DOI:
10.1039/C4SC02602K
(Edge Article)
Chem. Sci., 2015,
6, 275-282
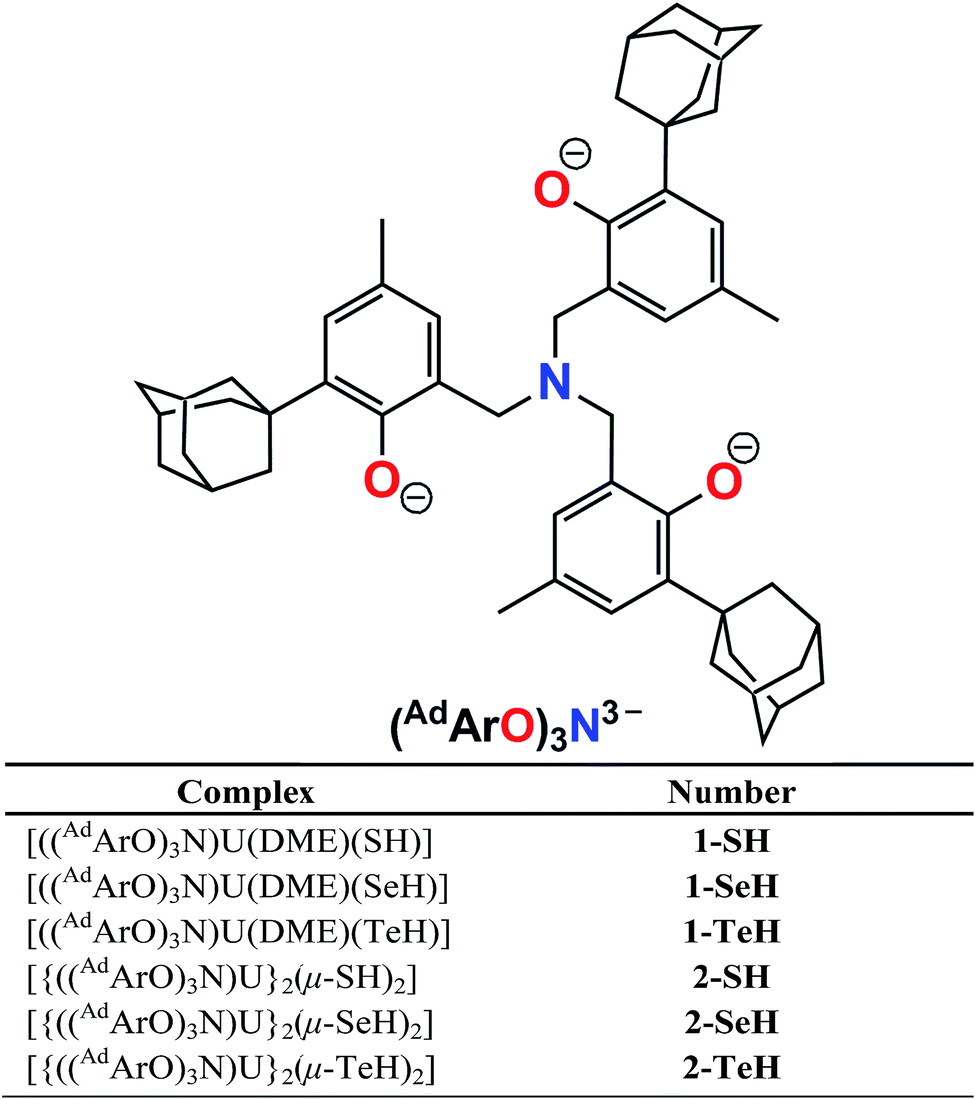
Reactivity of uranium(III) with H2E (E = S, Se, Te): synthesis of a series of mononuclear and dinuclear uranium(IV) hydrochalcogenido complexes†
Received
26th August 2014
, Accepted 29th September 2014
First published on 29th September 2014
Abstract
We report the syntheses, electronic properties, and molecular structures of a series of mono- and dinuclear uranium(IV) hydrochalcogenido complexes supported by the sterically demanding but very flexible, single N-anchored tris(aryloxide) ligand (AdArO)3N)3−. The mononuclear complexes [((AdArO)3N)U(DME)(EH)] (E = S, Se, Te) can be obtained from the reaction of the uranium(III) starting material [((AdArO)3N)UIII(DME)] in DME via reduction of H2E and the elimination of 0.5 equivalents of H2. The dinuclear complexes [{((AdArO)3N)U}2(μ-EH)2] can be obtained by dissolving their mononuclear counterparts in non-coordinating solvents such as benzene. In order to facilitate the work with the highly toxic gases, we created concentrated THF solutions that can be handled using simple glovebox techniques and can be stored at −35 °C for several weeks.
Introduction
Hydrochalcogenido complexes have received considerable interest in transition metal chemistry due to their potential applications as hydrodesulfurization and hydrogenation catalysts,1–9 as well as their relevance in bioinorganic chemistry.1,9–15 This class of compounds also serves as excellent precursors to a variety of homo- and hetero-bimetallic chalcogenido clusters with high nuclearity,9,16–25 some of which exhibit interesting optical properties.26,27 Furthermore, hydrochalcogenides have shown unusual reactivity such as Michael-type addition reactions with activated alkenes to afford the respective chalcogenolato complexes.19,28,29 In actinide chemistry, reports on chalcogenido complexes in general are rather scarce compared to the transition metals.30–48 Nevertheless, there has been a tremendous recent increase of interest in these types of compounds, which is motivated by the academic interest in synthesizing novel uranium–chalcogenide compounds and the fundamental importance of understanding the nature of covalent bonding between hard uranium ions and the soft chalcogenido ligands.49–52 In contrast to their transition metal chemistry, heavier main group 6 hydrochalcogenides of the actinides are exceedingly rare. In fact, the only reported compound is the hydrosulfido complex [(Cp*)3U(SH)] (Cp* = C5Me5) that was synthesized by Spirlet et al. from H2S or in a salt metathesis reaction from [(Cp*)3U(Br)] and NaSH, but no crystallographic evidence was provided.53
Herein, we report the synthesis of the first series of uranium hydrochalcogenido complexes that can be obtained as mononuclear complexes [((AdArO)3N)U(DME)(EH)] (1-EH, E = S, Se, Te) or as dinuclear complexes [{((AdArO)3N)U}2(μ-EH)2] (2-EH, E = S, Se, Te) depending on the choice of solvent (Chart 1). In this manner, either 1-EH or 2-EH can be synthesized from the uranium(III) starting material [((AdArO)3N)U(DME)] via reduction of H2E and the elimination of 0.5 equivalents of H2.
| Complex | Number |
|---|
| [((AdArO)3N)U(DME)(SH)] | 1-SH |
| [((AdArO)3N)U(DME)(SeH)] | 1-SeH |
| [((AdArO)3N)U(DME)(TeH)] | 1-TeH |
| [{((AdArO)3N)U}2(μ-SH)2] | 2-SH |
| [{((AdArO)3N)U}2(μ-SeH)2] | 2-SeH |
| [{((AdArO)3N)U}2(μ-TeH)2] | 2-TeH |
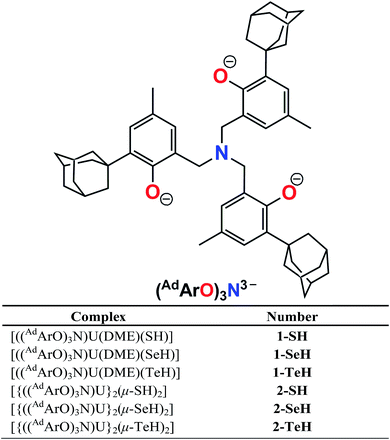 |
| | Chart 1 Complex formulas and numbering scheme of uranium complexes 1-EH and 2-EH (E = S, Se, Te), employing the chelating N-anchored ligand (AdArO)3N3− (top). | |
Results and discussion
Recently, we reported the synthesis of the uranium(III) complex [((AdArO)3N)U(DME)] supported by the flexible, N-anchored chelator (AdArO)3N3− (with (AdArO)3N3− = tris(2-hydroxy-3-adamantyl-5-methylbenzyl)amine).54 This reactive, low-valent complex can undergo reactions such as the activation of CO2 and its heterocumulene analogs COS and CS2,55–57 as well as the activation of elemental chalcogens.58 The starting material [((AdArO)3N)U(DME)] exhibits a half-step potential of −1.879 V for the uranium(III/IV) redox couple (see ESI†), supporting the fact that this complex is a potent reductant, which – in part – explains its observed reactivity with the H2E substrates. Furthermore, even uranium(IV) complexes supported by the (AdArO)3N3− ligand can show remarkable reactivity under the right conditions, such as the formation of polychalcogenido complexes through stepwise addition of stoichiometrically precise amounts of elemental chalcogen.59 H2S and its heavier congeners H2Se and H2Te are known and well-documented precursors for the synthesis of hydrochalcogenides, however, the resultant hydrochalcogenides are rarely stable compounds and subsequent reactions often lead to the formation of metal polychalcogenide clusters or binary metal salts instead.1,9 Furthermore, synthetic work is greatly complicated by the high toxicity and malodor of these gases. With the commercial availability of H2S as a solution in THF, reactions with this gas can be carried out under much simpler conditions. That said, no such reagents exist for its selenium and tellurium analogs. To solve these synthetic challenges, we created concentrated solutions of H2Se and H2Te by condensing the respective gas formed from Al2E3 (E = Se, Te) and sulfuric acid into a THF solution at −70 °C.60 These solutions can be stored under an inert atmosphere in a freezer at −35 °C for several weeks. This procedure greatly simplifies handling H2Se and H2Te.
Synthesis and molecular structures of mononuclear uranium(IV) hydrochalcogenido complexes 1-EH (E = S, Se, Te)
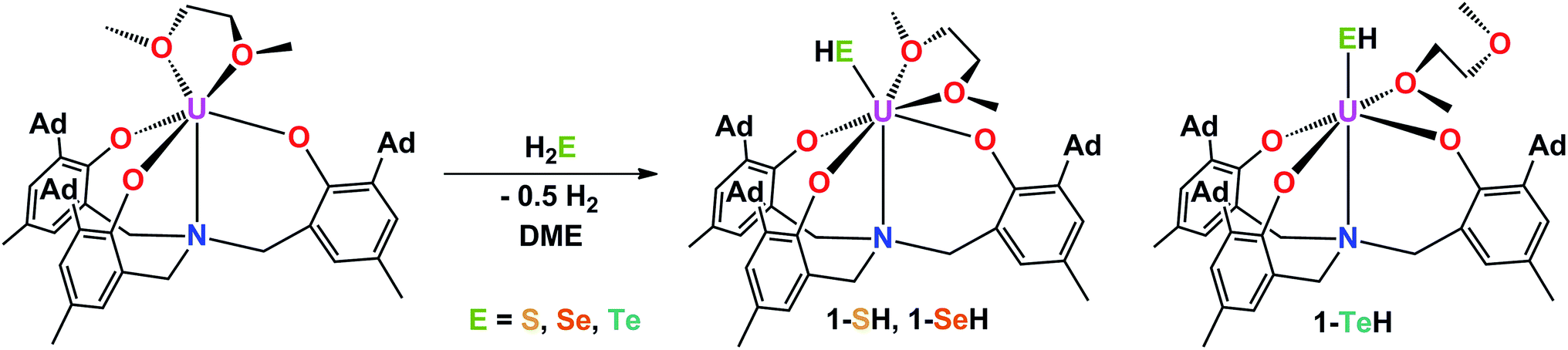
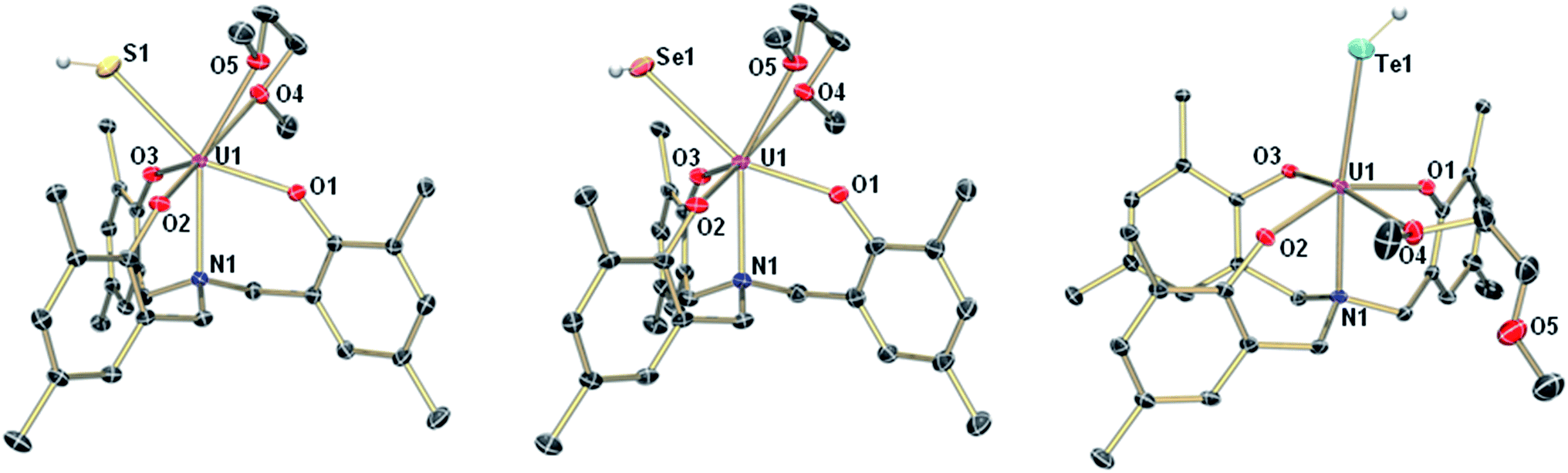
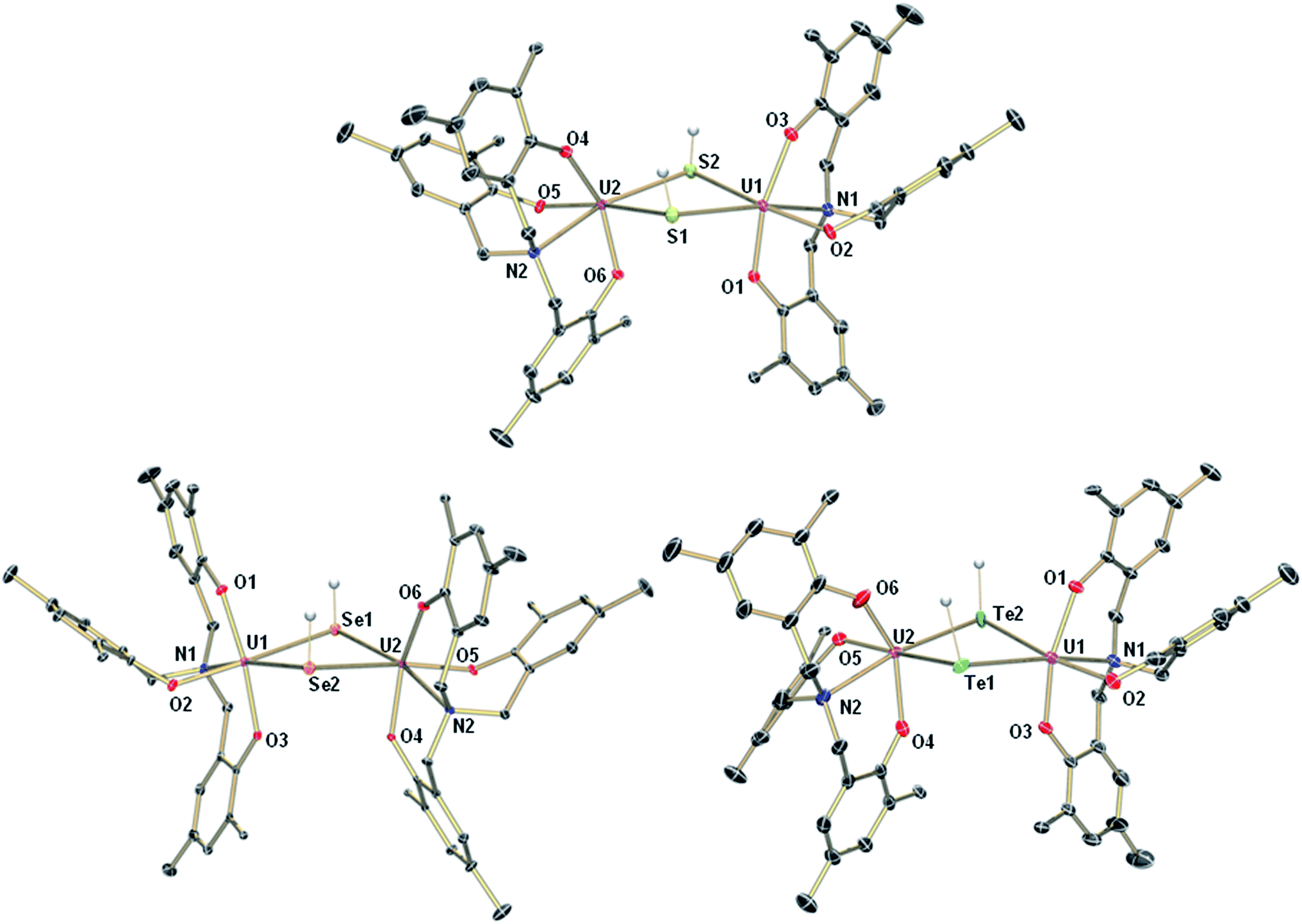
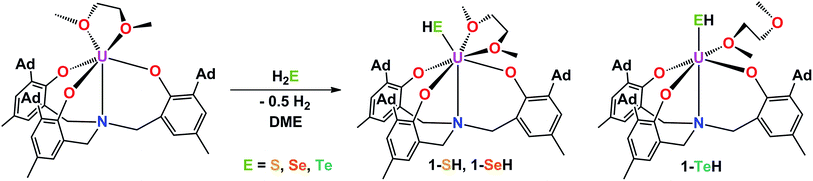
The mononuclear thiolato complex [((AdArO)3N)U(DME)(SH)] (1-SH) has been synthesized by reacting the uranium(III) starting material [((AdArO)3N)U(DME)] in THF with an excess amount of H2S via dropwise addition of a solution of H2S in THF until the reaction mixture turned green. Within five minutes, a light green precipitate formed and 1-SH was obtained in 70% yield after filtration and drying of the solids in vacuo (Scheme 1). Recrystallization of the solid via diffusion of n-hexane into a concentrated DME solution yielded crystals suitable for X-ray diffraction analysis. The solid state structure of 1-SH reveals a mononuclear complex [((AdArO)3N)U(DME)(SH)] with a seven-coordinate uranium center in a distorted, monocapped trigonal prismatic coordination environment, in which one molecule of DME is coordinated to the uranium center in a bidentate fashion (Fig. 1, left). The U–S bond distance of 2.797(1) Å is clearly longer than reported U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bonds (2.382(11)–2.481(1) Å) and is in good agreement with a U–S single bond (2.588(1)–2.907(3) Å, Table 1).58,59,61–65 Furthermore, the chalcogen-bound hydrogen could be located in the difference Fourier map and was subsequently treated using a riding model. The U–N distance of 2.616(2) Å and the U–Oavg. bonds of 2.171 Å are comparable to other complexes supported by the N-anchored ligand (AdArO)3N3−.54,58,59 Additionally, the SH− ligand is not coordinated in the axial position directly trans to the nitrogen anchor, which is clearly shown in the N–U–S bond angle of 136.24(4)°.
S double bonds (2.382(11)–2.481(1) Å) and is in good agreement with a U–S single bond (2.588(1)–2.907(3) Å, Table 1).58,59,61–65 Furthermore, the chalcogen-bound hydrogen could be located in the difference Fourier map and was subsequently treated using a riding model. The U–N distance of 2.616(2) Å and the U–Oavg. bonds of 2.171 Å are comparable to other complexes supported by the N-anchored ligand (AdArO)3N3−.54,58,59 Additionally, the SH− ligand is not coordinated in the axial position directly trans to the nitrogen anchor, which is clearly shown in the N–U–S bond angle of 136.24(4)°.
 |
| | Scheme 1 Synthesis of hydrochalcogenido complexes 1-EH (E = S, Se, Te). | |
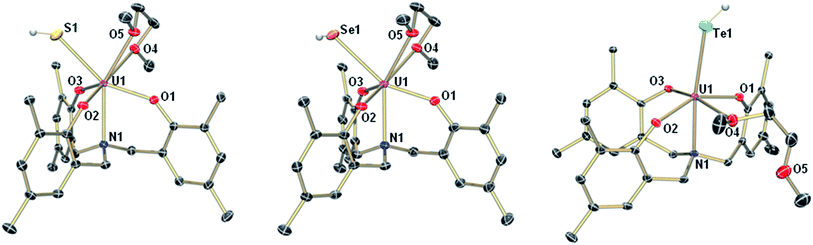 |
| | Fig. 1 Molecular structures of uranium hydrosulfido complex 1-SH (left), uranium hydroselenido complex 1-SeH (center), and uranium hydrotellurido complex 1-TeH (right). The chalcogen-bound H atoms in 1-SH and 1-TeH could be located in the difference Fourier map, the hydrogen in 1-SeH was placed in a position of optimized geometry. All other H atoms, the adamantyl groups, and co-crystallized solvent molecules are omitted for clarity. Thermal ellipsoids are at 50% probability. | |
Table 1 Selected bond distances (Å) and angles (°) for complexes 1-EH and 2-EH (E = S, Se, Te)
| Structural parameters |
1-SH
|
1-SeH
|
1-TeH
|
2-SH
|
2-SeH
|
2-TeH
|
| U–Oavg. |
2.169 |
2.170 |
2.130 |
2.121, 2.122 |
2.120, 2.122 |
2.119, 2.136 |
| U–ODME |
2.582(2) 2.616(2) |
2.579(3) 2.599(3) |
2.508(2) |
— |
— |
— |
| U–N |
2.616(2) |
2.605(3) |
2.575(2) |
2.546(2), 2.542(2) |
2.552(3), 2.547(3) |
2.558(3), 2.567(3) |
| U1–E1,2 |
2.797(1) |
2.936(1) |
3.122(1) |
2.882(1), 2.961(1) |
2.992(1), 3.090(1) |
3.145(2), 3.296(2) |
| U2–E1,2 |
— |
— |
— |
2.964(1), 2.877(1) |
3.094(1), 2.989(1) |
3.163(2), 3.119(2) |
| N–U–E |
136.24(4) |
133.44(7) |
173.25(4) |
166.14(4), 87.79(4), 88.57(5), 166.63(4) |
168.44(6), 87.25(6), 88.23(6), 169.00(6) |
171.00(7), 87.43(8), 94.94(9), 166.68(10) |
| U–E2–U |
— |
— |
— |
159.99 |
158.10 |
160.09 |
| E1⋯E2 |
— |
— |
— |
3.888 |
4.157 |
4.479 |
| U1⋯U2 |
— |
— |
— |
4.295 |
4.381 |
4.448 |
Applying a similar synthetic procedure, the mononuclear selenolato complex [((AdArO)3N)U(DME)(SeH)] (1-SeH) can be synthesized in 72% yield (Scheme 1). The molecular structure of 1-SeH, obtained from crystals grown from a concentrated DME solution, compares well to that of 1-SH (Fig. 1, center). Noteworthy, the asymmetric unit of the cell in 1-SeH contains two independent but structurally very similar molecules of the complex, and hence, only the values of 1-SeH A are given in Table 1 (for more details see ESI†). According to the 3σ criterion, the U–N (2.605(3) Å) and U–Oavg. bond lengths (2.170 Å) are the same as those observed in 1-SH. The U–Se bond of 2.936(1) Å is significantly longer than a distinctive USe double bond (2.533(1)–2.646(1) Å) and can be compared to other complexes with U–Se single bond distances varying from 2.719(1) to 3.125(1) Å.57–59,61–63,66,67 As in complex 1-SH, the N–U–Se angle is strongly bent with 133.44(7)°. In contrast to 1-SH, the selenium-bound hydrogen could not be located in the difference Fourier map, but was confirmed in 1H and 2H{1H} NMR experiments. The 1H NMR spectrum of 1-SeH recorded in pyridine-d5 shows a total of ten signals, ranging from −63 to 11 ppm, however, the unambiguous assignment of the hydrogen resonance of the SeH− ligand is severely hampered due to the complicated nature of the supporting ligand system. As a result, the deuterated analog of 1-SeH, namely [((AdArO)3N)U(DME)(SeD)] (1-SeD), was synthesized in a reaction of the uranium(III) starting material [((AdArO)3N)U(DME)] with D2Se. As expected, the 1H NMR spectrum of 1-SeD shows only nine signals, and thus, unequivocally designates the signal at −62.86 ppm to the SeH− hydrogen. Additional 2H{1H} NMR experiments in pyridine revealed a single deuterium signal at −62.69 ppm that originates from the SeD− moiety, further supporting the previous assignment in the 1H NMR spectrum and confirming the presence of the chalcogen-bound hydrogen.
In contrast to the closely related syntheses of 1-SH and 1-SeH, the preparation of the tellurium analog complex [((AdArO)3N)U(DME)(TeH)] (1-TeH), requires special precautions, since H2Te is very susceptible to light and quickly decomposes at temperatures above 0 °C. As a consequence, the reaction needs to be carried out in cooled solvents under the rigorous exclusion of light to obtain 1-TeH in 78% yield (Scheme 1). Remarkably, the resulting complex 1-TeH is both stable at elevated temperatures up to 80 °C and in the presence of light, which contrasts with the very few reported, usually rather unstable, transition metal complexes featuring a TeH− ligand.1,9 Single crystals suitable for X-ray diffraction were obtained via diffusion of n-hexane into a mixture of benzene and DME (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). The molecular structure of 1-TeH is distinctly different from those of 1-SH and 1-SeH, as it shows a six-coordinate uranium center, located in a distorted octahedral coordination environment (Fig. 1, right). Furthermore, and consequently, the DME solvent molecule is now coordinated in a monodentate fashion. The U–N (2.575(1) Å) and U–Oavg. bond lengths (2.130 Å) are slightly shorter than those observed in 1-SH and 1-SeH. The U–Te bond distance of 3.122(1) Å is in accordance with the formulation of a U–Te single bond (Table 1).58,61,62,67 The TeH− ligand is coordinated almost linearly trans to the N-anchor in the axial position and shows a N–U–Te angle of 173.25(4)°. Attempts to obtain the hydroxo complex [((AdArO)3N)U(DME)(OH)] from H2O in the same way as complexes 1-EH lead to the formation of the bridging oxo species [{((AdArO)3N)U(DME)}2(μ-O)] instead,54 even in the presence of an excess amount of water. The rate of forming the hydroxo complex [((AdArO)3N)U(DME)(OH)] appears to be much slower than the subsequent reaction to form [{((AdArO)3N)U(DME)}2(μ-O)]. The reason why H2O reacts so slowly with the uranium(III) starting material may be ascribed to the high stability of the O–H bond compared to the decreasingly stable E–H bonds in the hydrides of the heavier chalcogens.68
:3). The molecular structure of 1-TeH is distinctly different from those of 1-SH and 1-SeH, as it shows a six-coordinate uranium center, located in a distorted octahedral coordination environment (Fig. 1, right). Furthermore, and consequently, the DME solvent molecule is now coordinated in a monodentate fashion. The U–N (2.575(1) Å) and U–Oavg. bond lengths (2.130 Å) are slightly shorter than those observed in 1-SH and 1-SeH. The U–Te bond distance of 3.122(1) Å is in accordance with the formulation of a U–Te single bond (Table 1).58,61,62,67 The TeH− ligand is coordinated almost linearly trans to the N-anchor in the axial position and shows a N–U–Te angle of 173.25(4)°. Attempts to obtain the hydroxo complex [((AdArO)3N)U(DME)(OH)] from H2O in the same way as complexes 1-EH lead to the formation of the bridging oxo species [{((AdArO)3N)U(DME)}2(μ-O)] instead,54 even in the presence of an excess amount of water. The rate of forming the hydroxo complex [((AdArO)3N)U(DME)(OH)] appears to be much slower than the subsequent reaction to form [{((AdArO)3N)U(DME)}2(μ-O)]. The reason why H2O reacts so slowly with the uranium(III) starting material may be ascribed to the high stability of the O–H bond compared to the decreasingly stable E–H bonds in the hydrides of the heavier chalcogens.68
Synthesis and molecular structures of dinuclear uranium(IV) hydrochalcogenido complexes 2-EH (E = S, Se, Te)
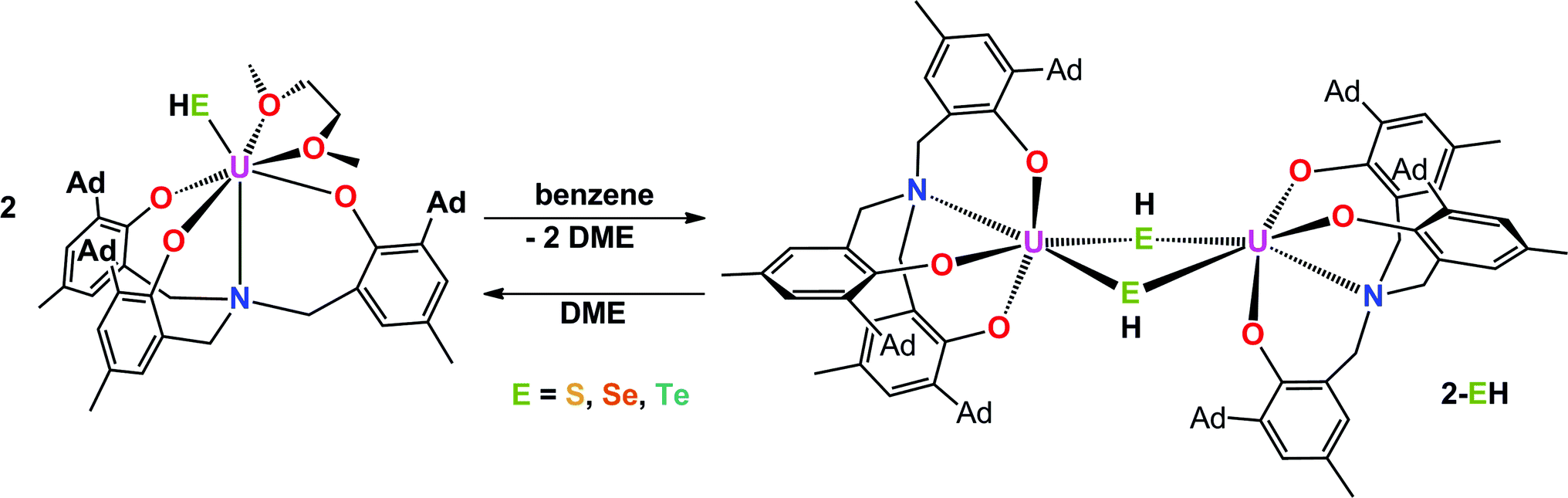
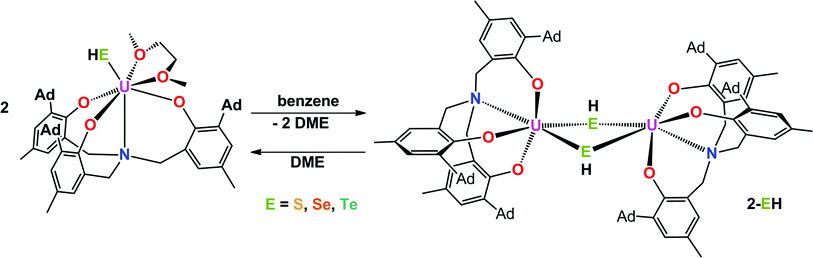
Interestingly, crystallization of 1-EH from non-coordinating solvents, such as benzene, leads to a change in coordination geometry. X-ray diffraction analysis on crystals grown from diffusion of n-hexane into saturated solutions of the complexes in benzene revealed the molecular structures of the dinuclear complexes [{((AdArO)3N)U}2(μ-EH)2] (E = S, 2-SH; Se, 2-SeH; Te, 2-TeH) (Scheme 2, Fig. 2). Each uranium center in 2-SH is now only six-coordinate and adopts a distorted octahedral coordination geometry. The U–S bond distances of 2.878(1)–2.964(1) Å are slightly longer than in 1-SH and are significantly longer than those observed in the dinuclear bis-μ-sulfido complex [Na(DME)3]2[{((AdArO)3N)U}2(μ-S)2] (2.688(2)–2.714(2) Å) or the bridging persulfido complexes [{(R2N)3U}2(μ-η2:η2-S2)] (R = SiMe3, 2.706(2)–2.923(2) Å) and [{((SiMe2NPh)3tacn)U}2(μ-η2:η2-S2)] (2.855(2)–2.907(3) Å).58,63,69 The SH− ligands with the shorter U–S distances are now coordinated in the axial positions trans to the nitrogen anchors, which is clearly visible in the N–U–S bond angles of 166.14(4)° and 166.63(4)°. The U–Oavg. (2.121 Å, 2.122 Å) and U–N bond distances (2.546(1) Å, 2.542(2) Å) are in good agreement with the respective structural parameters in complexes 1-EH (E = S, Se, Te). As in complexes 1-SH and 1-TeH, the chalcogen-bound hydrogens in complex 2-SH were located in the difference Fourier map.
 |
| | Scheme 2 Synthesis of dinuclear hydrochalcogenido complexes 2-EH (E = S, Se, Te). | |
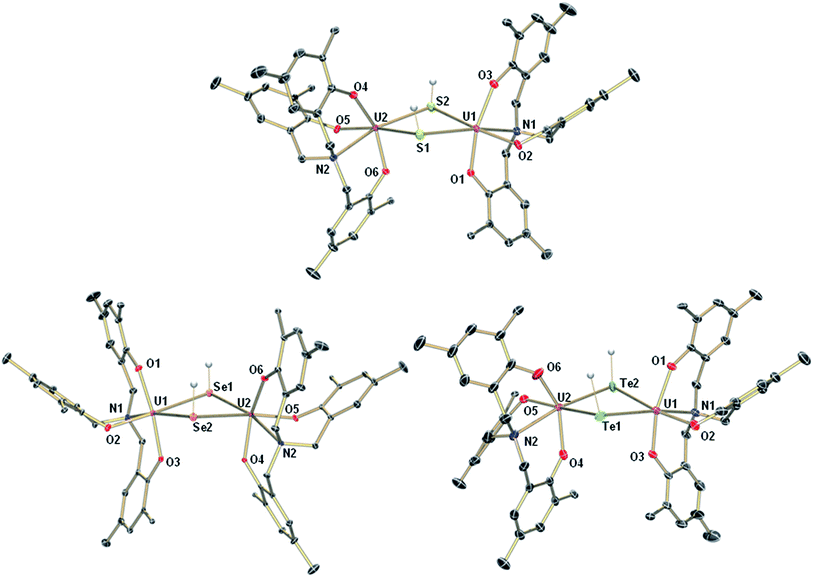 |
| | Fig. 2 Molecular structures of uranium hydrosulfido complex 2-SH (top), uranium hydroselenido complex 2-SeH (bottom left), and uranium hydrotellurido complex 2-TeH (bottom right). The chalcogen-bound H atoms in 2-SH, 2-SeH, and 2-TeH could be located in the difference Fourier map. All other H atoms, the adamantyl groups and co-crystallized solvent molecules are omitted for clarity. Thermal ellipsoids are at 50% probability. | |
The heavier chalcogenido complexes 2-SeH and 2-TeH exhibit the same molecular structures as 2-SH and feature comparable U–Oavg. and U–N bond lengths (see Table 1). The U–Se and U–Te bonds range from 2.989(1) to 3.094(1) Å and 3.119(2) to 3.296(2) Å, respectively, which is slightly longer compared to the bis-μ-chalcogenido complexes [Na(DME)3]2[{((AdArO)3N)U}2(μ-E)2] (E = Se, Te) (2.819(1)–2.866(1) Å for E = Se and 3.031(1)–3.112(1) Å for E = Te) and the bridging perselenido complex [{((AdArO)3N)U}2(μ-η2:η2-Se)(μ-DME)] (2.942(1)–3.079(1) Å).58,59 Furthermore, the N–U–E angles of 168.44(6)° and 169.00(6)° (2-SeH), as well as 171.00(7)° and 166.68(10)° (2-TeH) are very similar compared to 2-SH but are slightly less bent due to a greater distance between the uranium centers (d(U⋯U) = 4.296 Å (2-SH) < 4.381 Å (2-SeH) < 4.448 Å (2-TeH)), which is in accordance with the larger atomic radii of selenium and tellurium that lead to slightly higher steric repulsion within the complexes. Noteworthy, the mononuclear complexes 1-EH and their dinuclear counterparts 2-EH showed identical 1H NMR spectra in deuterated benzene. Complexes supported by the highly flexible N-anchored ligand (AdArO)3N3− as well as the often highly nucleophilic EH− functional groups show a strong tendency to form dinuclear species, hence, it is within expectation that 1-EH can dimerize to 2-EH in non-coordinating solvents, such as benzene, with the loss of coordinating solvent.1,54–59 This conclusion is further supported by the presence of broadened signals at approximately 3 ppm in all spectra, which are attributed to uncoordinated DME. Likewise, the dinuclear species 2-EH dissociates in DME to form the mononuclear complexes 1-EH.
Infrared spectroscopy of complexes 1-EH and 2-EH
IR spectroscopy can be a useful tool to determine the presence of an EH group, since the characteristic, but often rather weak IR-active ν(E–H) bands usually appear in the regions between 2300–2600 cm−1 (SH),70–76 2200–2500 cm−1 (SeH),74–82 and 1800–2000 cm−1 (TeH).74,76,83 However, complexes 1-SH and 2-SH, as well as 1-SeH, 1-SeD and 2-SeH, do not show any ν(E–H) bands at all, a fact that was also observed in several transition metal complexes.15,18,84–90 In contrast to this, complexes 1-TeH and 2-TeH both show one distinctive absorption band at 2000 cm−1 and 1998 cm−1, respectively, which is well in accordance with the scarce amount of data available for hydrotellurido complexes. Nevertheless, we could unambiguously identify the H atoms in all complexes 1-EH and 2-EHvia X-ray diffraction analysis and 2H{1H} NMR spectroscopic experiments.
UV/vis/NIR spectroscopy of complexes 1-EH and 2-EH
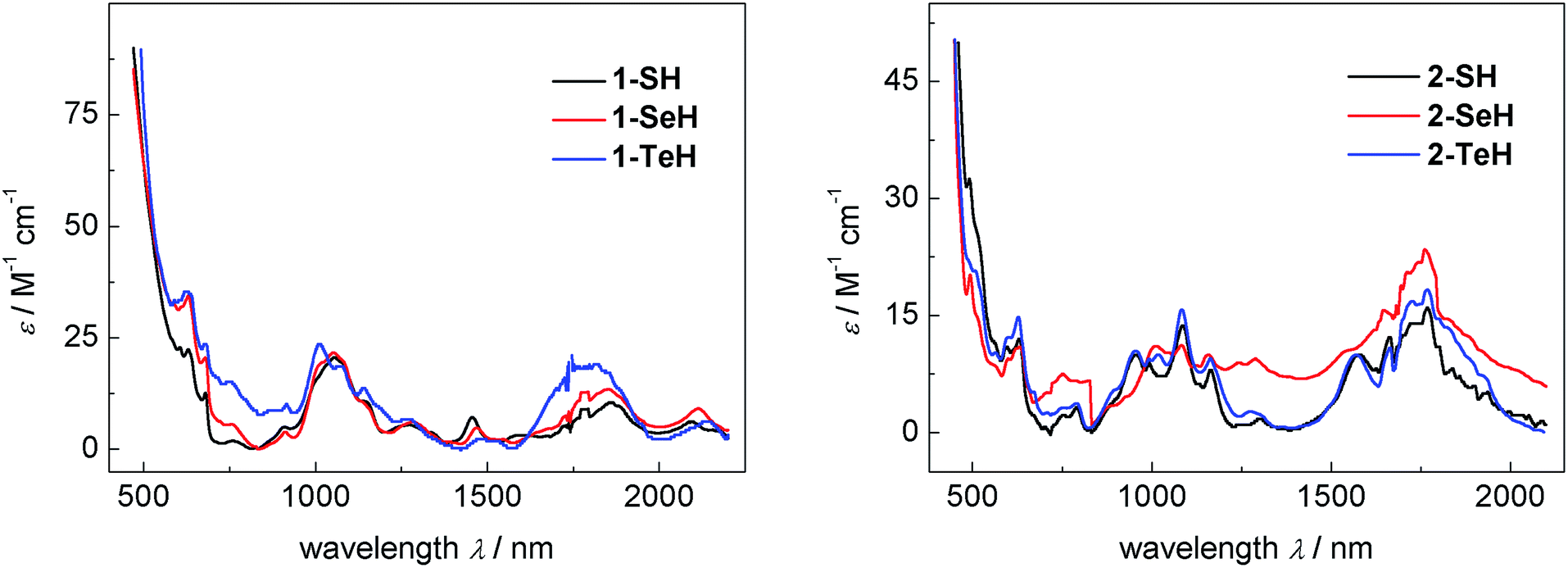
The vis/NIR spectra of complexes 1-EH, recorded in DME, are all very similar and show a number of low-intensity f–f transitions with small molar extinction coefficients of 10–35 M−1 cm−1 between 500 and 2200 nm (see Fig. 3, left), characteristic for tetravalent complexes of uranium.91 While the spectra of 1-SH and 1-SeH are almost identical, the spectrum of 1-TeH clearly shows the hypsochromic shift of two bands at 1011 nm (ε = 24 M−1 cm−1) and 1815 nm (ε = 20 M−1 cm−1). This observation can be rationalized by the discrepancy in the ligand-field splitting that should be expected due to the structural difference of 1-TeH compared to 1-SH and 1-SeH. The UV/vis region shows one intense charge-transfer band centered at 287 nm for each compound with molar extinction coefficients ranging from 14–19 × 103 M−1 cm−1 (see ESI, Fig. S25†). The electronic absorption spectra of complexes 2-EH, recorded in benzene, are all very similar as well, showing very weak f–f transitions from 480 to 2000 nm (ε = 5–32 M−1 cm−1, Fig. 3, right) and intense charge-transfer absorptions centered at 284 nm in the UV/vis region (ε = 12–14 × 103 M−1 cm−1, see ESI, Fig. S27†). For a better comparison, the molar extinction coefficients for complexes 2-EH were calculated per uranium center, resulting in much lower values than those in complexes 1-EH. This observation is indicative of weaker U–E bonds and lower covalency and is further supported by the longer U–E bonds in the molecular structures of complexes 2-EH.92,93 Furthermore, in contrast to complexes 1-EH, no hypsochromic shift was observed for any of the complexes 2-EH, however, 2-SH and 2-SeH exhibit an additional charge-transfer band at 395 and 406 nm, respectively (1-SH, ε = 2185 M−1 cm−1; 1-SeH, ε = 2455 M−1 cm−1), that could not be observed for 2-TeH. The spectra of compounds 1-EH and 2-EH are indicative for tetravalent uranium centers and are in contrast to the spectra anticipated for a U(V) 5f1 or a U(III) 5f3 species.91,94–97
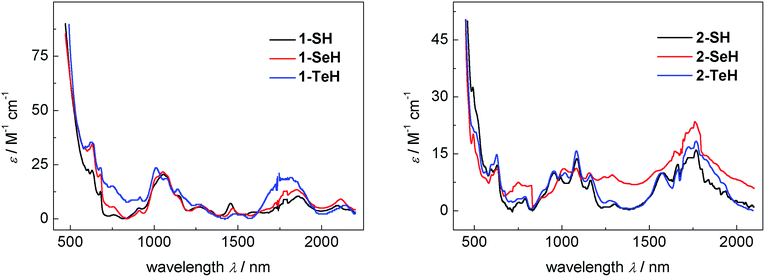 |
| | Fig. 3 Vis/NIR spectra of hydrochalcogenido complexes 1-SH (9.2 mM, black), 1-SeH (9.7 mM, red), and 1-TeH (4.1 mM, blue) recorded in DME at 25 °C (left) and vis/NIR spectra of hydrochalcogenido complexes 2-SH (2.0 mM, black), 2-SeH (6.6 mM, red), and 2-TeH (6.5 mM, blue) recorded in benzene at 25 °C (right). The molar extinction coefficients for complexes 2-EH were calculated per uranium center for a better comparison. | |
Magnetism of complexes 1-EH and 2-EH
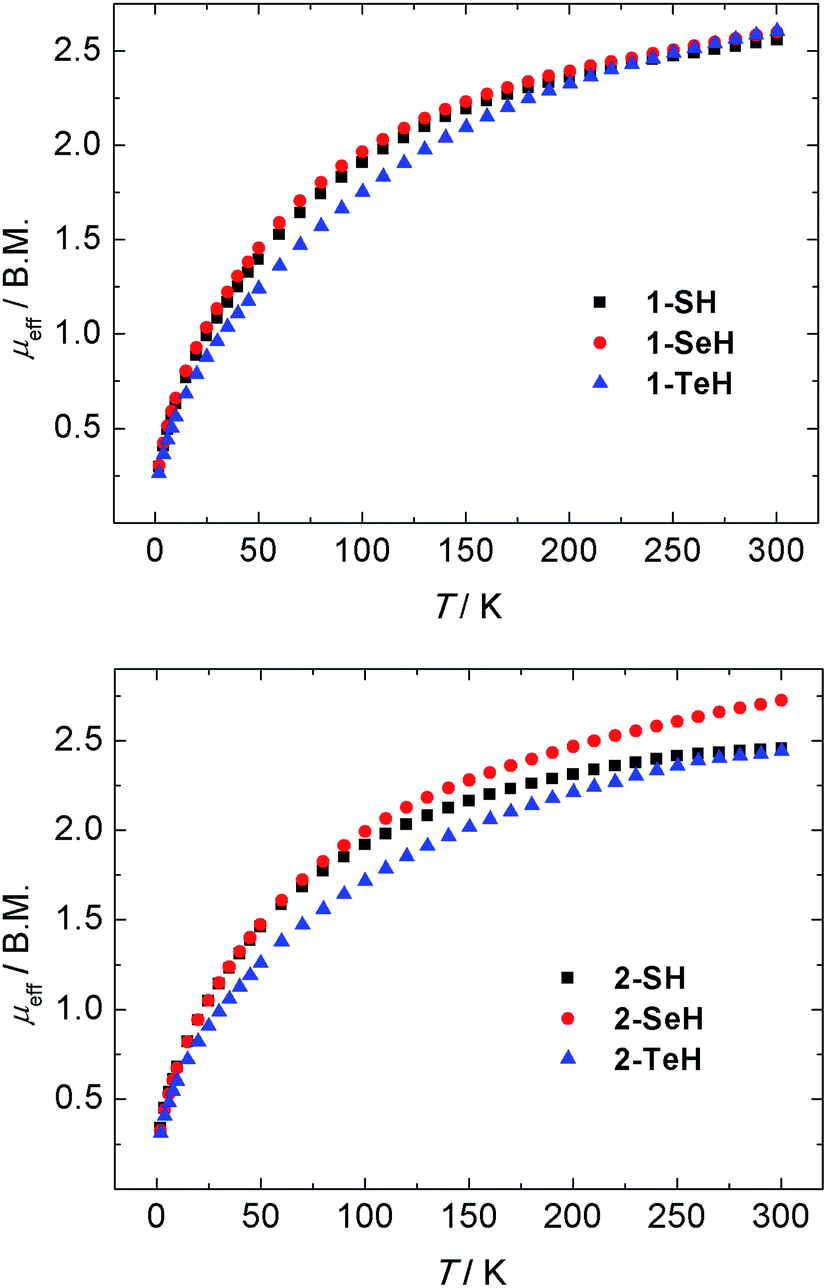
The oxidation states and electronic structures of the uranium centers in complexes 1-EH and 2-EH (E = S, Se, Te) were further characterized by temperature-dependent magnetic susceptibility measurements. SQUID magnetization measurements from 2–300 K of compounds 1-EH all reveal very similar magnetic moments of 2.56 (1-SH), 2.60 (1-SeH), and 2.60 B.M. (1-TeH) at 300 K, which decrease with decreasing temperature to 0.30 (1-SH), 0.30 (1-SeH), and 0.26 B.M. (1-TeH) at 2 K (Fig. 4, top). Likewise, the dinuclear complexes 2-EH show magnetic moments of 2.46 (2-SH), 2.65 (2-SeH), and 2.44 B.M. (2-TeH) at 300 K that decrease to 0.34 (2-SH), 0.33 (2-SeH), and 0.31 B.M. (2-TeH) at 2 K (Fig. 4, bottom). These observations are in agreement with tetravalent uranium centers with a non-magnetic 3H4 ground state.58,98,99 An interesting feature to note, is the difference in the plot of χMvs. T of 1-EH and 2-EH. While the mononuclear complexes 1-EH show temperature-independent paramagnetism (TIP) over a wide temperature range from 10 to 62 K (for 1-SH and 1-SeH) and 101 K (1-TeH), followed by a steady decrease of the molar susceptibility, this feature is slightly less pronounced in the dinuclear complexes 2-SH and 2-SeH (TIP below 50 K, see ESI†) and shows a significant difference in 2-TeH (TIP below 55 K).
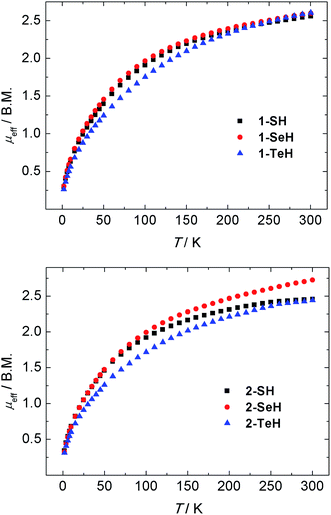 |
| | Fig. 4 Temperature-dependent SQUID magnetization data of hydrochalcogenido complexes 1-EH (top, E = S (black), Se (red), Te (blue)) and 2-EH (bottom, E = S (black), Se (red), Te (blue)) as a plot of μeffvs. T. Data were corrected for underlying magnetism. | |
Conclusion
The reaction of H2E (E = S, Se, Te) with a reducing metal center is a viable synthetic route to obtain metal hydrochalcogenido complexes, however, handling of these gases is greatly complicated due to their high toxicity. With the use of concentrated and cooled solutions of these gases in THF, we were able to synthesize the new uranium(IV) hydrochalcogenido complexes [((AdArO)3N)U(DME)(EH)] (1-EH, E = S, Se, Te), using simple glovebox techniques. These compounds yield the dinuclear complexes [{((AdArO)3N)U}2(μ-EH)2] (2-EH, E = S, Se, Te) in non-coordinating solvents, such as benzene, with the loss of coordinating DME. UV/vis/NIR spectroscopy and SQUID magnetization measurements further confirm that complexes 1-EH and 2-EH can be clearly distinguished both in solution and in the solid state. We are currently trying to access terminal mono-chalcogenido species from the herein presented hydrochalcogenido complexes, in order to establish a series of chalcogenido complexes with U–E single and UE double bonds with similar coordination environments. This series should provide an excellent opportunity to gain more insight into the covalency and f-orbital participation of the U–E bond in uranium chalcogenido complexes.
Acknowledgements
This work was supported by funds from the German Federal Ministry of Education and Research (support codes 02 NUK 012C and 02 NUK 020C). We greatly thank Alexander F. R. Kilpatrick and Dr. Andreas Scheurer for performing 77Se, 125Te, 2H{1H}, and COSY NMR experiments. We would also like to thank Dr. A. K. Abdul-Sada and Prof. Geoffrey N. Cloke for performing mass spectrometry experiments.
Notes and references
- M. Peruzzini, I. D. L. Rios and A. Romerosa, Prog. Inorg. Chem., 2007, 49, 169–453 CrossRef.
- R. R. Chianelli, T. A. Pecoraro, T. R. Halbert, W. H. Pan and E. I. Stiefel, J. Catal., 1984, 86, 226–230 CrossRef CAS.
- R. J. Angelici, Acc. Chem. Res., 1988, 21, 387–394 CrossRef CAS.
- M. R. DuBois, Chem. Rev., 1989, 89, 1–9 CrossRef CAS.
- R. A. Sánchez-Delgado, J. Mol. Catal., 1994, 86, 287–307 CrossRef.
- M. D. Curtis and S. H. Druker, J. Am. Chem. Soc., 1997, 119, 1027–1036 CrossRef CAS.
- C. Bianchini and A. Meli, Acc. Chem. Res., 1998, 31, 109–116 CrossRef CAS.
- E. J. M. Hensen, G. M. H. J. Lardinois, V. H. J. de Beer, J. A. R. van Veen and R. A. van Santen, J. Catal., 1999, 187, 95–108 CrossRef CAS.
- S. Kuwata and M. Hidai, Coord. Chem. Rev., 2001, 213, 211–305 CrossRef CAS.
- T. Yamamura, G. Christou and R. H. Holm, Inorg. Chem., 1983, 22, 939–949 CrossRef CAS.
- J. H. Enemark and C. G. Young, Adv. Inorg. Chem., 1993, 40, 1–88 CrossRef.
- R. Hille, Chem. Rev., 1996, 96, 2757–2816 CrossRef CAS PubMed.
- R. G. Matthews, Acc. Chem. Res., 2001, 34, 681–689 CrossRef CAS PubMed.
- G. Parkin, Chem. Rev., 2004, 104, 699–768 CrossRef CAS PubMed.
- M. M. Ibrahim, J. Seebacher, G. Steinfeld and H. Vahrenkamp, Inorg. Chem., 2005, 44, 8531–8538 CrossRef CAS PubMed.
- D. P. Klein, G. M. Kloster and R. G. Bergman, J. Am. Chem. Soc., 1990, 112, 2022–2024 CrossRef CAS.
- K. Hashizume, Y. Mizobe and M. Hidai, Organometallics, 1996, 15, 3303–3309 CrossRef CAS.
- J. Ruiz, V. Rodríguez, C. Vicente, J. M. Martí, G. López and J. Pérez, Inorg. Chem., 2001, 40, 5354–5360 CrossRef CAS PubMed.
- H. Kato, H. Seino, Y. Mizobe and M. Hidai, Inorg. Chim. Acta, 2002, 339, 188–192 CrossRef CAS.
- F. Takagi, H. Seino, Y. Mizobe and M. Hidai, Organometallics, 2002, 21, 694–699 CrossRef CAS.
- S. Nagao, H. Seino, M. Hidai and Y. Mizobe, J. Organomet. Chem., 2003, 669, 124–134 CrossRef CAS.
- L. A. Oro, M. A. Ciriano, J. J. Pérez-Torrente, M. A. Casado and M. A. F. Hernandez-Gruel, C. R. Chim., 2003, 6, 47–57 CrossRef CAS.
- W.-Y. Yeh, H. Seino, T. Amitsuka, S. Ohba, M. Hidai and Y. Mizobe, J. Organomet. Chem., 2004, 689, 2338–2345 CrossRef CAS.
- Q.-F. Zhang, Z. Yu, A. Rothenberger, D. Fenske and W.-H. Leung, Inorg. Chim. Acta, 2007, 360, 1568–1574 CrossRef CAS.
- J. J. Pérez-Torrente, M. V. Jiménez, M. A. F. Hernandez-Gruel, M. J. Fabra, F. J. Lahoz and L. A. Oro, Chem.–Eur. J., 2009, 15, 12212–12222 CrossRef PubMed.
-
C.-C. Ou and C.-S. Yang in Advanced Topics on Crystal Growth, ed. S. O. Ferreira, InTech, 2013, ch. 13 Search PubMed.
- O. Fuhr, S. Dehnen and D. Fenske, Chem. Soc. Rev., 2013, 42, 1871–1906 RSC.
- F. Takagi, H. Seino, M. Hidai and Y. Mizobe, J. Chem. Soc., Dalton Trans., 2002, 3603–3610 RSC.
- F. Takagi, H. Seino, Y. Mizobe and M. Hidai, Can. J. Chem., 2001, 79, 632–634 CrossRef CAS.
- D. L. Perry, A. Zalkin, H. Ruben and D. H. Templeton, Inorg. Chem., 1982, 21, 237–240 CrossRef CAS.
- J. G. Brennan, R. A. Andersen and A. Zalkin, Inorg. Chem., 1986, 25, 1761–1765 CrossRef CAS.
- D. A. Wrobleski, D. T. Cromer, J. V. Ortiz, T. B. Rauchfuss, R. R. Ryan and A. P. Sattelberger, J. Am. Chem. Soc., 1986, 108, 174–175 CrossRef CAS.
- A. C. Sutorik and M. G. Kanatzidis, J. Am. Chem. Soc., 1991, 113, 7754–7755 CrossRef CAS.
- L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245–4254 CrossRef CAS.
- A. C. Sutorik and M. G. Kanatzidis, Polyhedron, 1997, 16, 3921–3927 CrossRef CAS.
- C. Lescop, T. Arliguie, M. Lance, M. Nierlich and M. Ephritikhine, J. Organomet. Chem., 1999, 580, 137–144 CrossRef CAS.
- L. Ventelon, C. Lescop, T. Arliguie, M. Ephritikhine, P. C. Leverd, M. Lance and M. Nierlich, Chem. Commun., 1999, 659–660 RSC.
- B. Liang, L. Andrews, N. Ismail and C. J. Marsden, Inorg. Chem., 2002, 41, 2811–2813 CrossRef CAS PubMed.
- A. J. Gaunt, B. L. Scott and M. P. Neu, Inorg. Chem., 2006, 45, 7401–7407 CrossRef CAS PubMed.
- W. J. Evans, M. K. Takase, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2008, 28, 236–243 CrossRef.
- C. R. Graves, B. L. Scott, D. E. Morris and J. L. Kiplinger, Chem. Commun., 2009, 776–778 RSC.
- X. Wang, L. Andrews and C. J. Marsden, Inorg. Chem., 2009, 48, 6888–6895 CrossRef CAS PubMed.
- W. J. Evans, E. Montalvo, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2010, 49, 222–228 CrossRef CAS PubMed.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183–13196 CrossRef CAS PubMed.
- N. A. Siladke, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 3507–3516 CrossRef CAS PubMed.
- R. Thomson, C. Graves, B. Scott and J. Kiplinger, J. Chem. Crystallogr., 2011, 41, 1241–1244 CrossRef CAS.
- D. E. Smiles, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2013, 136, 96–99 CrossRef PubMed.
- H. S. La Pierre and K. Meyer, Prog. Inorg. Chem., 2014, 58, 303–416 CrossRef CAS.
- C. Madic, M. Lecomte, P. Baron and B. Boullis, C. R. Phys., 2002, 3, 797–811 CrossRef CAS.
- T. Mehdoui, J.-C. Berthet, P. Thuery and M. Ephritikhine, Chem. Commun., 2005, 2860–2862 RSC.
- K. I. M. Ingram, N. Kaltsoyannis, A. J. Gaunt and M. P. Neu, J. Alloys Compd., 2007, 444–445, 369–375 CrossRef CAS.
- K. I. M. Ingram, M. J. Tassell, A. J. Gaunt and N. Kaltsoyannis, Inorg. Chem., 2008, 47, 7824–7833 CrossRef CAS PubMed.
- M.-R. Spirlet, J. Rebizant, C. Apostolidis, E. Dornberger, B. Kanellakopulos and B. Powietzka, Polyhedron, 1996, 15, 1503–1508 CrossRef CAS.
- O. P. Lam, S. C. Bart, H. Kameo, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137–3139 RSC.
- O. P. Lam, F. W. Heinemann and K. Meyer, Angew. Chem., Int. Ed., 2011, 50, 5965–5968 CrossRef CAS PubMed.
- O. P. Lam, L. Castro, B. Kosog, F. W. Heinemann, L. Maron and K. Meyer, Inorg. Chem., 2012, 51, 781–783 CrossRef CAS PubMed.
- O. P. Lam, S. M. Franke, F. W. Heinemann and K. Meyer, J. Am. Chem. Soc., 2012, 134, 16877–16881 CrossRef CAS PubMed.
- O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC.
- S. M. Franke, F. W. Heinemann and K. Meyer, Chem. Sci., 2014, 5, 942–950 RSC.
-
M. Baudler, G. Brauer, F. Fehér, F. Huber, R. Klement, W. Kwasnik, P. W. Schenk, M. Schmeisser and R. Steudel, Handbuch der Präparativen Anorganischen Chemie, ed. G. Brauer, Ferdinand Enke Verlag, Stuttgart, Germany, 3rd edn, 1975, Vol. 1 Search PubMed.
- J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed.
- J. L. Brown, S. Fortier, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 15468–15475 CrossRef CAS PubMed.
- J. L. Brown, G. Wu and T. W. Hayton, Organometallics, 2012, 32, 1193–1198 CrossRef.
- E. M. Matson, M. D. Goshert, J. J. Kiernicki, B. S. Newell, P. E. Fanwick, M. P. Shores, J. R. Walensky and S. C. Bart, Chem.–Eur. J., 2013, 19, 16176–16180 CrossRef CAS PubMed.
- W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568–3576 CrossRef CAS.
- L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, Inorg. Chem., 2009, 48, 11615–11623 CrossRef CAS PubMed.
- W. J. Evans, M. K. Takase, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2008, 28, 236–243 CrossRef.
-
A. F. Hollemann and N. Wiberg, in Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, Germany, 101st edn, 1995 Search PubMed.
- C. Camp, M. A. Antunes, G. Garcia, I. Ciofini, I. C. Santos, J. Pecaut, M. Almeida, J. Marcalo and M. Mazzanti, Chem. Sci., 2014, 5, 841–846 RSC.
- D. E. Schwarz, J. A. Dopke, T. B. Rauchfuss and S. R. Wilson, Angew. Chem., Int. Ed., 2001, 40, 2351–2353 CrossRef CAS.
- J. I. Pinkas, I. Císařová, M. Horáček, J. I. Kubišta and K. Mach, Organometallics, 2011, 30, 1034–1045 CrossRef CAS.
- D. C. Gerbino, E. Hevia, D. Morales, M. E. N. Clemente, J. Perez, L. Riera, V. Riera and D. Miguel, Chem. Commun., 2003, 328–329 RSC.
- S. J. Smith, C. M. Whaley, T. B. Rauchfuss and S. R. Wilson, Inorg. Chem., 2005, 45, 679–687 CrossRef PubMed.
- W. A. Howard and G. Parkin, J. Am. Chem. Soc., 1994, 116, 606–615 CrossRef CAS.
- M. Di Vaira, P. Stoppioni and M. Peruzzini, J. Organomet. Chem., 1987, 333, C53–C56 CrossRef CAS.
- M. Di Vaira, M. Peruzzini and P. Stoppioni, Inorg. Chem., 1991, 30, 1001–1007 CrossRef CAS.
- B. S. Mandimutsira, S. J. Chen, R. A. Reynolds III and D. Coucouvanis, Polyhedron, 1997, 16, 3911–3920 CrossRef CAS.
- J. Wallick, C. G. Riordan and G. P. A. Yap, J. Am. Chem. Soc., 2013, 135, 14972–14974 CrossRef CAS PubMed.
- H. Seino, Y. Mizobe and M. Hidai, Organometallics, 2000, 19, 3631–3639 CrossRef CAS.
- H. Seino, Y. Mizobe and M. Hidai, New J. Chem., 2000, 24, 907–911 RSC.
- D. M. Smith and J. A. Ibers, Polyhedron, 1998, 17, 2105–2108 CrossRef CAS.
- C. Cui, H. W. Roesky, H. Hao, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 2000, 39, 1815–1817 CrossRef CAS.
- O. Blacque, H. Brunner, M. M. Kubicki, B. Nuber, B. Stubenhofer, J. Wachter and B. Wrackmeyer, Angew. Chem., Int. Ed., 1997, 36, 352–353 CrossRef CAS.
- F. Sécheresse, J. Lefebvre, J. C. Daran and Y. Jeannin, Inorg. Chim. Acta, 1981, 54, L175–L176 CrossRef.
- F. Sécheresse, J. M. Manoli and C. Potvin, Inorg. Chem., 1986, 25, 3967–3971 CrossRef.
- T. R. Gaffney and J. A. Ibers, Inorg. Chem., 1982, 21, 2857–2859 CrossRef CAS.
- D. R. English, D. N. Hendrickson, K. S. Suslick, C. W. Eigenbrot Jr and W. R. Scheidt, J. Am. Chem. Soc., 1984, 106, 7258–7259 CrossRef CAS.
- J. Vicente, M.-T. Chicote, P. González-Herrero, P. G. Jones and B. Ahrens, Angew. Chem., Int. Ed., 1994, 33, 1852–1853 CrossRef.
- M. Rombach and H. Vahrenkamp, Inorg. Chem., 2001, 40, 6144–6150 CrossRef CAS PubMed.
- J. W. Pavlik, B. C. Noll, A. G. Oliver, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2010, 49, 1017–1026 CrossRef CAS PubMed.
-
L. Guokui and J. V. Beitz, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Dordrecht, The Netherlands, 3rd edn, 2006, Vol. 3, ch. 18 Search PubMed.
- D. E. Morris, R. E. Da Re, K. C. Jantunen, I. Castro-Rodriguez and J. L. Kiplinger, Organometallics, 2004, 23, 5142–5153 CrossRef CAS.
- C. R. Graves, P. Yang, S. A. Kozimor, A. E. Vaughn, D. L. Clark, S. D. Conradson, E. J. Schelter, B. L. Scott, J. D. Thompson, P. J. Hay, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 5272–5285 CrossRef CAS PubMed.
- J. Selbin and H. J. Sherrill, Inorg. Chem., 1974, 13, 1235–1239 CrossRef CAS.
- S. J. Kraft, U. J. Williams, S. R. Daly, E. J. Schelter, S. A. Kozimor, K. S. Boland, J. M. Kikkawa, W. P. Forrest, C. N. Christensen, D. E. Schwarz, P. E. Fanwick, D. L. Clark, S. D. Conradson and S. C. Bart, Inorg. Chem., 2011, 50, 9838–9848 CrossRef CAS PubMed.
- O. P. Lam, C. Anthon, F. W. Heinemann, J. M. O'Connor and K. Meyer, J. Am. Chem. Soc., 2008, 130, 6567–6576 CrossRef CAS PubMed.
- T. J. Marks, Prog. Inorg. Chem., 1979, 25, 223–333 CrossRef CAS.
- S. J. Zuend, O. P. Lam, F. W. Heinemann and K. Meyer, Angew. Chem., Int. Ed., 2011, 50, 10626–10630 CrossRef CAS PubMed.
- A.-C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic and experimental details, spectroscopic data for 1H NMR, SQUID and UV/vis and detailed X-ray crystallographic data in CIF format. CCDC 1020771–1020776. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02602k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a