
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Merging of the photocatalysis and copper catalysis in metal–organic frameworks for oxidative C–C bond formation†
Dongying
Shi
a,
Cheng
He
a,
Bo
Qi
a,
Cong
Chen
a,
Jingyang
Niu
b and
Chunying
Duan
*a
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: cyduan@dlut.edu.cn
bCollege of Chemistry and Chemical Engineering, Henan University, Kaifeng, 475004, P. R. China
First published on 30th October 2014
Abstract
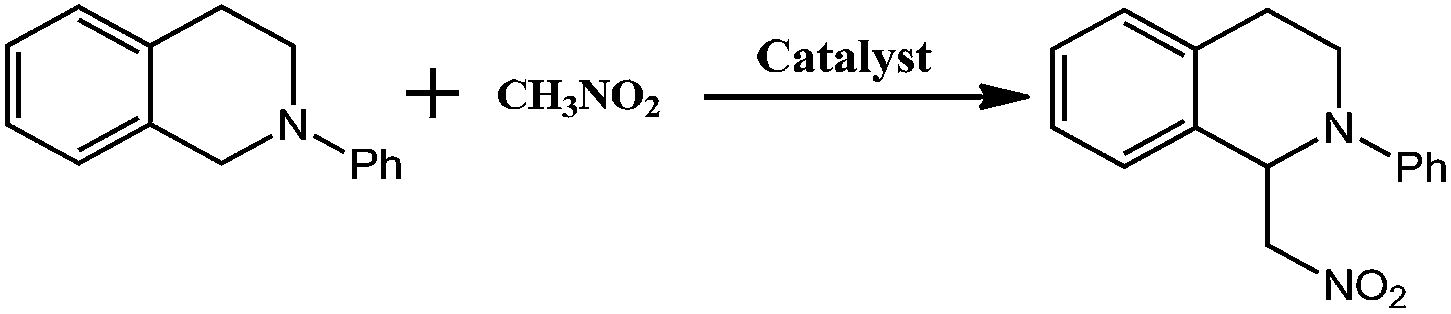
The direct formation of new C–C bonds through photocatalytic oxidative coupling from low reactive sp3 C–H bonds using environmentally benign and cheap oxygen as oxidant is an important area in sustainable chemistry. By incorporating the photoredox catalyst [SiW11O39Ru(H2O)]5− into the pores of Cu-based metal–organic frameworks, a new approach for merging Cu-catalysis/Ru-photocatalysis within one single MOF was achieved. The direct CuII–O–W(Ru) bridges made the two metal catalyses being synergetic, enabling the application on the catalysis of the oxidative coupling C–C bond formation from acetophenones and N-phenyl-tetrahydroisoquinoline with excellent conversion and size-selectivity. The method takes advantage of visible light photoredox catalysis to generate iminium ion intermediate from N-phenyl-tetrahydroisoquinoline under mild conditions and the easy combination with Cu-catalyzed activation of nucleophiles. Control catalytic experiments using similar Cu-based sheets but with the photoredox catalytic anions embedded was also investigated for comparison.
Introduction
The direct formation of new C–C bonds through oxidative coupling reactions from the lower active sp3 C–H bonds using oxygen as oxidant is an important area in sustainable chemistry.1 Among the reported promising examples, the oxidative activation of the C–H bonds adjacent to nitrogen atom in tertiary amines represents a powerful strategy, giving valuable, highly reactive iminium ion intermediates for further functionalization.2 Recent investigations also revealed that the visible light photoredox catalysis was a promising approach to such reaction sequences3 with respect to the development of new sustainable and green synthetic methods. It was also postulated that the combination of the photocatalysis and the metal catalysis within a dual catalytic transformation is attractive to circumvent the potential side reactions relative to the highly active intermediates that exist in the photocatalysis.4 The hurdles that need to be overcome include the careful adaptation and the fine tuning of the reaction rates of the two catalytic cycles,5 beside the appropriate choice of the metal catalysis and photocatalysis.6Metal–organic frameworks are hybrid solids with infinite network structures built from organic bridging ligands and inorganic connecting nodes. Besides the potential applications in many diverse areas,7 MOFs are ideally suited for catalytic conversions, since they can impose size and shape selective restriction through readily fine-tuned channels or pores,8 providing precise knowledge about the pore structure, the nature and distribution of catalytically active sites.9 In comparison to the heterogeneous catalytic systems that have been examined earlier, the design flexibility and framework tunability resulting from the huge variations of metal nodes and organic linkers allow the introduction of more than two independent catalyses in one single MOF.10 The combination of photocatalysis with the metal ions or organocatalysis was expected to be a promising approach to create synergistic catalysts.11
By incorporating a ruthenium(III) substituted polyoxometalate [SiW11O39Ru(H2O)]5− within the pores of copper(II)-bipyridine MOFs, herein, we reported a new approach to merge the visible light photocatalytic aerobic oxidation and copper(II) catalytic coupling reaction within one MOF (Scheme 1). We envisioned that the ruthenium-containing fragments possibly worked as oxidative photocatalyst to generate the iminium ion from N-phenyl-tetrahydroisoquinolines,12 whereas the Cu-based MOF potentially activated the nucleophiles, as it was shown in the oxidative C–C bond coupling.13
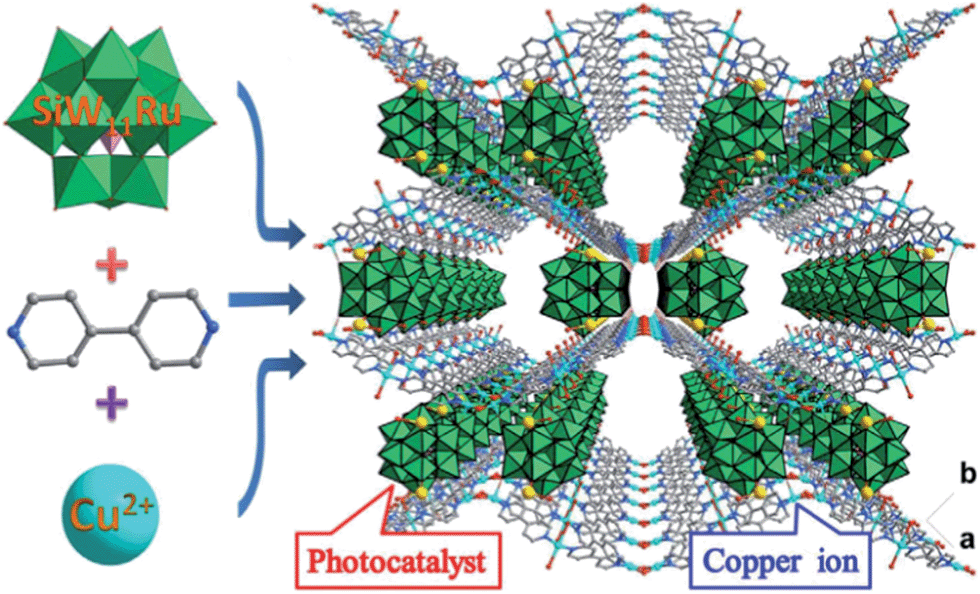 | ||
| Scheme 1 Synthetic procedure for the 3D CR–BPY1 MOF that is composed of wavy-like Cu–BPY sheets and [SiW11O40Ru]7− anions showing the combination of the dual catalytic units and channels for chemical transformations. | ||
Results and discussion
Synthesis and characterizations of CR–BPY1
Solvothermal reaction of 4,4′-bipyridine (BPY), Cu(NO3)2·3H2O and K5[SiW11O39Ru(H2O)]·10H2O gave CR–BPY1 in a yield of 52%. Elemental analyses and powder X-ray analysis indicated the pure phase of its bulk sample. Single-crystal structural analysis revealed that CR–BPY1 crystallized in a space group P421m. Two crystallographically independent copper(II) ions are connected by BPY ligands and μ2-water bridges alternatively to produce 2D wavy-like Cu–BPY sheets (Fig. S5, ESI†). The Cu(2) atom adopted a six-coordinate octahedral geometry with four nitrogen atoms from four BPY ligands positioned in the equatorial plane and two water molecules occupied the axial positions. The Cu(1) atom displayed a five-coordinate square pyramidal geometry with two μ2-water groups and two nitrogen atoms of BPY ligands positioned in the basal plane, and a terminal oxygen atom of the depronated [SiW11O39Ru(H2O)]5− polyoxoanion occupied the vertex position. The ruthenium atom disordered in the twelve equivalent positions within a depronated [SiW11O39Ru(H2O)]5−.14 The availability of vacant d-orbitals on the metal atoms adjacent to the heteroatom allows the polyoxometalate matrix to function as a π-acceptor ligand.15While these copper atoms were connected by the BPY ligands to form two-dimension square grid at first, adjacent sheets were connected together using the deprotonated [SiW11O39Ru(H2O)]5− polyoxoanion by CuII–O–W(Ru) bridges to generate a 3D framework. Two symmetric-related frameworks further interpenetrated each other perpendicularly to consolidate the robust structure (Fig. 1), in which the opening of the pores was reduced to 10.0 Å × 5.3 Å. To the best of our knowledge, CR–BPY1 represents the first example of MOFs which are comprised of ruthenium substituted polyoxometalate [SiW11O39Ru(H2O)]5−. As the noble metal substituted polyoxometalates exhibited excellent photoreactivity in various catalytic oxidation processes of organic substrates,16 such kinds of MOFs potentially allow the combination of photocatalysis and MOF-based heterogeneous catalysis to achieve synthetically useful organic transformations. Moreover, the directly bridging of the copper and ruthenium by CuII–O–W(Ru) provided a promising way to achieve the synergistic catalysis between photocatalyst and metal catalyst.
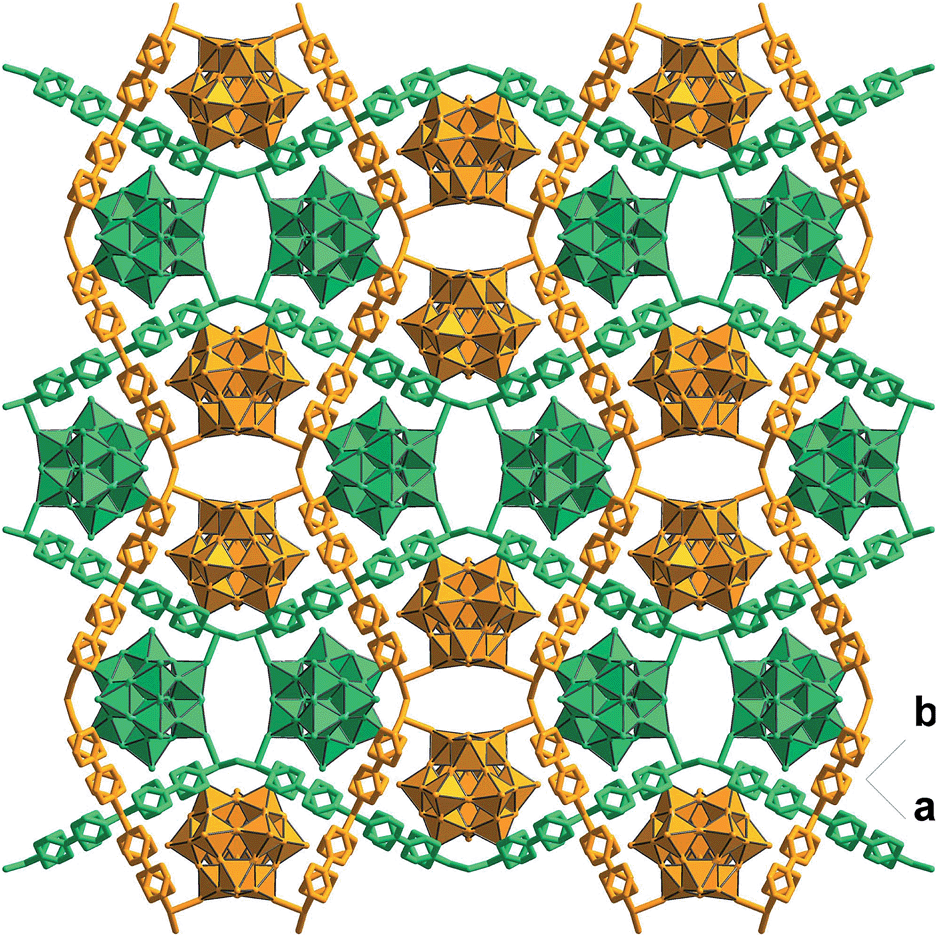 | ||
| Fig. 1 Crystal structure of CR–BPY1 with the 3D framework generated by the covalently linking of the wavy-like 2D sheets (drawn in stick-ball model) and the [SiW11O39Ru(H2O)]5− (drawn in polyhedron) by CuII–O–W(Ru) bridges, showing the interpenetration of two symmetric frameworks. | ||
Confocal fluorescence microscopy has attracted much attention in biological imaging. It may provide a way to analyse relatively thick porous materials, because it offers the advantage of increased penetration depth (>500 mm).17 The assessment of guest-accessible volume in MOFs can be reliably done by using confocal fluorescence microscopy with a tool-box of dyes with a wide range of sizes. It would be applicable to any porous materials, whose single-crystal structures are not available, or non-crystalline materials.18 Dye uptake investigation was carried out by soaking CR–BPY1 in a methanol solution of 2′,7′-dichlorofluorescein. It gave the quantum uptake equivalent to 5% of the MOF weight (Fig. S11, ESI†).19 The confocal laser scanning microscopy exhibited strong green fluorescence (λex = 488 nm) assignable to the emission of the fluorescein dye (Fig. 2), confirming the successful uptake of the dye molecules inside the crystals of the MOF.20 Furthermore, the rather uniform distribution of the dye molecules throughout the crystal suggested that the dyes penetrated deeply into the crystal rather than staying on the external surface. Without guest water molecules, the effective free volume of CR–BPY1 was estimated to be 29.0% by PLATON software.21
 | ||
| Fig. 2 Confocal images of empty (a and c) and soaked (b and d) 2′,7′-dichlorofluorescein dye. Brightfield images (a and b) and confocal images (c and d) detected at λem = 510–610 nm, exited by λex = 488 nm through a 405/488 nm filter. (e) The 3D reconstruction of the soaked 2′,7′-dichlorofluorescein dye (b). Three images at the end of axes in (e) exemplify the X, Y and Z-axis projections of the soaked dye. | ||
CR–BPY1 exhibited an absorption band centered at 398 nm in the solid state UV-vis absorption spectrum (Fig. S1, ESI†), assignable to the transitions of [SiW11O39Ru(H2O)]5−.22 Upon excitation at this band, CR–BPY1 did not exhibit any obvious emission, however, progressive addition of the N-phenyl-tetrahydroisoquinoline into the dichloromethane suspension of CR–BPY1 up to 0.50 mM caused the appearance of the RuII-relative emission band at about 422 nm (Fig. 3a).23 The results suggested that CR–BPY1 oxidized N-phenyl-tetrahydroiso-quinoline to form the RuII species and the iminium intermediate.24 Electrospray ionization mass spectrometry of the CH2Cl2 suspension containing N-phenyl-tetrahydroisoquinoline and CR–BPY1 after 3 hours light irradiation exhibited an intense peak at m/z = 208. This peak was assignable to the relative imine ion, confirming that CR–BPY1 oxidized N-phenyl-tetrahydroisoquinoline to form the RuII species and the iminium intermediate (Fig. S13, ESI†). The electron paramagnetic resonance (EPR) of CR–BPY1 exhibited the characteristic signal of CuII with g = 2.14 (Fig. 3c). Solid state electrochemical measurements (Fig. 3d) exhibited a broad redox band centred at −186 mV (vs. SCE) relative to the overlap of the CuII/CuI and RuIII/RuII redox couples. The potentials were comparable to these CuII and RuIII-containing catalysts,25 and enabled CR–BPY1 to prompt the oxidative coupling of N-phenyl-tetrahydroisoquinoline with nucleophiles under light.26 It seems that CR–BPY1 adsorbed the N-phenyl-tetrahydroisoquinoline in its pores and activated the substrate to form the iminium intermediate.
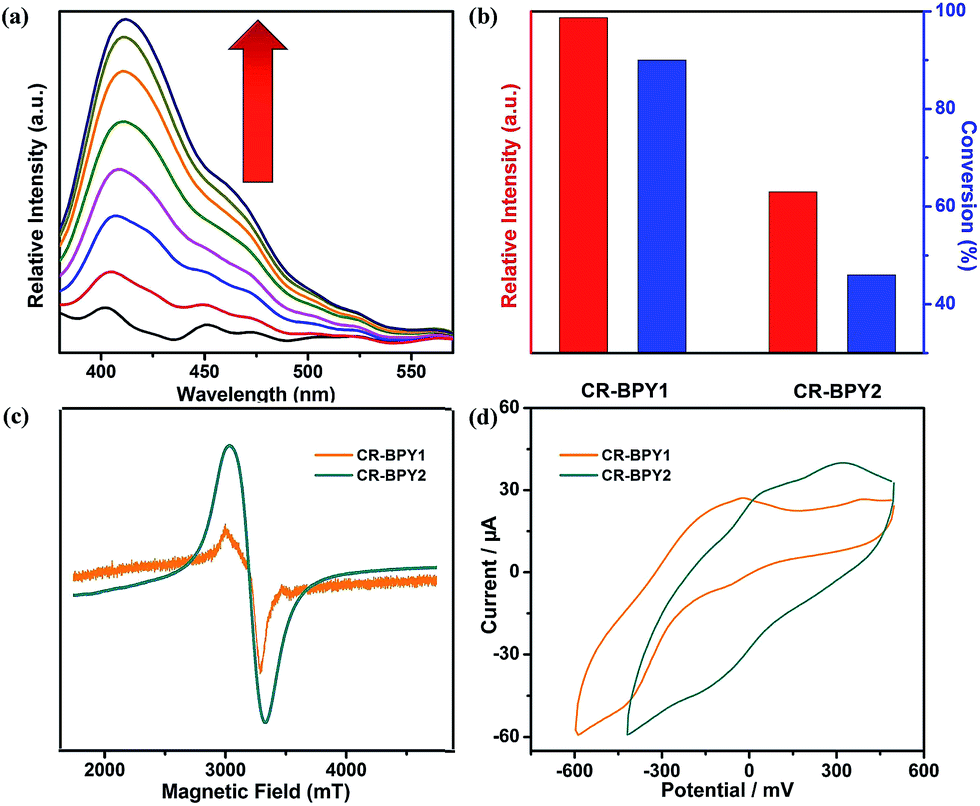 | ||
| Fig. 3 (a) Family of emission spectra of CR–BPY1 (0.1% in weight) in CH2Cl2 suspension upon addition of N-phenyl-tetrahydroisoquinoline up to 0.50 mM, excitation at 362 nm. (b) The luminescence intensities of CR–BPY1 and CR–BPY2 upon addition of the same amount of N-phenyl-tetrahydroisoquinoline up to 0.50 mM at room temperature in CH2Cl2; the catalytic activities of CR–BPY1 and CR–BPY2 using N-phenyl-tetrahydroisoquinoline and nitromethane as the coupling partners. (c) EPR spectra of CR–BPY1 (g = 2.1438) and CR–BPY2 (g = 2.1320) in solid state at 77 K, respectively. (d) Solid state cyclic voltammetry of CR–BPY1 and CR–BPY2, respectively, scan rate: 50 mV s−1. | ||
Catalysis details of CR–BPY1
The catalysis was examined initially using N-phenyl-tetrahydroisoquinoline and nitromethane as the coupling partners, along with a common fluorescent lamp (18 W) as the light source. The resulting reaction gave a yield of 90% after 24 hours irradiation. The removal of CR–BPY1 by filtration after 18 hours shut down the reaction, and the filtrate afforded only 12% additional conversion for another 18 hours at the same reaction conditions. The observation suggested that CR–BPY1 was a true heterogeneous catalyst.27 Solids of CR–BPY1 could be isolated from the reaction suspension by simple filtration alone and reused at least three times with moderate loss of activity (from 90% to 82% of yield after three cycles). The index of XRD patterns of CR–BPY1 filtrated off from the reaction mixture suggested the maintenance of the crystallinity (Fig. S14, ESI†). With the size of the microcrystals reduced to 2 μm by grinding CR–BPY1 crystals for 20 min, the time of the reaction giving the same conversion to that of the as-synthesized materials was reduced by about 10% (Fig. S15, ESI†). It seems that the MOF-based particles having well-defined size were really helpful for the catalytic reactions, but the size of the crystals did not dominate the catalysis directly.Control experiments for the C–C coupling reaction of N-phenyl-tetrahydroisoquinoline and nitromethane were carried out and summarized in Table 1. Almost no conversion was observed when the reaction was conducted in the dark (entry 7), while a very slow background reaction was observed in the absence of catalyst (entry 6), which demonstrated that both the light and the photocatalyst are required for efficient conversion to the coupling products. In addition, using the same equiv. of copper(II) salts or/and K5[SiW11O39Ru(H2O)] as catalysts, respectively gives conversions of 39%, 25% and 42% in homogeneous fashion (entry 3–5). These results suggested that the direct connection of copper(II) ions to [SiW11O40Ru]7− anions not only enabled the dual catalysts to individually activate N-phenyl-tetrahydroisoquinoline and nitromethane, but also enforced the proximity between the potential intermediates i.e. the iminium ion and nucleophile, avoiding the unwanted side reactions or reverse reactions.28
|
|
||
|---|---|---|
| Entry | Catalystsa | Conversionb (%) |
| a Reaction conditions: N-phenyl-tetrahydroisoquinoline (0.25 mmol), 1 mol% catalyst, 2.0 mL nitromethane, 18 W fluorescent lamp at room temperature. b The conversions after 24 hour irradiation were determined by 1H NMR of crude products. | ||
| 1 | CR–BPY1 | 90 |
| 2 | CR–BPY2 | 46 |
| 3 | Cu(NO3)2·3H2O | 39 |
| 4 | K5[SiW11O39Ru(H2O)] | 25 |
| 5 | K5[SiW11O39Ru(H2O)], Cu(NO3)2·3H2O | 42 |
| 6 | No catalyst | 20 |
| 7 | CR–BPY1, no light | <10 |
Although several examples of photocatalysts and metal copper catalysts have been reported to prompt the oxidative coupling C–C bond formation, CR–BPY1 represents a new example of a heterogeneous bimetal catalyst that merges the copper catalyst and the ruthenium(III) substituted polyoxometalate catalyst within one single material. The high modularity of the systems allows an easy adaptation of the concept of dual catalysis, and represents an example of combining dual catalysis to achieve synthetically useful organic transformation. In this case, our catalytic systems were extended with other pronucleophiles, i.e. substituted acetophenones. As shown in Fig. 4, 1H NMR of desolvated CR–BPY1 solids immersed in a dichloromethane solution of acetophenone exhibited that CR–BPY1 could adsorb about 2 equiv. of acetophenone per copper(II) moiety. IR spectrum of CR–BPY1 impregnated with a dichloromethane solution of acetophenone revealed a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at 1679 cm−1. The red shift from 1685 cm−1 (free acetophenone) suggested the adsorption and the activation of the acetophenone in the channels of the MOFs. It is hypothesized that the interactions between the copper ions in CR–BPY1 and the CO groups of acetophenone possibly gave an active nucleophile.29
O stretching vibration at 1679 cm−1. The red shift from 1685 cm−1 (free acetophenone) suggested the adsorption and the activation of the acetophenone in the channels of the MOFs. It is hypothesized that the interactions between the copper ions in CR–BPY1 and the CO groups of acetophenone possibly gave an active nucleophile.29
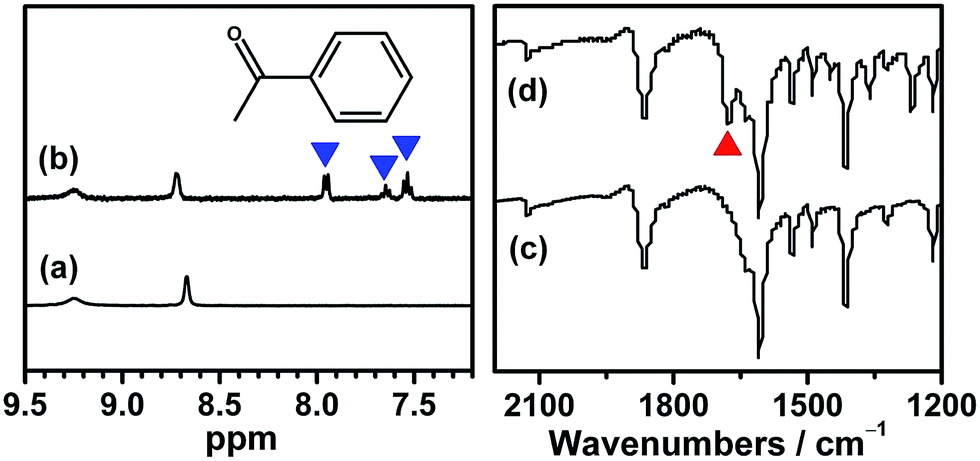 | ||
| Fig. 4 1H NMR in DCl/DMSO and solid state IR spectra of the desolvated CR–BPY1 (a) and (c), respectively, and of the desolvated CR–BPY1 impregnated in a dichloromethane solution of acetophenone (b) and (d), respectively, showing the absorbency and activation of substrate in the MOF. The blue and red triangle represented the signals of acetophenone in the NMR and IR spectra, respectively. | ||
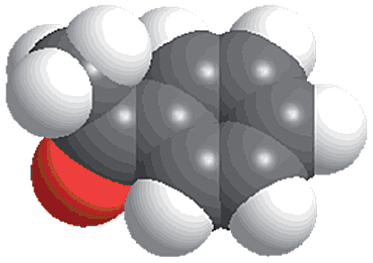
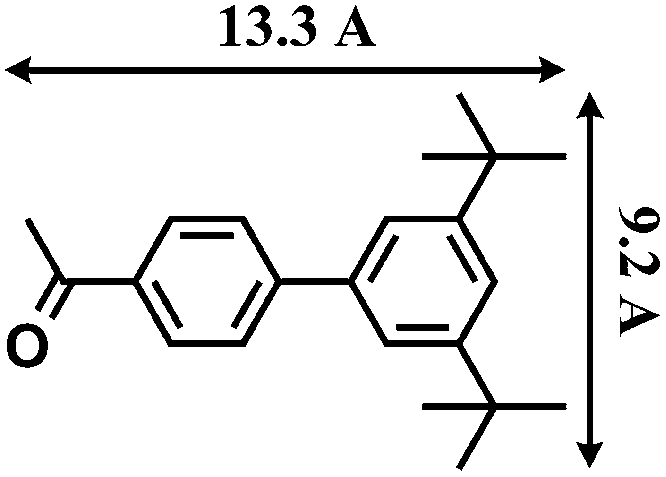
The reactions were carried out in the presence of a common used secondary amine, L-proline, as an organic co-catalyst to activate the ketones.30 In the case of the acetophenone as reactant with a fluorescent lamp (18 W) as the light source; the catalytic reaction gave a yield of 72%. Control experiments demonstrated that the use of K5[SiW11O39Ru(H2O)] or copper(II) salts as catalysts, only gave less than 25% of the conversions, respectively. The results indicated the significant contribution of cooperative effects of the individual parts within one single MOF. From the mechanistic point of view, the ruthenium(III) of the polyoxometalate [SiW11O39Ru(H2O)]5− interacted with N-phenyl-tetrahydroisoquinoline to form iminium ions, whereas the copper atoms coordinated to the acetophenones weakly to form the enol intermediate that worked as active nucleophile for the oxidative coupling C–C bond formation. At the same time, the presence of copper ions could enhance the activation of N-phenyl-tetrahydroisoquinoline, benefiting the synergistic catalysis between photocatalyst and metal catalyst. Importantly, in contrast to the smooth reactions of substrates 1–3, the C–C coupling reaction in the presence of bulky ketone (1-(3′,5′-di-tert-butyl [1,1′-biphenyl]-4-yl)-ethanone) 4, gave less than 10% conversion under the same reaction conditions (Table 2, entry 4). The negligible adsorption by immersing CR–BPY1 into a dioxane solution of substrate 4, coupled with the fact that the size of substrate 4 was larger than that of the channels,31 revealed that 4 was too large to be adsorbed in the channels. Furthermore, it is suggested that the synergistic catalytic coupling reaction indeed occurred in the channels of the MOF, not on the external surface.
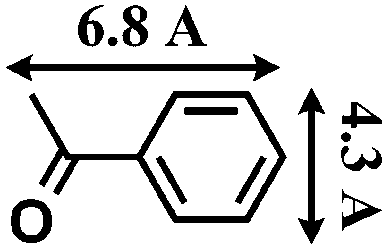
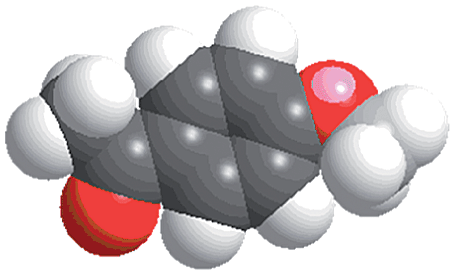
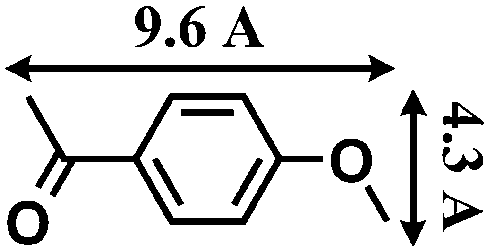
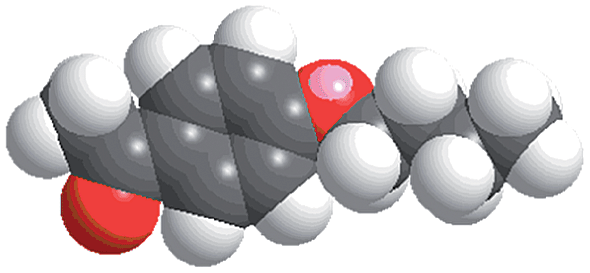
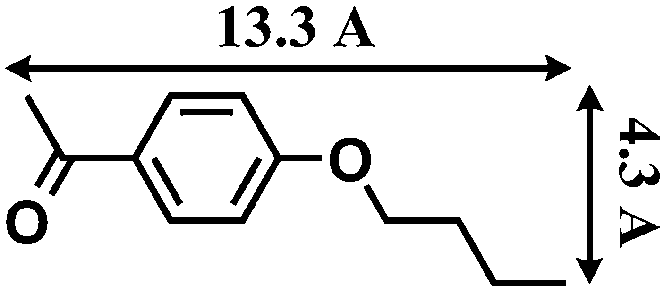
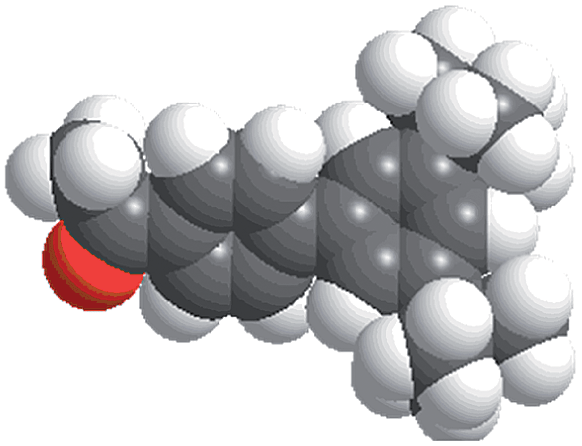
Synthesis and catalytic characterizations of CR–BPY2
To further investigate the synergistic interactions between the inorganic copper and [SiW11O39Ru(H2O)]5− anion, a reference compound CR–BPY2 was assembled using the same starting components but different synthetic conditions (hierarchical diffusion). CR–BPY2 was synthesized by a diffusion method in a test tube by laying a solution of 4,4′-bipyridine in acetonitrile onto the solution of K5[SiW11O39Ru(H2O)]·10H2O and Cu(NO3)2·3H2O in water for several days in a yield of 59%. Elemental analyses and powder X-ray analysis indicated the pure phase of its bulk sample. Single-crystal structural analysis revealed that CR–BPY2 crystallized in the orthorhombic lattice with a space group Pccn. Two crystallographically independent copper(II) ions connected four BPY bridges alternatively to produce a 2D sheet (Fig. 5), which were further stacked paralleled along the crystallographic a axis to form the 3D structure with embedded [SiW11O39Ru(H2O)]5− (Fig. S8, ESI†). The copper(II) ions resided in an octahedral geometries with the equatorial plane which was defined by four nitrogen atoms of BPY ligands, and the axial positions were occupied by two water molecules (Fig. S7, ESI†). Without guest water molecules, the effective free volume of CR–BPY2 was also estimated to be 33.9% by PLATON software, which is quite larger than that of CR–BPY1. These results suggested that the pore of CR–BPY2 is larger enough to adsorb the substrates. Since [SiW11O39Ru(H2O)]5− polyoxoanions were embedded in the channels, it is thus an excellent reference for investigating the catalytic activity on the same coupling reaction.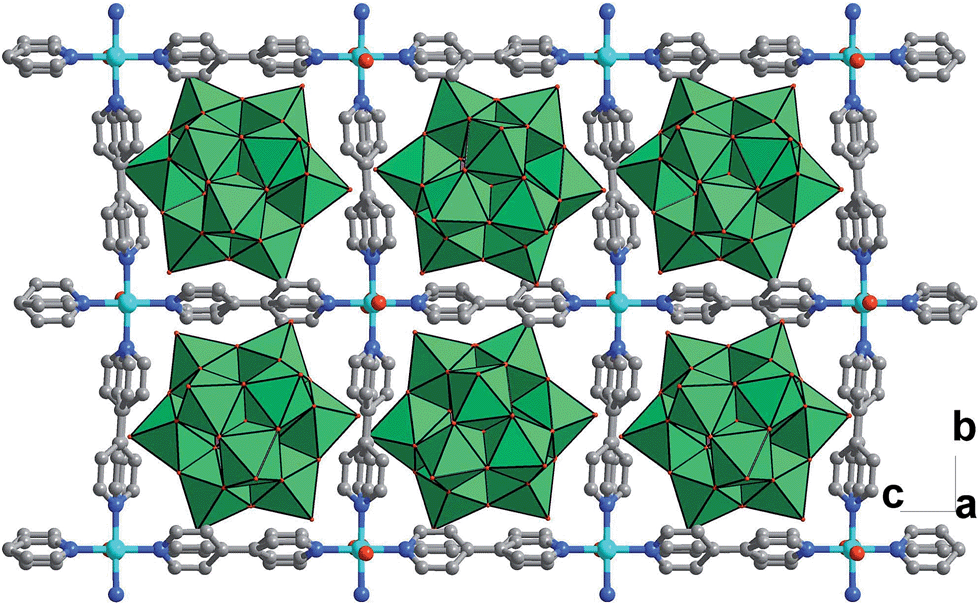 | ||
| Fig. 5 Crystal structure of CR–BPY2 showing the stacking pattern of the grid-like sheets with embedded [SiW11O39Ru(H2O)]5−. The atoms of copper, tungsten, carbon, nitrogen and oxygen were drawn in cyan, green, gray, dark blue and red, respectively. | ||
CR–BPY2 also exhibited an absorption band centered at 398 nm in the solid state UV-vis absorption spectrum. Upon excitation at this band, CR–BPY2 did not exhibit obvious emission, however, progressive addition of the N-phenyl-tetrahydroisoquinoline into the dichloromethane suspension of CR–BPY2 up to 0.50 mM caused the appearance of the RuII-relative emission band at about 422 nm, suggesting that CR–BPY2 oxidized N-phenyl-tetrahydroisoquinoline to form the RuII species. The EPR of CR–BPY2 exhibited the characteristic signal of CuII (g = 2.13).32 The sharper peak shape compared to that of CR–BPY1 might be one of the indicator of isolated CuII ions in CR–BPY2. No metal–metal interactions were found corresponding to the CuII ions in CR–BPY2. Solid state electrochemical measurements exhibited two redox peaks corresponding to the CuII/CuI and RuIII/RuII redox couples, with the redox potential calculated at 75 mV and 84 mV (vs. SCE). The potentials enabled CR–BPY2 to prompt the oxidative coupling of N-phenyl-tetrahydroisoquinoline with nucleophiles under light. However, the separated redox peaks also suggested that these CuI and RuIII ions did not interacted directly. It seems that CR–BPY2 adsorbed the N-phenyl-tetrahydroisoquinoline in its pores and was a convincing reference to investigate the synergistic action between CuII–O–W(Ru) bimetal of CR–BPY1.
The catalytic activities of CR–BPY2 in the C–C coupling reactions were examined under the same conditions using nitromethane and N-phenyl-tetrahydroisoquinoline as the reactants. About 1 mol% loading amount of the catalyst gave rise to a 46% conversion, which was superior to the case when copper(II) salts and the K5[SiW11O39Ru(H2O)] were employed as catalysts, indicating the significance of the two constitute parts for CR–BPY2 as a photocatalyst. However, the catalytic activities of CR–BPY2 were significantly weaker than that of CR–BPY1 (Fig. 3b). It should be concluded that the direct bridging of the copper and ruthenium by CuII–O–W(Ru) provided a promising way to achieve the synergistic catalysis between photocatalyst and metal catalyst, and the high reaction efficiency in the reactions was dominated by the spacious environment of the channels, like those of CR–BPY1.
Conclusions
In a summary, we reported the new example of copper MOFs containing the ruthenium substituted polyoxometalate with the aim of merging the synergistic Cu-catalysis/Ru-photocatalysis in a single MOF. CR–BPY1 exhibited perpendicularly inter-penetrated structure and the catalytic sites positioned in the robust pores of MOFs. Luminescence titration and IR spectra of the MOF-based material revealed the adsorbance and activation of N-phenyl-tetrahydroisoquinoline and acetophenone, by the ruthenium center and copper ions, respectively. The direct connection of copper(II) ions to [SiW11O40Ru(H2O)]5− not only provided the possibility of the dual catalysts to individually activate the substrates, but also enforced the proximity between the intermediates, avoiding the unwanted side reactions or reverse reactions. CR–BPY1 exhibited high activity for the photocatalytic oxidative coupling C–C bond formation with excellent size-selectivity, suggesting the catalytic reactions occurred in the channels of the MOF, and not on the external surface.Experimental section
General methods and materials
All chemicals were of reagent grade quality obtained from commercial sources and used without further purification. 1H NMR was measured on a Varian INOVA-400 spectrometer with chemical shifts reported as ppm (in DMSO-d6 or CDCl3, TMS as internal standard). The elemental analyses (EA) of C, H and N were performed on a Vario EL III elemental analyzer. Inductively coupled plasma (ICP) analyses were performed on a NexION 300D spectrometer. The powder XRD diffractograms were obtained on a Riguku D/MAX-2400 X-ray diffractometer with Cu sealed tube (λ = 1.54178 Å). IR spectra were recorded as KBr pellets on a NEXUS instrument. Solid UV-vis spectra were recorded on a U-4100 spectrometer. Liquid UV-vis spectra were performed on a TU-1900 spectrophotometer. Solid fluorescent spectra were measured on a JASCO FP-6500 instrument. The excitation and emission slits were both 3 nm wide. Solid state cyclic voltammograms were measured on a Zahner PP211 instrument by using a carbon paste working electrode. Thermogravimetric analyses (TGA) were carried out at a ramp rate of 5 °C min−1 in nitrogen flow with a SDTQ600 instrument. Electron paramagnetic resonance (EPR) spectra were recorded on a EMX-10/12 spectrometer. Scanning electron microscopy (SEM) images were taken with a NOVA NanoSEM 450 microscope. All confocal laser scanning microscopy (CLSM) micrographs were collected by Olympus Fluoview FV1000. Products were purified by flash column chromatography on 200–300 mesh silica gel SiO2.Synthesis of CR–BPY1
K5[SiW11O39Ru(H2O)]·10H2O33 were prepared according to the literature methods and characterized by IR spectroscopy and UV-vis spectroscopy, respectively. A mixture of K5[SiW11O39Ru(H2O)]·10H2O (70.0 mg, 0.02 mmol), 4,4′-bipyridine (19.3 mg, 0.12 mmol) and Cu(NO3)2·3H2O (30.4 mg, 0.12 mmol) in mixed water (5.0 mL) and acetonitrile (3.0 mL) was stirred and its pH value was adjusted to 4.4 with 1 mol L−1 HAc. The resulting suspension was sealed in a 25 mL Teflon-lined reactor and kept at 120 °C for three days. After cooling the autoclave to room temperature, brownish black prismatic single crystals were separated, washed with water and air-dried (yield: ca. 52% based on K5[SiW11O39Ru(H2O)]·10H2O). EA and ICP calcd (%) for C40H60N8O54SiW11RuCu3K: C 12.32, H 1.55, N 2.87, Cu 4.84, W 51.92, Ru 2.61; found: C 12.48, H 1.57, N 2.83, Cu 4.91, W 52.32, Ru 2.51. IR (KBr pellet; cm−1): 3395 (br, s), 1865 (w), 1608 (s), 1413 (m), 1219 (w), 1068 (w), 1008 (w), 957 (m), 913 (s), 881 (m), 788 (s).Synthesis of CR–BPY2
The CR–BPY2 was synthesized by a diffusion method in a test tube. A mixture of acetonitrile and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10.0 mL) was gently layered on the top of a solution of K5[SiW11O39Ru(H2O)]·10H2O (70.0 mg, 0.02 mmol) and Cu(NO3)2·3H2O (9.1 mg, 0.04 mmol) in water (5 mL). A solution of 4,4′-bipyridine (12.5 mg, 0.08 mmol) in acetonitrile (5 mL) was added carefully as the third layer. Brownish black block single crystals were separated after four weeks, washed with acetonitrile and water, and dried in air (yield: ca. 59% based on K5[SiW11O39Ru(H2O)]·10H2O). EA and ICP calcd (%) for C40H58N8O52SiW11RuCu2K: C 12.63, H 1.54, N 2.95, Cu 3.31, W 53.24, Ru 2.68; found: C 12.88, H 1.69, N 2.89, Cu 3.39, W 53.41, Ru 2.58. IR (KBr pellet; cm−1): 3404 (br, s), 1610 (s), 1415 (m), 1220 (w), 1075 (w), 1005 (w), 955 (m), 911 (s), 872 (m), 784 (s).
:1, 10.0 mL) was gently layered on the top of a solution of K5[SiW11O39Ru(H2O)]·10H2O (70.0 mg, 0.02 mmol) and Cu(NO3)2·3H2O (9.1 mg, 0.04 mmol) in water (5 mL). A solution of 4,4′-bipyridine (12.5 mg, 0.08 mmol) in acetonitrile (5 mL) was added carefully as the third layer. Brownish black block single crystals were separated after four weeks, washed with acetonitrile and water, and dried in air (yield: ca. 59% based on K5[SiW11O39Ru(H2O)]·10H2O). EA and ICP calcd (%) for C40H58N8O52SiW11RuCu2K: C 12.63, H 1.54, N 2.95, Cu 3.31, W 53.24, Ru 2.68; found: C 12.88, H 1.69, N 2.89, Cu 3.39, W 53.41, Ru 2.58. IR (KBr pellet; cm−1): 3404 (br, s), 1610 (s), 1415 (m), 1220 (w), 1075 (w), 1005 (w), 955 (m), 911 (s), 872 (m), 784 (s).
X-ray crystallography
Data of CR–BPY1 and CR–BPY2 were collected on a Bruker Smart APEX CCD diffractometer with graphite-monochromated Mo-Kα (λ = 0.71073 Å) using the SMART and SAINT programs.34 Their structures were determined and the heavy atoms were found by direct methods using the SHELXTL-97 program package.35 Crystallographic data for them are summarized in Table 3. Except some partly occupied solvent water molecules, the other non-hydrogen atoms were refined anisotropically. Hydrogen atoms within the ligand backbones were fixed geometrically at their positions and allowed to ride on the parent atoms. In both of the two structures, the ruthenium atoms were disordered in the equivalent positions of tungsten atoms. For CR–BPY2, several bond distances constraints were used to help the refinement on the BPY moiety, and thermal parameters on adjacent oxygen atoms of the polyoxometalate anion were restrained to be similar.| CR–BPY1 | CR–BPY2 | |
|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = [∑w(|Fo|2 − |Fc|2)/∑w(Fo2)2]1/2; w = 1/[σ2(Fo2) + (xP)2 + yP], P = (Fo2 + 2Fc2)/3, where x = 0.1000, y = 0.0000 for CR–BPY1; x = 0.0874, y = 678.0471 for CR–BPY2. | ||
| Empirical formula | C40H60N8O54 SiW11RuCu3K | C40H58N8O52 SiW11RuCu2K |
| M, g mol−1 | 3898.19 | 3800.63 |
| Crystal system | Tetragonal | Orthorhombic |
| Space group | P421m | Pccn |
| a, Å | 24.415(3) | 17.866(1) |
| b, Å | 24.415(3) | 22.192(2) |
| c, Å | 15.337(4) | 22.284(2) |
| V, Å3 | 9142(3) | 8835.3(10) |
| Z | 4 | 4 |
| D calcd, g cm−3 | 2.777 | 2.857 |
| T, K | 296(2) | 296(2) |
| Refl. collected/unique | 61657/10865 Rint = 0.1270 |
42658/7765 Rint = 0.0688 |
| μ, mm−1 | 14.762 | 15.044 |
| GOOF | 0.972 | 1.022 |
| R 1 (I > 2σ(I)) | 0.0509 | 0.0753 |
| wR2b (I > 2σ(I)) | 0.1256 | 0.1918 |
| R 1 (all data) | 0.1156 | 0.0971 |
| wR2b (all data) | 0.1708 | 0.2042 |
| Diff peak and hole, e Å−3 | 2.937/−1.494 | 5.265/−4.627 |
Acknowledgements
We gratefully acknowledge financial support from the Natural Science Foundation of China (21025102 and 21171029), the Basic Research Program of China (Grant no. 2013CB733700) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1213).Notes and references
- (a) C. J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed; (b) S. I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 44, 6931–6933 CrossRef CAS PubMed.
- (a) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed; (b) M. Z. Wang, C. Y. Zhou, M. K. Wong and C. M. Che, Chem.–Eur. J., 2010, 16, 5723–5735 CrossRef CAS PubMed.
- (a) M. Rueping, C. Vila, R. M. Koenigs, K. Poscharny and D. C. Fabry, Chem. Commun., 2011, 47, 2360–2362 RSC; (b) C. Wang, Z. G. Xie, K. E. deKrafft and W. B. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- (a) C. Zhong and X. Shi, Eur. J. Org. Chem., 2010, 2999–3025 CrossRef CAS; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- (a) M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem.–Eur. J., 2012, 18, 5170–5174 CrossRef CAS PubMed; (b) A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Helv. Chim. Acta, 1981, 64, 2175–2182 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS PubMed; (b) D. Feng, W. C. Chung, Z. Wei, Z. Y. Gu, H. L. Jiang, Y. P. Chen, D. J. Darensbourg and H. C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- (a) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed; (b) L. Yang, S. Kinoshita, T. Yamada, S. Kanda, H. Kitagawa, M. Tokunaga, T. Ishimoto, T. Ogura, R. Nagumo, A. Miyamoto and M. Koyama, Angew. Chem., Int. Ed., 2010, 49, 5348–5351 CrossRef CAS PubMed.
- (a) Q. X. Han, C. He, M. Zhao, B. Qi, J. Y. Niu and C. Y. Duan, J. Am. Chem. Soc., 2013, 135, 10186–10189 CrossRef CAS PubMed; (b) C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2009, 131, 1883–1888 CrossRef CAS PubMed; (c) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS PubMed; (d) M. A. Nasalevich, M. van der Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919–4926 RSC.
- (a) D. Jiang, T. Mallat, F. Krumeich and A. Baiker, J. Catal., 2008, 257, 390–395 CrossRef CAS PubMed; (b) D. Jiang, A. Urakawa, M. Yulikov, T. Mallat, G. Jeschke and A. Baiker, Chem.–Eur. J., 2009, 15, 12255–12262 CrossRef CAS PubMed.
- (a) L. Q. Ma and W. B. Lin, Top. Curr. Chem., 2010, 293, 175–205 CrossRef CAS; (b) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- S. I. Murahashi, T. Nakae, H. Terai and N. Komiya, J. Am. Chem. Soc., 2008, 130, 11005–11012 CrossRef CAS PubMed.
- (a) Z. P. Li and C. J. Li, J. Am. Chem. Soc., 2005, 127, 3672–3673 CrossRef CAS PubMed; (b) E. Boess, D. Sureshkumar, A. Sud, C. Wirtz, C. Farès and M. Klussmann, J. Am. Chem. Soc., 2011, 133, 8106–8109 CrossRef CAS PubMed.
- M. Sadakane, D. Tsukuma, M. H. Dickman, B. Bassil, U. Kortz, M. Higashijima and W. Ueda, Dalton Trans., 2006, 4271–4276 RSC.
- M. Bonchio, M. Carraro, A. Sartorel, G. Scorrano and U. Kortz, J. Mol. Catal. A: Chem., 2006, 251, 93–99 CrossRef CAS PubMed.
- (a) N. V. Izarova, M. T. Pope and U. Kortz, Angew. Chem., Int. Ed., 2012, 51, 9492–9510 CrossRef CAS PubMed; (b) V. Kogan, Z. Aizenshtat, R. Popovitz-Biro and R. Neumann, Org. Lett., 2002, 4, 3529–3532 CrossRef CAS PubMed.
- (a) W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1369–1377 CrossRef CAS PubMed; (b) F. Helmchen and W. Denk, Nat. Methods, 2005, 2, 932–940 CrossRef CAS PubMed.
- (a) S. Han, T. M. Hermans, P. E. Fuller, Y. Wei and B. A. Grzybowski, Angew. Chem., Int. Ed., 2012, 51, 2662–2666 CrossRef CAS PubMed; (b) M. Ma, A. Gross, D. Zacher, A. Pinto, H. Noei, Y. Wang, R. A. Fischer and N. Metzler-Nolte, CrystEngComm, 2011, 13, 2828–2832 RSC.
- (a) L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed; (b) Q. R. Fang, G. S. Zhu, Z. Jin, Y. Y. Ji, J. W. Ye, M. Xue, H. Yang, Y. Wang and S. L. Qiu, Angew. Chem., Int. Ed., 2007, 46, 6638–6642 CrossRef CAS PubMed.
- (a) S. M. Shin, M. S. Lee, J. H. Han and N. Jeong, Chem. Commun., 2014, 50, 289–291 RSC; (b) L. J. Zhang, Y. Jian, J. Wang, C. He, X. Z. Li, T. Liu and C. Y. Duan, Dalton Trans., 2012, 41, 10153–10155 RSC.
- A. L. Spek, PLATON99, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1999 Search PubMed.
- (a) A. M. Khenkin, I. Efremenko, L. Weiner, J. M. L. Martin and R. Neumann, Chem.–Eur. J., 2010, 16, 1356–1364 CrossRef CAS PubMed; (b) M. Murakami, D. Hong, T. Suenobu, S. Yamaguchi, T. Ogura and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 11605–11613 CrossRef CAS PubMed.
- P. J. DeLaive, J. T. Lee, H. W. Sprintschnik, H. Abruna, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1977, 99, 7094–7097 CrossRef CAS.
- S. I. Murahashi and D. Zhang, Chem. Soc. Rev., 2008, 37, 1490–1501 RSC.
- (a) M. Higashijima, Chem. Lett., 1999, 1093–1094 CrossRef CAS; (b) M. Sadakane and M. Higashijima, Dalton Trans., 2003, 659–664 RSC; (c) R. C. Weast, Handbook of Chemistry and Physics 1988, 69th edn, 1988–1989, p. D-151 Search PubMed.
- (a) C. P. Anderson, D. J. Salmon, T. J. Meyer and R. C. Young, J. Am. Chem. Soc., 1977, 99, 1980–1982 CrossRef CAS; (b) A. Yu, Z. Gu, D. Chen, W. He, P. Tan and J. Xiang, Catal. Commun., 2009, 11, 162–166 CrossRef CAS PubMed.
- S. Horike, M. Dincă, K. Tamaki and J. R. Long, J. Am. Chem. Soc., 2008, 130, 5854–5855 CrossRef CAS PubMed.
- X. W. Gao, Q. Y. Meng, M. Xiang, B. Chen, K. Feng, C. H. Tung and L. Z. Wu, Adv. Synth. Catal., 2013, 355, 2158–2164 CrossRef CAS.
- P. Y. Wu, C. He, J. Wang, X. J. Peng, X. Z. Li, Y. L. An and C. Y. Duan, J. Am. Chem. Soc., 2012, 134, 14991–14999 CrossRef CAS PubMed.
- P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138–6171 CrossRef CAS PubMed.
- The molecular size (13.3 Å × 9.2 Å) was calculated by using the program Chem3D.
- (a) X. M. Zhang, M. L. Tong, M. L. Gong, H. K. Lee, L. Luo, K. F. Li, Y. X. Tong and X. M. Chen, Chem.–Eur. J., 2002, 8, 3187–3194 CrossRef CAS; (b) S. Reinoso, P. Vitoria, L. S. Felices, L. Lezama and J. M. Gutiérrez-Zorrilla, Inorg. Chem., 2006, 45, 108–118 CrossRef CAS PubMed.
- M. Sadakane, D. Tsukuma, M. H. Dickman, B. Bassil, U. Kortz, M. Higashijima and W. Ueda, Dalton Trans., 2006, 4271–4276 RSC.
- (a) SMART, Data collection software (version 5.629), Bruker AXS Inc., Madison, WI, 2003 Search PubMed; (b) SAINT, Data reduction software (version 6.45), Bruker AXS Inc., Madison, WI, 2003 Search PubMed.
- G. M. Sheldrick, SHELX-97, Program for crystal structure solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 997028 and 997029. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02362e |
| This journal is © The Royal Society of Chemistry 2015 |