
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDivergent regioselectivity in photoredox-catalyzed hydrofunctionalization reactions of unsaturated amides and thioamides†
Peter D.
Morse
and
David A.
Nicewicz
*
Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-3290, USA. E-mail: nicewicz@unc.edu; Web: http://www.chem.unc.edu/people/faculty/nicewicz/
First published on 23rd September 2014
Abstract
A direct method to construct 2-oxazolines and 2-thiazolines from corresponding allylic amides and thioamides is reported. The redox-neutral intramolecular hydrofunctionalization is enabled by a dual catalyst system comprised of 9-mesityl-N-methyl acridinium tetrafluoroborate and phenyl disulphide and exhibits complete selectivity for the anti-Markovnikov regioisomeric products. The cyclization of allylic thioamides is postulated to operate via a modified mechanism in which oxidation of the thioamide, rather than the alkene, is responsible for the observed reactivity.
Introduction
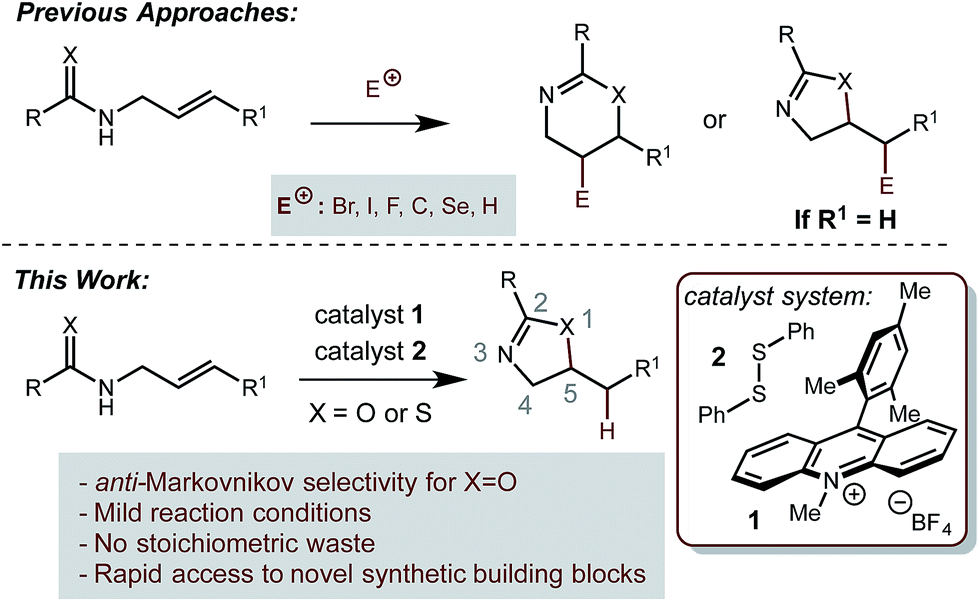
Oxazolines and thiazolines are prevalent motifs found in a variety of naturally and unnaturally occurring small molecules. Many natural products bearing these moieties have been found to posses potent levels of bioactivity, including antiobiotic,1,2 anti-tumor,3 anti-inflammatory,4 and anti-fungal5 activity. Additionally, these structures appear frequently in the architecture of ligands for asymmetric catalysis, with two of the most prevalent examples being the bisoxazoline6 and phosphinooxazoline7 classes.Many methods have been developed to synthesize oxazolines and thiazolines.8,9 One general method, frequently employed in the synthesis of chiral ligands, involves the condensation of an appropriate β-amino alcohol onto an aldehyde.10 These amino alcohols can be derived from the corresponding amino acid and this method has the advantage of providing rapid access to enantiopure material. However, substitution patterns accessible by this strategy tend to be limited by the availability of the β-amino alcohol substrates. Varying the functionality at the 4-position, as well as obtaining substitution at the 5-position both require additional manipulations of the starting material (Scheme 1).
 | ||
| Scheme 1 Prior methods for the synthesis of oxazolines and thiazolines by intramolecular cyclization. | ||
The cyclization of a pendant amide or thioamide nucleophile onto a degree of unsaturation represents a very direct and broadly applicable method for the synthesis of oxazolines and thiazolines. This strategy is especially well-suited to situations when substitution at the 5-position is desired. One general strategy that has been successfully employed for this purpose is the activation of an alkene by an electrophilic species, followed by nucleophilic attack, to produce oxidative cyclization adducts.11–14 While such products are highly useful in a variety of circumstances, due to the incorporation of a new synthetic handle, additional steps would be required to arrive at the formal hydrofunctionalization product.
Direct, catalytic hydrofunctionalization by amide and thioamide nucleophiles presents a significant challenge to existing methodology. A variety of metal-catalyzed redox-neutral cyclizations of propargyl amides have been reported in recent years, furnishing oxazolines bearing an exo-alkene.15–19 However, in order to employ olefins as substrates, strong acids such as sulfuric or p-toluenesulphonic acid are required.11 The use of strong acid limits the potential functional group tolerance of these reactions, and additionally results in the exclusive formation of Markovnikov regioisomeric products. To the best of our knowledge, there currently exists no alternative catalytic method for carrying out the hydrofunctionalization of alkenes with amide and thioamide nucleophiles.
Our laboratory is interested in the use of organic photoredox catalysis20 to accomplish challenging bond constructions under operationally mild conditions. We are specifically interested in the single electron oxidation of olefins as a general and orthogonal strategy for their activation towards nucleophilic attack. Towards this goal, we have disclosed a dual catalyst system for the anti-Markovnikov hydrofunctionalization of alkenes. In this system, the Fukuzumi acridinium photooxidant21 is used in conjugation with a redox-active hydrogen atom donor, such as thiophenol. This system has been demonstrated to work well with a variety of nucleophiles.22–26 Based on this precedent, we hypothesized that amides and thioamides could function as nucleophiles in an analogous fashion, furnishing the corresponding oxazolines and thiazolines as products. Successful implementation of this strategy would provide a general and mild route to these important classes of molecules that avoids the generation of any stoichiometric waste products. Additionally, this method would well complement acid-catalyzed methods by providing access to products arising from anti-Markovnikov selectivity.
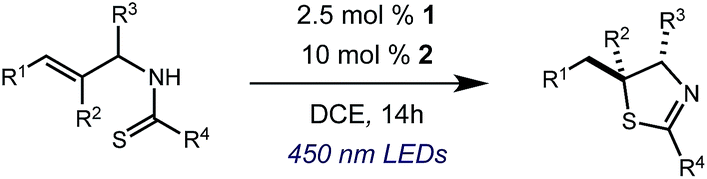
Results and discussion
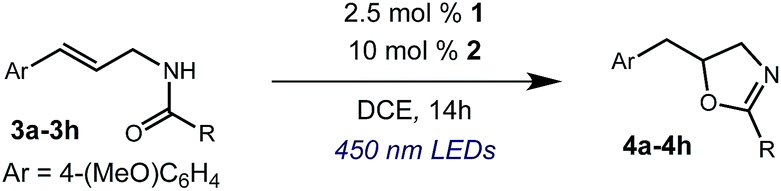
We began our investigation using 3a as a model substrate for the desired hydrofunctionalization. The 9-mesityl-N-methyl acridinium tetrafluoroborate salt (1) was selected as the photoredox catalyst for this study, based on its high excited state reduction potential (Ered1/2 = +2.06 V vs. SCE).27 A screen of potential catalytic hydrogen atom donors indicated that thiophenol served as an excellent hydrogen atom source, and provided access to the desired product 4a in >95% yield by NMR and 82% yield after isolation. Phenyl disulphide was screened as well, and was shown to give comparable results. Its role in the proposed mechanism of the transformation will be discussed later. We chose to continue the study employing phenyl disulphide as the hydrogen atom transfer catalyst for practical reasons – it is a bench-stable solid that is odourless, as opposed to thiophenol, which is a pungent, toxic liquid which must be stored under nitrogen and added via syringe (Table 1).
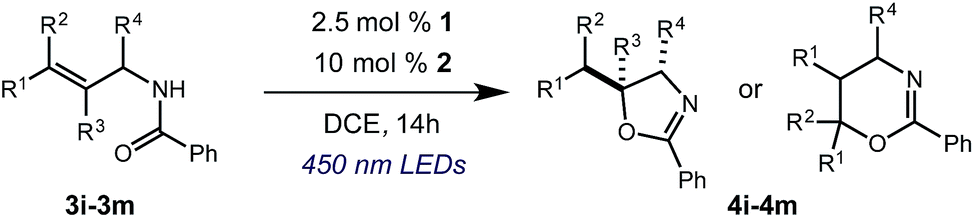
After establishing optimal conditions, we then sought to explore the scope of substitution patterns that would be tolerated on the amide portion of the substrate. When run for 14 hours, we found that substrates bearing a variety of aromatic and aliphatic groups could be converted into the desired oxazoline products in good yields. However, strongly electron withdrawing groups such as trifluoromethyl and 2-pyridinyl amides yielded no detectable product formation, presumably due to their diminished nucleophilicity.
We were particularly interested in the cyclization of the substrate bearing a bromide at the ortho position of the phenyl ring, as the product could then be transformed into a PHOX-type ligand by installing an aryl phosphine group using copper-catalyzed coupling.28 Under normal reaction conditions, modest yields of the desired product could be obtained, albeit accompanied by significant levels of non-selective degradation of the substrate. However, when phenyl disulphide was replaced by 20 mol% 4-methoxythiophenol, the yield was substantially improved. Tuning the electronic properties of the hydrogen atom donor could potentially influence a number of steps in the catalytic cycle, and so the origin of this beneficial effect on yield is not well understood at this time.
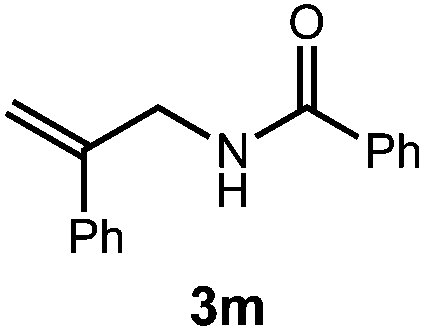
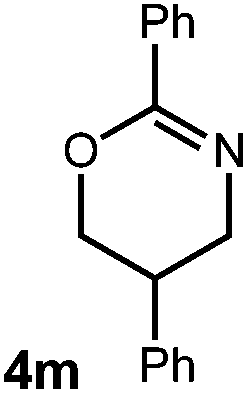
A variety of substitution patterns on the alkene portion of the substrate were also well-tolerated under the reaction conditions, and we observed that trisubstituted aliphatic alkenes could be employed. Substrates bearing a pre-existing stereocenter cyclized with modest levels of diastereoselectivity. Additionally, we were able to show, using substrate 3m, that 6-membered ring formation is also viable using this methodology. We propose that the regioselectivity of this transformation is governed by the thermodynamics of forming the more stable of the two possible radical intermediates following reversible nucleophilic attack by the amide on the cation radical (Scheme 2) (Table 2).
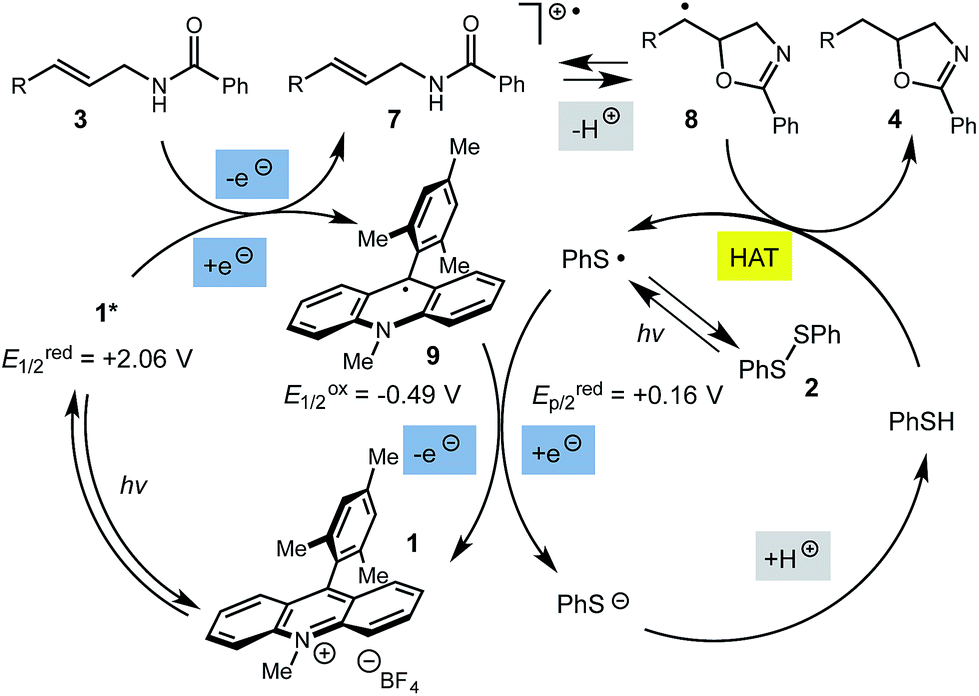 | ||
| Scheme 2 Proposed catalytic cycle. | ||
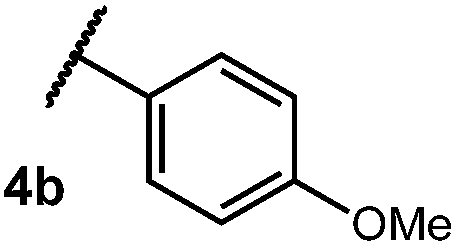
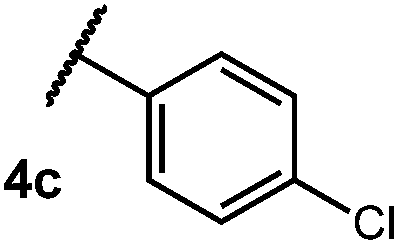
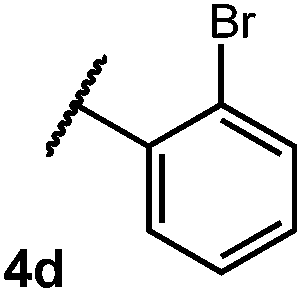



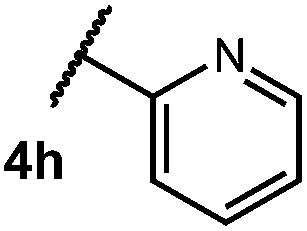
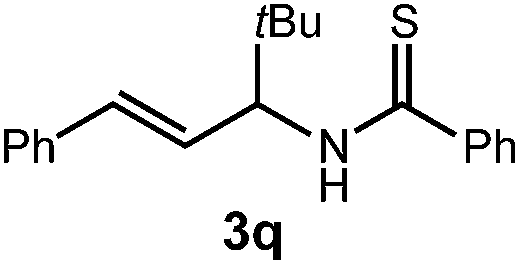
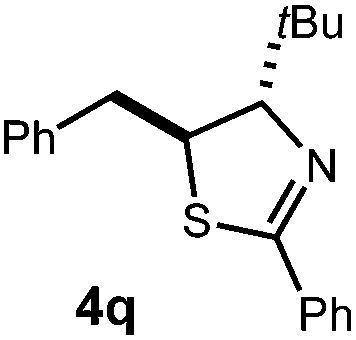
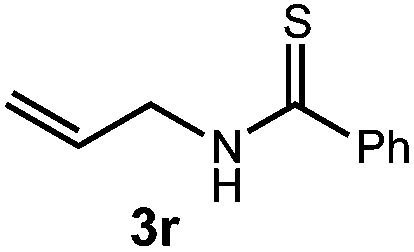
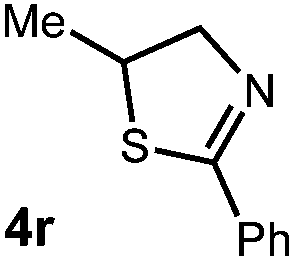
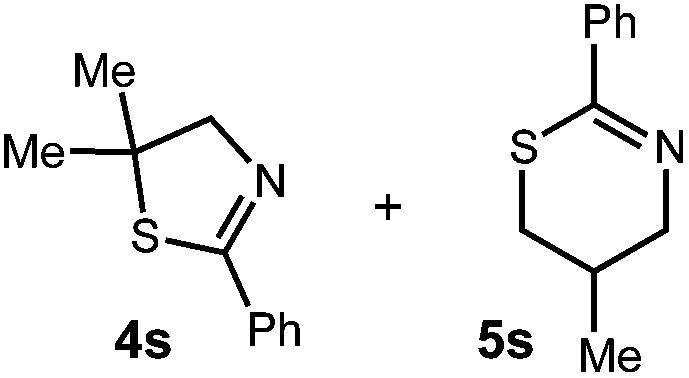
We then turned our attention to the cyclization of thioamides to the corresponding thiazolines. Here again, we found that our optimal conditions provided the desired products in good yields. We were further intrigued by the possibility that in this case, the thioamide functional group itself was acting as the oxidizable moiety. If this were the case, it would present the possibility of using terminal and disubstituted alkenes as substrates, which would be otherwise unreactive due to their high oxidation potentials. Indeed, cyclic voltammetry measurements of substrate 3r indicated that it possessed a half wave oxidation potential of +1.53 V vs. SCE, whereas the corresponding amide was greater than +2.5 V vs. SCE.29 Given that this is well within the range of oxidation capable by the Fukuzumi catalyst, the substrate was submitted to standard reaction conditions. It was found to smoothly convert to the corresponding thiazoline, albeit in this case with formal Markovnikov selectivity. However substrate 3s, bearing a vinyl methyl group, furnished a mixture of Markovnikov and anti-Markovnikov adducts. The selectivity observed in these cases indicates that a different mechanism than that proposed in Scheme 2 is in operation, as the product formation is not governed by the stabilities of radical intermediates. Control experiments with the corresponding amides of 3r and 3s showed no reactivity, confirming our hypothesis that the presence of the thioamide is necessary in these cases. However, in entries 3n–3p we cannot rule out the possibility that alkene oxidation is occurring, as electron rich styrene derivatives are reported to have similarly low oxidation potentials.22 We also considered the possibility that direct excitation of either the thioamide or phenyl disulfide by a photon could be responsible for the observed cyclization. A control experiment in which 9-mesityl-N-methyl acridinium was omitted resulted in the formation of the formation of small quantities of an oxidative cyclization product plus other unidentified by-products (Fig. 1). However, no traces of the hydrofunctionalization product were detected, indicating that the Fukuzumi photooxidant is necessary for the desired reactivity (Table 3).
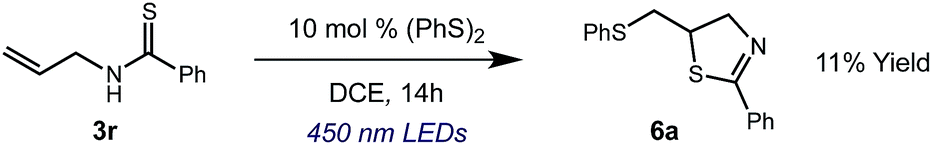 | ||
| Fig. 1 Exclusion of the photooxidant results in alternative reactivity, but no hydrofunctionalization is observed. | ||
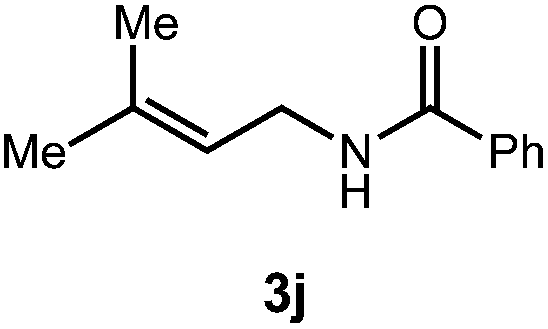
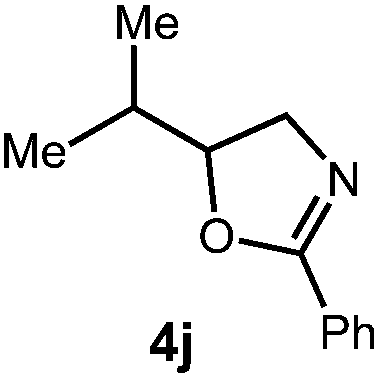
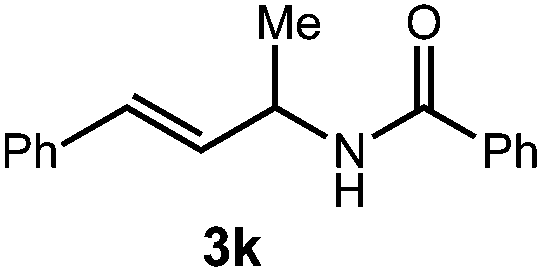
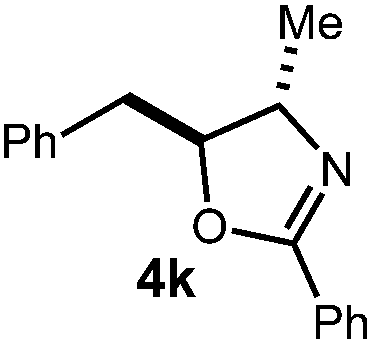
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: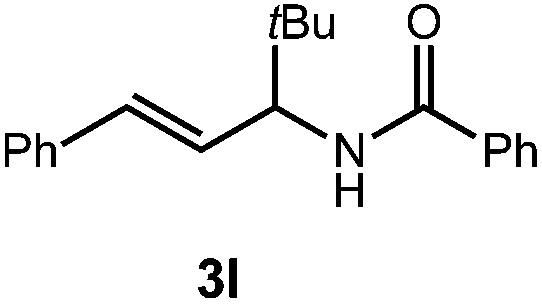
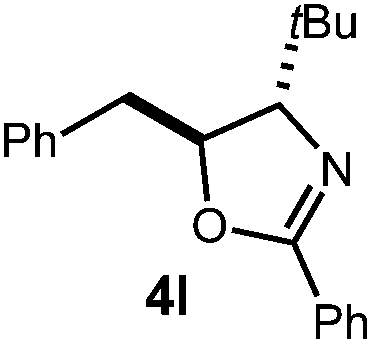
The mechanism for this transformation is believed to occur in a manner analogous to our previously reported olefin hydrofunctionalization reactions (Scheme 2). Excitation of the acridinium salt generates the active single electron oxidant (1*), which accepts an electron from the allylic amide substrate, generating cation-radical intermediate 7. Reversible cyclization of the amide onto 7, followed by proton loss affords radical 8. The final oxazoline adduct is formed via hydrogen atom transfer from thiophenol to 8. The resultant thiyl radical is presumed to re-oxidize the acridine radical (9), regenerating the active catalyst. Protonation of the resulting thiolate anion regenerates the active hydrogen atom donor, closing the catalytic cycle. Key to the catalytic cycle is the formation of the oxidizing equivalent of thiyl radical, which is presumably formed via direct homolysis of phenyl disulfide by light.
While this mechanism explains the anti-Markovnikov reactivity observed for the unsaturated amides, it is inconsistent with the regioselectivity observed in the cyclization of the unsaturated thioamides. In the case of these substrates, we hypothesize that oxidation of the sulfur atom occurs to generate a cation radical (10), which is likely deprotonated to form radical 11 (eqn (1)), which then cyclizes onto the pendant olefin. We favour this mechanism due to the fact that aliphatic alkenes such as 3r and 3s are typically outside the oxidation range (Ep/2 > +2.2 V vs. SCE) of 1* and are unreactive in this context and that radical 11 should display a kinetic regioselectivity preference for cyclization which is observed in the reactions of thioamides 3r and 3s (ref. 30) (Table 4).
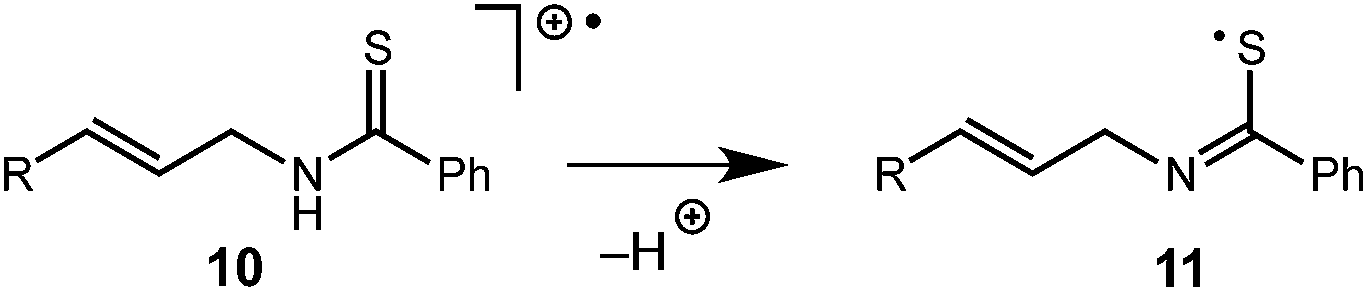 | (1) |
Conclusions
In conclusion, we have developed a mild and efficient method for the synthesis of novel oxazolines and thiazolines that requires no stoichiometric reagents and operates under very mild conditions. A variety of substitution patterns are well tolerated, and anti-Markovnikov selectivity is exclusively observed when amides are employed as the nucleophile. This selectivity is proposed to result from the formation of more thermodynamically stable radical intermediates over the course of the reaction. The reactivity of the unsaturated thioamides in this case provides the corresponding thiazolines with a preference for the formation of the 5-exo over 6-endo adducts, presumably lending support for the oxidation of the thioamide group as being the operative mechanism.Acknowledgements
This work was supported by Award no. R01 GM098340 from the National Institute of General Medical Sciences.Notes and references
- M. C. Pirrung, L. N. Tumey, A. L. McClerren and C. R. H. Raetz, J. Am. Chem. Soc., 2003, 125, 1575–1586 CrossRef CAS PubMed.
- T. Kline, N. H. Andersen, E. A. Harwood, J. Bowman, A. Malanda, S. Endsley, A. L. Erwin, M. Doyle, S. Fong, A. L. Harris, B. Mendelsohn, K. Mdluli, C. R. H. Raetz, C. K. Stover, P. R. Witte, A. Yabannavar and S. Zhu, J. Med. Chem., 2002, 45, 3112–3129 CrossRef CAS PubMed.
- S. Carmeli, R. E. Moore, G. M. L. Patterson, T. H. Corbett and F. A. Valeriote, J. Am. Chem. Soc., 1990, 112, 8195–8197 CrossRef CAS.
- K. C. Nicolaou, D. E. Lizos, D. W. Kim, D. Schlawe, R. G. de Noronha, D. A. Longbottom, M. Rodriquez, M. Bucci and G. Cirino, J. Am. Chem. Soc., 2006, 128, 4460–4470 CrossRef CAS PubMed.
- H. B. Bode, H. Irschik, S. C. Wenzel, H. Reichenbach, R. Müller and G. Höfle, J. Nat. Prod., 2003, 66, 1203–1206 CrossRef CAS PubMed.
- A. K. Ghosh, P. Mathivanan and J. Cappiello, Tetrahedron: Asymmetry, 1998, 9, 1–45 CrossRef CAS.
- G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336–345 CrossRef CAS PubMed.
- P. Wipf, Chem. Rev., 1995, 95, 2115–2134 CrossRef CAS.
- A.-C. Gaumont, M. Gulea and J. Levillain, Chem. Rev., 2009, 109, 1371–1401 CrossRef CAS PubMed.
- K. Schwekendiek and F. Glorius, Synthesis, 2006, 2006, 2996–3002 CrossRef PubMed.
- L. Engman, J. Org. Chem., 1991, 56, 3425–3430 CrossRef CAS.
- Y.-M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928–12931 CrossRef CAS PubMed.
- E. Cahard, N. Bremeyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2013, 52, 9284–9288 CrossRef CAS PubMed.
- B. C. Lemercier and J. G. Pierce, Org. Lett., 2014, 16, 2074–2076 CrossRef CAS PubMed.
- C. Jin, J. P. Burgess, J. A. Kepler and C. E. Cook, Org. Lett., 2007, 9, 1887–1890 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. C. Blanco Jaimes, A. M. Schuster and F. Rominger, J. Org. Chem., 2012, 77, 6394–6408 CrossRef CAS PubMed.
- A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391–4394 CrossRef CAS PubMed.
- M. Harmata and C. Huang, Synlett, 2008, 2008, 1399–1401 CrossRef PubMed.
- X. Meng and S. Kim, Org. Biomol. Chem., 2011, 9, 4429–4431 CAS.
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577–18580 CrossRef CAS PubMed.
- T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS PubMed.
- A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 10334–10337 CrossRef CAS PubMed.
- T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198–6201 CrossRef CAS PubMed.
- D. J. Wilger, J.-M. M. Grandjean, T. R. Lammert and D. A. Nicewicz, Nat. Chem., 2014, 6, 720–726 CAS.
- A. C. Benniston, A. Harriman, P. Li, J. P. Rostron, H. J. van Ramesdonk, M. M. Groeneveld, H. Zhang and J. W. Verhoeven, J. Am. Chem. Soc., 2005, 127, 16054–16064 CrossRef CAS PubMed.
- K. Tani, D. C. Behenna, R. M. McFadden and B. M. Stoltz, Org. Lett., 2007, 9, 2529–2531 CrossRef CAS PubMed.
- See ESI† for experimental details.
- J. Hartung, R. Kneuer, C. Rummey and G. Bringmann, J. Am. Chem. Soc., 2004, 126, 12121–12129 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc02331e |
| This journal is © The Royal Society of Chemistry 2015 |