kim and J. M. Huibregtse, Mol. Cell. Biol., 2009, 29, 3307 CrossRef CAS PubMed.
- Z. P. Gates, J. R. Stephan, D. J. Lee and S. B. H. Kent, Chem. Commun., 2013, 49, 786 RSC.
- S. Vijay-Kumar, C. E. Bugg, K. D. Wilkinson and W. J. Cook, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3582 CrossRef CAS.
- M. J. Ellison and M. Hochstrasser, J. Biol. Chem., 1991, 266, 21150 CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266(5186), 776 CAS.
- P. E. Dawson and S. B. Kent, Annu. Rev. Biochem., 2000, 69, 923 CrossRef CAS PubMed.
- R. J. Ingham, G. Gish and T. Pawson, Oncogene, 2004, 23, 1972 CrossRef CAS PubMed.
- E. Maspero, E. Valentini, S. Mari, V. Cecatiello, P. Soffientini, S. Pasqualato and S. Polo, Nat. Struct. Mol. Biol., 2013, 20, 696 CAS.
- D. M. Wenzel, A. Lissounov, P. S. Brzovic and R. E. Klevit, Nature, 2011, 474, 105 CrossRef CAS PubMed.
- A. Ordureau, S. A. Sarraf, D. M. Duda, J. M. Heo, M. P. Jedrykowski, V. O. Sviderskiy, J. L. Olszewski, J. T. Koerber, T. Xie, S. Beausoleil, J. A. Wells, S. P. Gygi, B. A. Schulman and J. W. Harper, Mol. Cell, 2014, 56, 360 CrossRef CAS PubMed.
- E. Maspero, S. Mari, E. Valentini, A. Musacchio, A. Fish, S. Pasqualato and S. Polo, EMBO Rep., 2011, 12, 342 CrossRef CAS PubMed.
- C. M. Pickart, E. M. Kasperek, R. Beal and A. Kim, J. Biol. Chem., 1994, 269, 7115 CAS.
- A. Ernst, G. Avvakumov, J. Tong, Y. Fan, Y. Zhao, P. Alberts, A. Persaud, J. R. Walker, A. M. Neculai, D. Neculai, A. Vorobyov, P. Garg, L. Beatty, P. K. Chan, Y. C. Juang, M. C. Landry, C. Yeh, E. Zeqiraj, K. Karamboulas, A. Allali-Hassani, M. Vedadi, M. Tyers, J. Moffat, F. Sicheri, L. Pelletier, D. Durocher, B. Raught, D. Rotin, J. Yang, M. F. Moran, S. Dhe-Paganon and S. S. Sidhu, Science, 2013, 339, 590 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc02340d |
| This journal is © The Royal Society of Chemistry 2015 |