DOI:
10.1039/C4SC02555E
(Edge Article)
Chem. Sci., 2015,
6, 1027-1034
Dehydrogenation of formic acid by Ir–bisMETAMORPhos complexes: experimental and computational insight into the role of a cooperative ligand†
Received
22nd August 2014
, Accepted 21st October 2014
First published on 22nd October 2014
Abstract
The synthesis and tautomeric nature of three xanthene-based bisMETAMORPhos ligands (La–Lc) is reported. Coordination of these bis(sulfonamidophosphines) to Ir(acac)(cod) initially leads to the formation of IrI(LH) species (1a), which convert via formal oxidative addition of the ligand to IrIII(L) monohydride complexes 2a–c. The rate for this step strongly depends on the ligand employed. IrIII complexes 2a–c were applied in the base-free dehydrogenation of formic acid, reaching turnover frequencies of 3090, 877 and 1791 h−1, respectively. The dual role of the ligand in the mechanism of the dehydrogenation reaction was studied by 1H and 31P NMR spectroscopy and DFT calculations. Besides functioning as an internal base, bisMETAMORPhos also assists in the pre-assembly of formic acid within the Ir-coordination sphere and aids in stabilizing the rate-determining transition state through hydrogen-bonding.
Introduction
Enzyme active sites are a major source of inspiration for scientists in the field of synthetic chemistry and homogeneous catalysis, because of the high activities and selectivities achieved in chemical transformations and the conceptual strategies employed by these systems.1 For instance, weak but highly directional hydrogen bonding stands out as a key element used by enzymes to selectively pre-assemble and pre-activate substrates and stabilize transition states. Inspired by this, functional ligands decorated with H-bond donor and/or acceptor groups have been used to construct supramolecular ligand structures, enabling modular ligand families2 to be utilized in transition metal catalysis and also for substrate pre-organization and pre-activation via specific ligand–substrate interactions.3
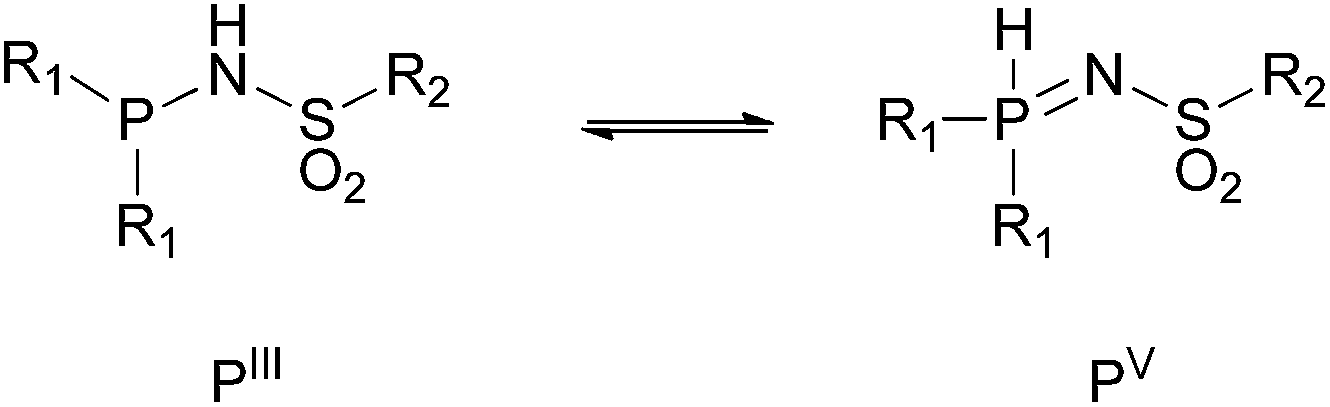
In our quest for novel systems able to undergo hydrogen-bonding interactions to steer reactivity we have developed sulfonamidophosphine (METAMORPhos) ligands. These ligands are based on a PNSO2 scaffold that displays NH–P/N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PH tautomerism (see Fig. 1). They have been employed for mono- and bimetallic Rh-catalyzed hydrogenation, Ru-based heterolytic H2 cleavage and [2 + 2 + 2] cycloaddition reactions.4
PH tautomerism (see Fig. 1). They have been employed for mono- and bimetallic Rh-catalyzed hydrogenation, Ru-based heterolytic H2 cleavage and [2 + 2 + 2] cycloaddition reactions.4
 |
| | Fig. 1 General composition of METAMORPhos ligands with their two tautomeric forms [NH–P (PIII) and NPH (PV)] in equilibrium. | |
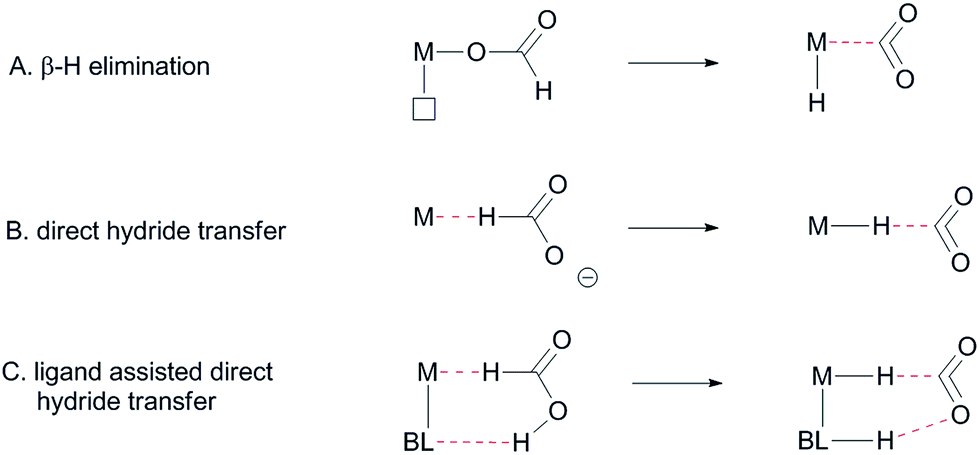
Formate dehydrogenase metalloenzymes have successfully been employed for the reduction of CO2 and oxidation of formate. Although there is no complete consensus concerning the mechanism of either the reduction or the oxidation, hydrogen-bonding and proton-shuttling are suggested to play a crucial role in the way in which these enzymes operate.5 We recently described our initial results with an Ir-catalyst bearing a bisMETAMORPhos ligand in the base-free catalytic dehydrogenation of formic acid, a reaction that has attracted much recent interest in the context of hydrogen storage/release systems.4f Typically, formic acid (HCOOH) dehydrogenation catalysts require the addition of sub-stoichiometric amounts of base (e.g. 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of HCOOH:NEt3). However, this significantly reduces the overall hydrogen weight percentage of the reaction mixture.6 One promising strategy to circumvent the use of exogenous base is to employ catalyst systems bearing cooperative ligands to access metal–ligand bifunctional pathways for substrate activation.7 Hydrogen-bonding interactions between ligand and substrate as (additional) tools to enhance reactivity have previously been proposed both in the hydrogenation of CO2 and in the aluminium catalysed dehydrogenation of HCOOH.8 These interactions were also suggested to play a role in the formation of half-sandwich ruthenium and rhodium hydrides from formic acid.9 Several studies on the mechanism of HCOOH dehydrogenation have compared conventional β-hydride elimination with direct hydride-transfer (Fig. 2A and B).10 Given the potential H-bonding abilities of METAMORPhos ligands and the protic nature of formic acid, we wondered if biomimetic non-covalent interactions between the ligand backbone and formic acid could play a role in this catalytic system. Herein we show that the proton-responsive ligand not only acts as an internal base (Fig. 2C), but that its hydrogen-bonding abilities steer substrate pre-assembly and stabilize catalytic transition states.
:2 ratio of HCOOH:NEt3). However, this significantly reduces the overall hydrogen weight percentage of the reaction mixture.6 One promising strategy to circumvent the use of exogenous base is to employ catalyst systems bearing cooperative ligands to access metal–ligand bifunctional pathways for substrate activation.7 Hydrogen-bonding interactions between ligand and substrate as (additional) tools to enhance reactivity have previously been proposed both in the hydrogenation of CO2 and in the aluminium catalysed dehydrogenation of HCOOH.8 These interactions were also suggested to play a role in the formation of half-sandwich ruthenium and rhodium hydrides from formic acid.9 Several studies on the mechanism of HCOOH dehydrogenation have compared conventional β-hydride elimination with direct hydride-transfer (Fig. 2A and B).10 Given the potential H-bonding abilities of METAMORPhos ligands and the protic nature of formic acid, we wondered if biomimetic non-covalent interactions between the ligand backbone and formic acid could play a role in this catalytic system. Herein we show that the proton-responsive ligand not only acts as an internal base (Fig. 2C), but that its hydrogen-bonding abilities steer substrate pre-assembly and stabilize catalytic transition states.
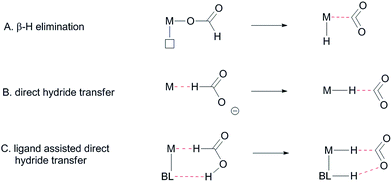 |
| | Fig. 2 Different proposed mechanisms for the dehydrogenation of formic acid, BL: bifunctional ligand. | |
We will present the synthesis of three bisMETAMORPhos ligands as well as their coordination to iridium, compare the activity of these systems in HCOOH dehydrogenation and explain the dual role of the ligand framework during catalysis by means of both experimental and computational results.
Results and discussion
Synthesis of ligands and complexes
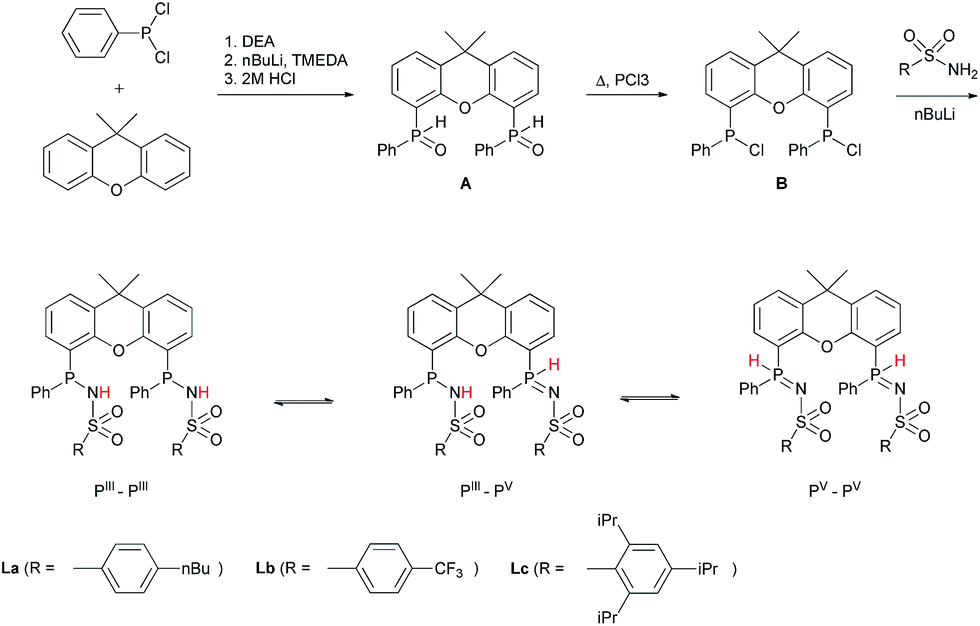
METAMORPhos ligands are prepared by a simple condensation reaction between a sulfonamide of choice and a chlorophosphine. They show high stability to both oxidation (at phosphorus) and hydrolysis (of the P–N bond). We attribute this stability to their prototropic character, resulting in an equilibrium between the PIII and PV oxidation states that is influenced by R1 and R2.4e In order to gain insight into the effect of ligand modification on the coordination and degree of tuning in HCOOH dehydrogenation catalysis, we prepared bisMETAMORPhos ligands La–Lcvia a three-step synthetic protocol (Scheme 1) that results in selective formation of only the respective meso-isomers, see ESI.†.
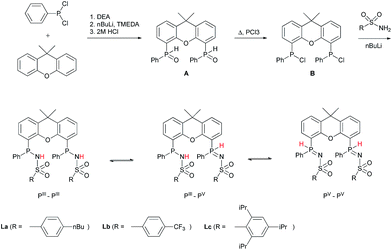 |
| | Scheme 1 The synthesis of bisMETAMORPhos ligands La–Lc and their three tautomeric forms (PIII–PIII, PIII–PV and PV–PV). | |
All three ligands La–Lc display PIII/PV tautomerism, but in different ratios for the three possible forms. For ligand La only the PIII–PIII and PIII–PV tautomers were observed in CD2Cl2 in a ratio of 1:0.4, as determined by 31P NMR spectroscopy. For both ligands Lb and Lc all three possible tautomers were observed, with ratios (PIII–PIII:PIII–PV:PV–PV) of 1:1.8:0.2 and 1:1.9:0.1 for Lb and Lc, respectively. These data show that the overall PIII/PV ratio is not only determined by the acidity of the N–H bond and basicity of the phosphorus atom but is also greatly influenced by the steric bulk of the substituent on the sulfon group. The PV tautomer provides stability towards oxidation and hydrolysis, even to the extent that La–Lc can be conveniently purified by column chromatography. Upon coordination to a metal center, the tautomeric behaviour of these ligands is lost.
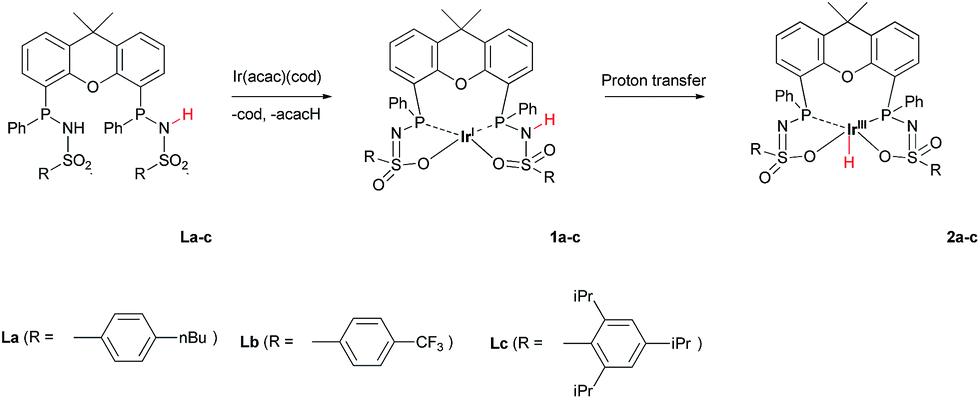
The addition of La–Lc to IrI(acac)(cod) generated complexes [IrI(LH)] 1a–cvia a single proton-transfer from the ligand to acetylacetonate and displacement of cyclooctadiene. These species show symmetric 1H and 31P NMR spectra, irrespective of the specific ligand substitution pattern, which likely originates from highly fluxional behaviour between the protonated and deprotonated ligand arms. Formal oxidative addition of the remaining ligand –NH group in 1a–1c generates the corresponding IrIII(H)(L) complexes 2a–c (Scheme 2), see also ESI.†
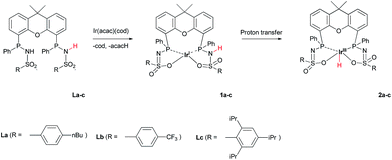 |
| | Scheme 2 Synthesis of complexes 1a–c and 2a–c from IrI(acac)(cod) and La–c. | |
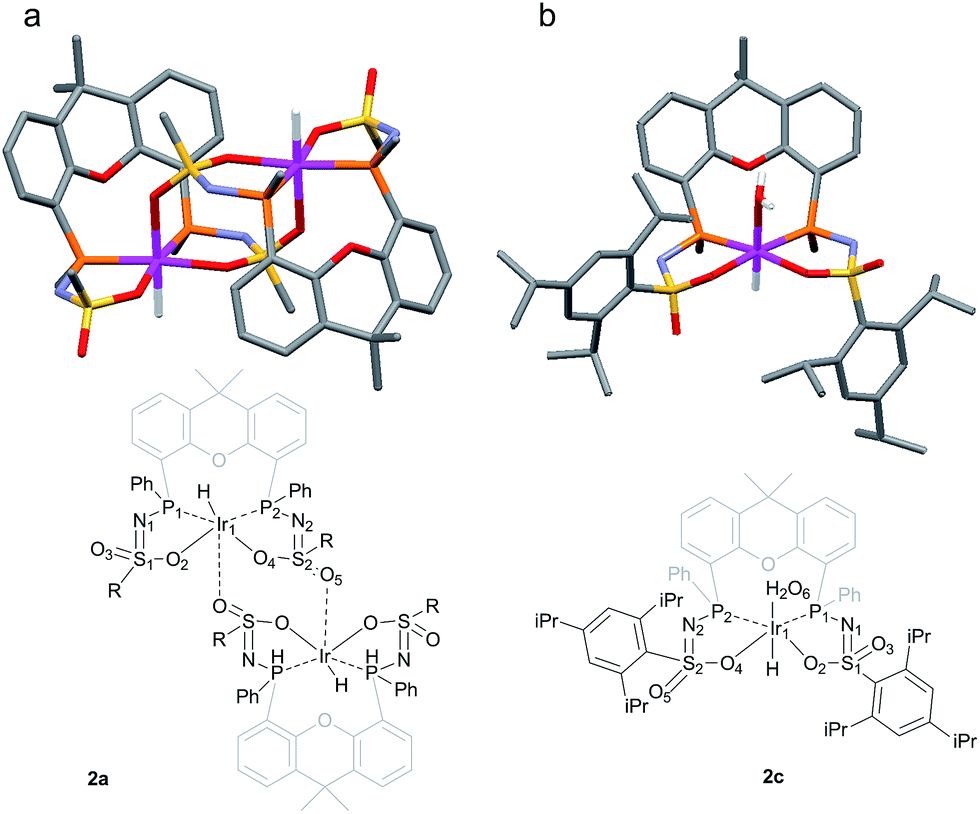
The rate for this overall proton-transfer step varies significantly for ligands 1a–c. IrIII complex 2a (with La) was obtained quantitatively after 30 hours at room temperature, but no formation of complex 2b was observed under the same conditions. This species could only be obtained after heating the mixture for 40 hours at 70 °C. In contrast, the conversion of 1c into 2c was significantly faster than the conversion of 1a into 2a, and full conversion was already observed after 16 hours at room temperature. We previously reported the molecular structure of 2a to be dimeric in the solid state (2a2), with the ‘vacant’ axial site coordinated by an oxygen from the sulfonamide of a second equivalent of 2a (Fig. 3a).4f The molecular structure determination for the more bulky analogue 2c revealed a slightly distorted octahedral mononuclear IrIII hydride complex, with axial coordination of a water molecule trans to the hydride to complete the octahedral coordination environment of IrIII (see Fig. 3b). The steric hindrance of the isopropyl groups in complex 2c seems to effectively prevent the formation of dinuclear complexes, as was also found by modelling studies. This finding also supports the previously proposed mononuclear configuration in solution, based on diffusion NMR data.4f The N–S bond lengths of 1.554(2) Å are in agreement with deprotonated sulfonamide fragments.4f The Ir–Owater bond length was found to be 2.2427(18) Å, which is in accordance with trans-IrIII(H)(OH2) complexes described in literature.11
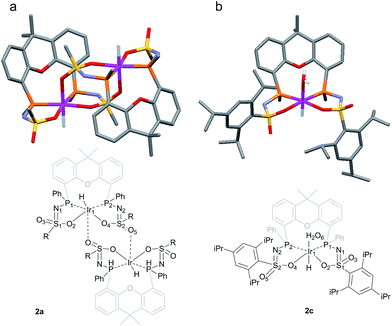 |
| | Fig. 3 (a) Schematic representation and molecular structure of 2a.4f (b) Schematic representation and molecular structure of 2c.14 Solvent molecules and H-atoms omitted for clarity, except for hydride and hydrogens of the aqua ligand. Color labels: purple: iridium, orange: phosphorus, blue: nitrogen, yellow: sulfur, red: oxygen. Selected bond lengths (Å) and angles (°) for 2c: N1–S1 1.554(2), N2–S2 1.554(2), S1–O2 1.4974(18), S2–O4 1.5093(18), S1–O3 1.4541(18), S2–O5 1.439(2), Ir1–P1 2.2311(6), Ir1–P2 2.2315(6), Ir1–O2 2.1639(18), Ir1–O4 2.1752(17), Ir1–O6 (H2O) 2.2427(18), P1–Ir1–P2 104.54(2). | |
Dehydrogenation studies with complexes 2a–c
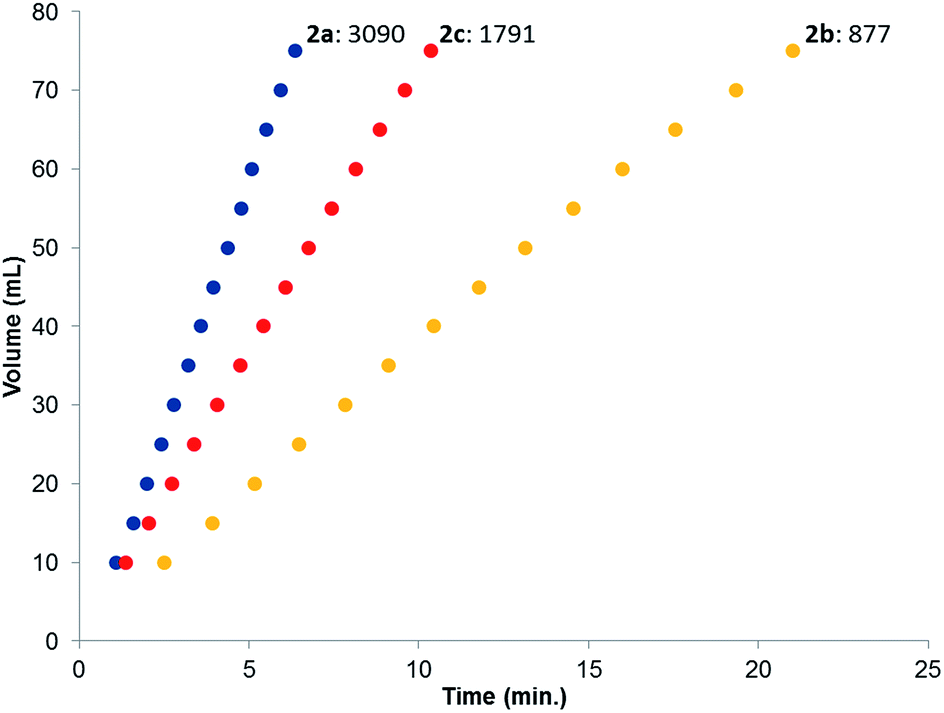
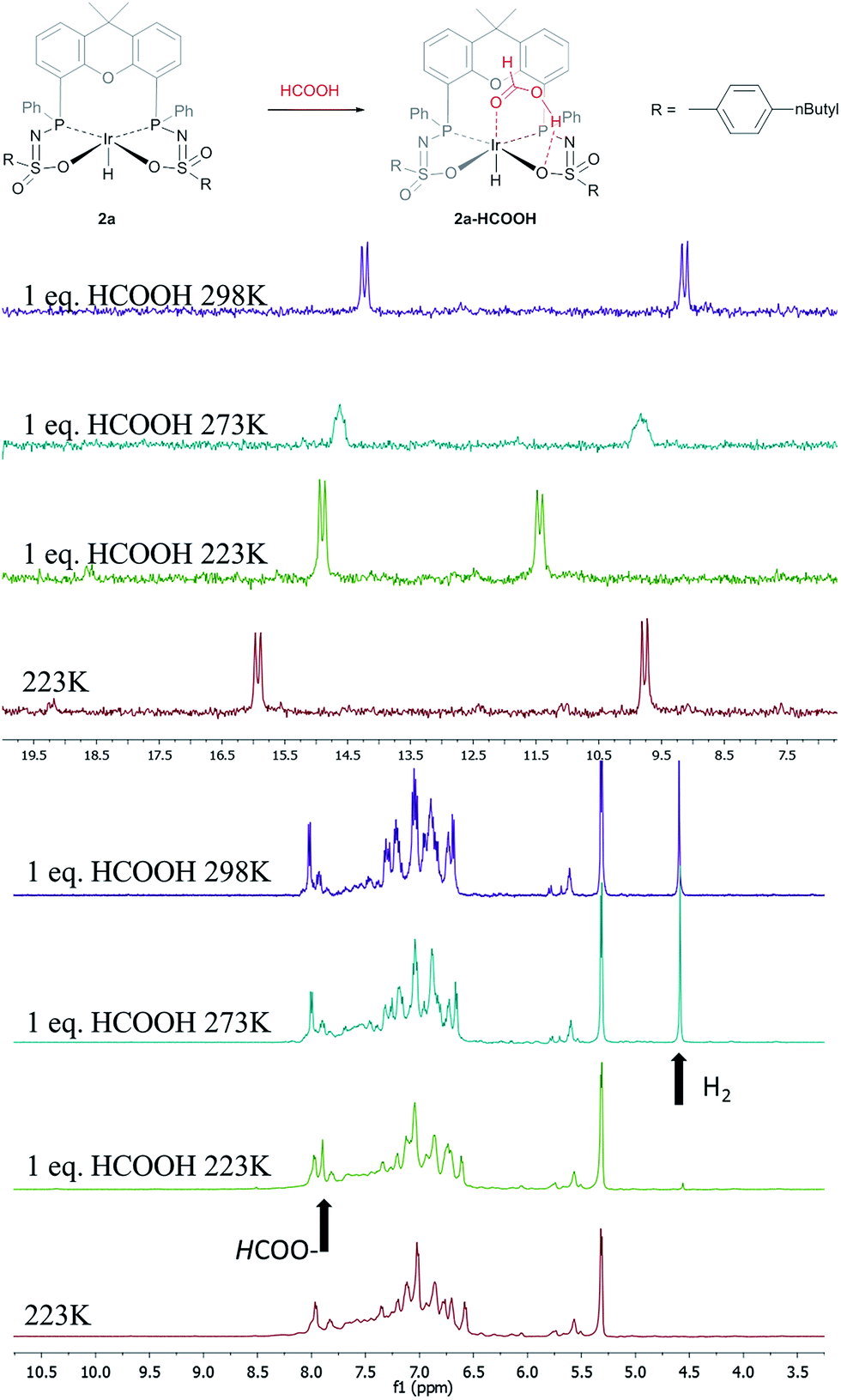
The catalytic dehydrogenation of HCOOH was investigated with complexes 2a–c.‡ Catalysis was performed in toluene at 85 °C in the absence of base to generate dehydrogenation curves that are shown in Fig. 4, see also ESI.† The turnover frequencies (TOFs) of 3090 (2a), 877 (2b) and 1791 h−1 (2c) reveal a correlation with the electronic nature of the sulfonamide organic side-group, i.e. high activity is obtained with an electron-donating group in the para-position (2a, n-butyl), whereas an electron-withdrawing group in the para-position (2b, CF3) results in much lower activity (see Computational section for explanation). Also with sterically encumbered complex 2c a lower activity was obtained than with 2a. Variable temperature (VT) NMR spectroscopy was performed to obtain mechanistic insight and detect relevant intermediates.§ The addition of an equimolar amount of HCOOH to complex 2a at 223 K led to a significant downfield shift (1.7 ppm) of one of the phosphorus signals (see Fig. 5, top). This might indicate (partial) protonation of one of the ligand arms. Upon increasing the temperature to 298 K the original 31P NMR spectrum for species 2a is instantaneously restored with no observation of any intermediates.¶ Addition of one equivalent of HCOOH to 2a results in an upfield shift in the 1H NMR spectrum for the formate proton of 0.13 ppm at 223 K (Fig. 5, right) compared to free HCOOH, which is an indication of HCOOH coordination to the axial vacant site (Fig. 5, bottom). Increasing the temperature leads to decreasing HCOOH signals together with the formation of H2.
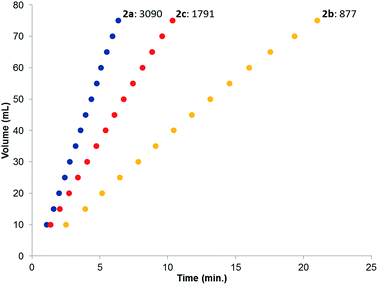 |
| | Fig. 4 Dehydrogenation curves of 2a–c (TOFs are determined between 4–30% conversion); total measured volume (mL) consists of both H2 and CO2. | |
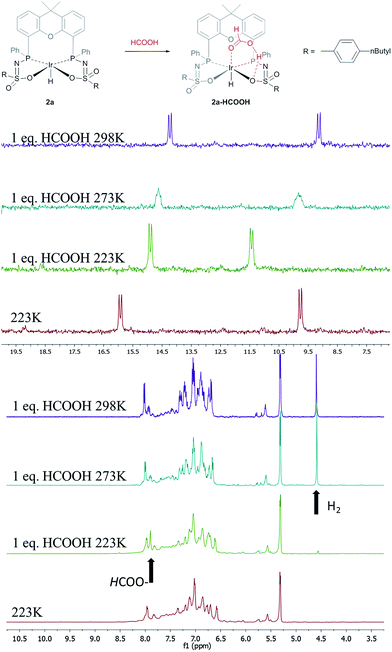 |
| | Fig. 5 Proposed coordination of HCOOH to complexes 2 to form a 2a–HCOOH adduct (top). VT 31P NMR spectra of 2a with one equivalent of HCOOH (left). VT 1H NMR spectra of 2a with one equivalent of HCOOH; arrows indicate HCOO–proton and H2 formed (right). | |
Computational investigation into β-hydride elimination
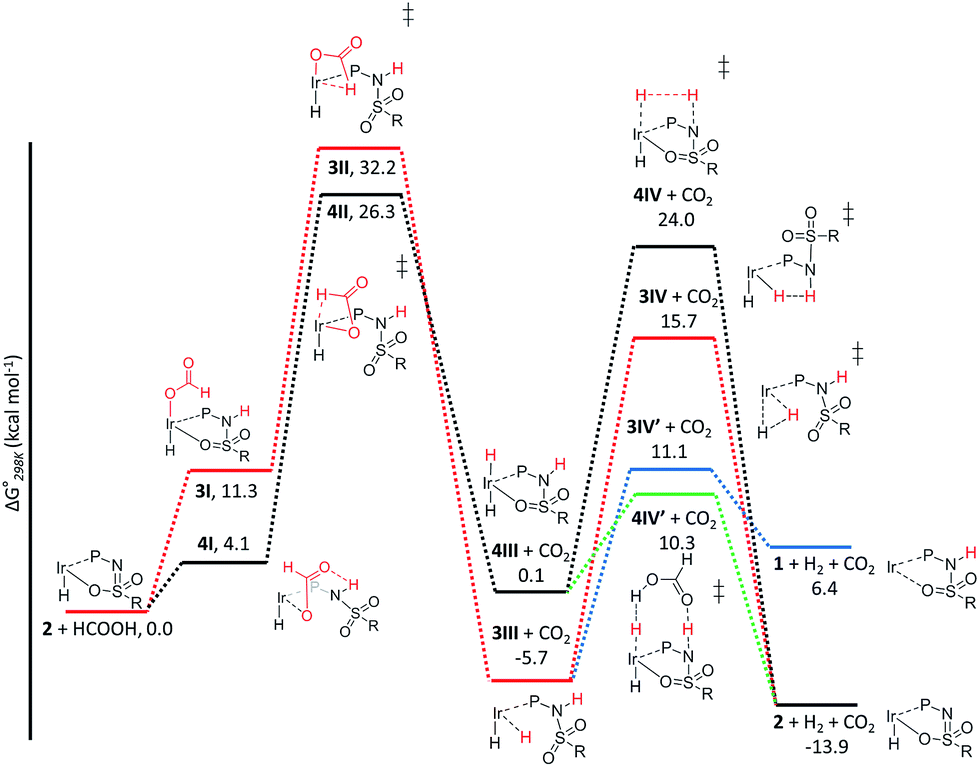
The C–H bond cleavage in the dehydrogenation of HCOOH, typically the rate determining step in the catalytic cycle, can either occur via β-hydride elimination or via (ligand-assisted) direct hydride-transfer of the formate hydrogen (HCOOH) atom (see Fig. 2A–C). Both possible mechanisms were computationally investigated using DFT in order to shed light on the potential role of the proton-responsive bisMETAMORPhos ligand. The obtained energy profiles for β-hydride elimination towards an equatorial and an axial coordination site of the catalyst are shown in Fig. 6 (only the relevant part of the computed structures is shown).||
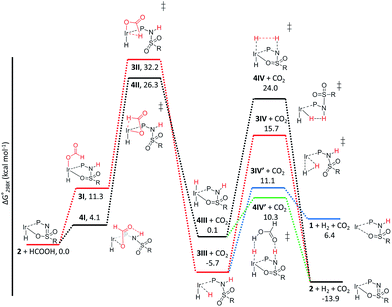 |
| | Fig. 6 Potential energy diagram (DFT, BP86, def2-TZVP) for the dehydrogenation of formic acid via β-hydride elimination at the equatorial (red) and axial (black) positions; ΔG0298K is in kcal mol−1. | |
The first step in the energy profile towards equatorial β-hydride elimination (red profile) is complete proton-transfer of HCOOH to the cooperative ligand, resulting in the formation of κ1-formate complex 3I, which is endergonic by 11.3 kcal mol−1. In this structure the formate C–H bond is pre-organized for β-hydride elimination via high barrier transition state 3II (ΔG‡ = 32.2 kcal mol−1).** This produces cis-dihydride structure 3III, which is an overall exergonic process (−5.7 kcal mol−1). Structure 3III can release H2via protonation of an iridium hydride by the ligand (N–H or O–H) or reductive elimination of the two hydride units. Protonation of the hydride by the N–H moiety of the ligand in transition state 3IV has a rather high barrier (red profile, 21.4 kcal mol−1). No transition state was found for protonation of the hydride via an O–H group. Transition state 3IV is potentially stabilized by axial coordination of the ligand via the sulfon group, which is in proximity to the iridium (2.42 Å). This stabilization cannot occur upon protonation via the O–H moiety, which potentially explains why no transition state could be found. The reductive elimination of H2 forms IrI structure 1 (similar to the initially formed complexes 1a–c that lead to the formation of 2a–c) via transition state 3IV′, which is 4.6 kcal mol−1 lower in energy (blue energy profile, 11.1 kcal mol−1) than 3IV. After release of H2 the formed structure 1 is 6.4 kcal mol−1 higher in energy than the starting structure 2. This is in agreement with experimental observations, as iridium(I) complexes 1a–c eventually transform to the thermodynamically more stable iridium(III)-hydride complexes 2a–c. The above described pathway to cis-dihydride species 3IIIvia transition state 3II (Fig. 6, red line) seems unlikely to occur, as the activation barrier obtained for C–H cleavage is rather high (32.2 kcal mol−1). The β-hydride elimination towards the axial position was found to be energetically more favorable (Fig. 6, black energy profile). Deprotonation of HCOOH by the ligand and formation of κ1-formate structure 4I is endergonic by 4.1 kcal mol−1. The protonated ligand showed a N–H⋯O hydrogen bonding interaction between the ligand and the coordinated formate. β-Hydride elimination from 4Ivia transition state 4II to yield 4III has an activation barrier of 26.3 kcal mol−1. The trans-dihydride structure 4III is slightly endergonic by 0.1 kcal mol−1. Protonation of the Ir–H bond by a ligand N–H group to release H2 from 4IIIvia transition state 4IV has a high barrier of 24.0 kcal mol−1. This is most likely related to the significant structural reorganization needed to bring the N–H proton in proximity to the hydride. Under catalytic conditions an excess of HCOOH is present, so we decided to investigate whether an additional equivalent of HCOOH could mediate the proton transfer from the ligand to the Ir–H. Indeed, a transition state was obtained wherein protonation of the Ir–H with HCOOH occurred simultaneously with reprotonation of HCOOH by the ligand N–H (Fig. 6, green profile). This transition state (4IV′) turned out to be 13.7 kcal mol−1 lower in energy (10.3 kcal mol−1) compared to transition state 4IV. Similar second-sphere interactions were previously proposed in the heterolysis of H2 assisted by exogenous water.12 In our case, HCOOH provides a perfect geometrical fit between Ir–H and the N–H moiety of the ligand for second-sphere assisted proton transfer to occur.
Computational investigation into direct hydride-transfer
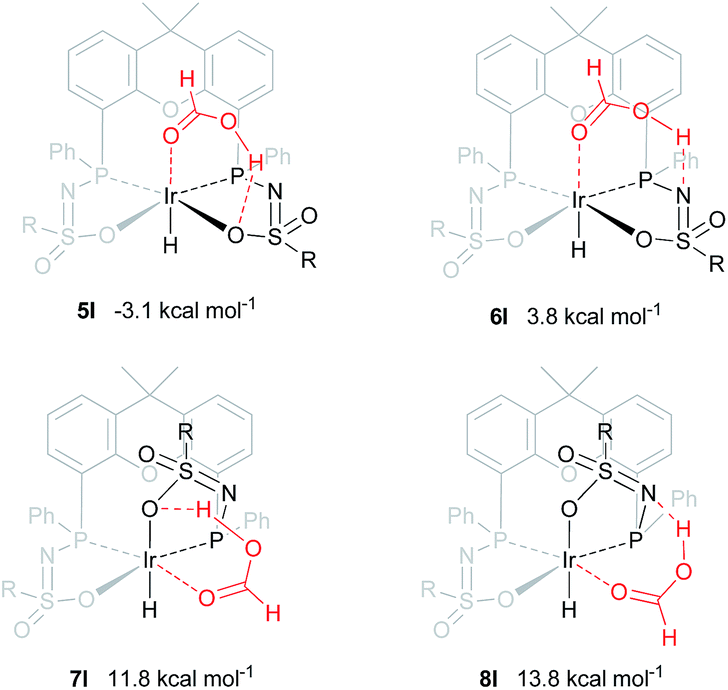
An alternative mechanism for the dehydrogenation of formic acid could involve a direct hydride-transfer of the formate hydrogen (HCOOH) to the Ir-center, subsequent to, or in concert with, proton transfer of the acidic HCOOH proton to the ligand scaffold. In this mechanism a single metal coordination site is sufficient for effective turnover. To test the validity of such a pathway, we computed different HCOOH–2 adducts. Adducts with either axial or equatorial coordination were all found to exhibit stabilizing hydrogen-bonding interactions with either the nitrogen atom or the coordinated oxygen atom in the ligand scaffold, resulting in structures 5I–8I (see Fig. 7). Axial coordination of HCOOH (5I; interaction with O) is the only structure found to be exergonic, by 3.07 kcal mol−1, compared to the free complex plus HCOOH. Formation of structure 6I, with an N–H interaction, is endergonic by 3.82 kcal mol−1. Equatorial coordination of HCOOH, leading to structures 7I or 8I, is associated with a significant energy penalty of 11.8 (7I) or 13.8 (8I) kcal mol−1 compared to formation of the axial adducts.
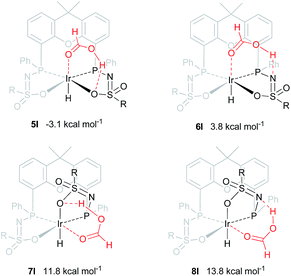 |
| | Fig. 7 DFT optimized geometries of formic acid adducts 5I–8I (BP86, def2-TZVP, ΔG0298K in kcal mol−1), R = phenyl. | |
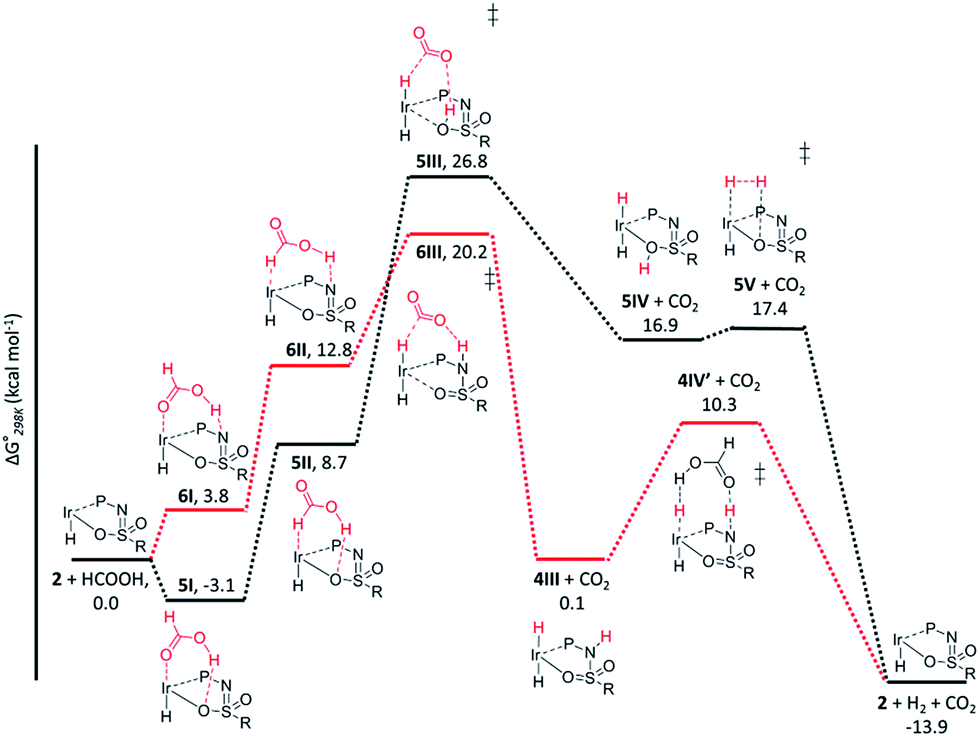
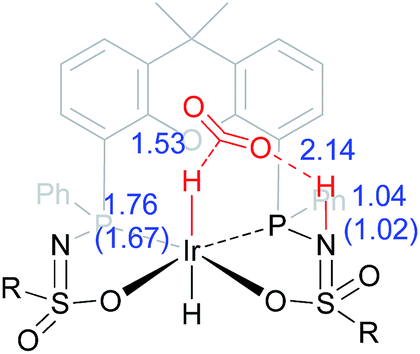
The dehydrogenation of HCOOH via a direct hydride-transfer pathway was first investigated using the axial HCOOH adducts 5I and 6I. Starting from the energetically most stable structure 5I (see black energy profile in Fig. 8), an endergonic rearrangement to 5II was found (+11.8 kcal mol−1), which orients the formate hydrogen in a favorable position for direct hydride-transfer to the metal. In transition state 5III (ΔG‡ = 26.8 kcal mol−1 and ΔG‡ = 29.9 kcal mol−1 with respect to 5I) HCOOH is fully deprotonated by the ligand. This enables facile expulsion of CO2 leading to a 6-membered Ir⋯H⋯C⋯O⋯H⋯O transition state. After release of CO2, dihydride complex 5IV is formed in an overall endergonic process (+16.9 kcal mol−1), whereafter protonation of the iridium-hydride via transition state 5V (+17.4 kcal mol−1) releases H2. The HCOOH dehydrogenation pathway was also investigated starting from structure 6I (red profile in Fig. 8). The rearrangement from 6I to 6II (similar to the rotation of 5I to 5II) is endergonic by 12.8 kcal mol−1. However, transition state 6III was found to be significantly lower in energy (ΔG‡ = 20.2 kcal mol−1) compared to 5III (ΔG‡ = 26.8 kcal mol−1), leading to formation of structure 4IIIvia an unusual 8-membered Ir⋯H⋯C⋯O⋯H⋯N⋯S⋯O transition state, see Fig. 6. Structures 6I–III are all stabilized by hydrogen bonding interactions between HCOOH/CO2 and the ligand pre-assembling HCOOH and stabilizing the transition state. The same role of the ligand was found in the energy profile starting from structure 5I. The Ir–H and N–H bonds in transition state 6III are elongated compared to those found in 4III (Ir–H: 1.76 Å vs. 1.67 Å; N–H: 1.04 Å vs. 1.02 Å), which indicates a late transition state (Fig. 9). A similar interaction was proposed by Hazari et al. for CO2 insertion of an Ir–H stabilized by hydrogen bonding between a ligand-based N–H group and CO2.13 Calculations performed on structures lacking these H-bonding interactions (by pointing the N–H fragment outwards) yielded unstable structures and no transition states could be found. After CO2 release from 6III, structure 4III is formed, which releases H2 with the assistance of another molecule of HCOOH (structure 4IV′), regenerating the catalyst as described above (Fig. 6, green profile). Direct hydride-transfer was also investigated starting from structures 7I and 8I, but these energy profiles were found to be significantly higher than for 5I and 6I, see ESI.†
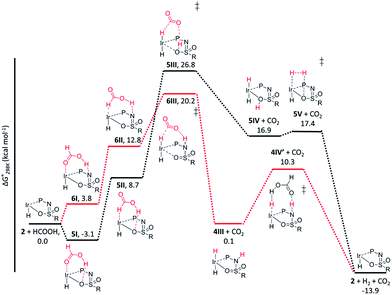 |
| | Fig. 8 Potential energy diagram (DFT, BP86, def2-TZVP) for dehydrogenation of formic acid by 5I (black profile) and 6I (red profile); ΔG0298K is in kcal mol−1. | |
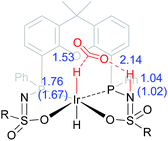 |
| | Fig. 9 Transition state 6III with stabilizing hydrogen-bond between ligand and CO2. Relevant computed bond lengths (in Å) are compared to values found computationally for structure 4III (in parentheses). | |
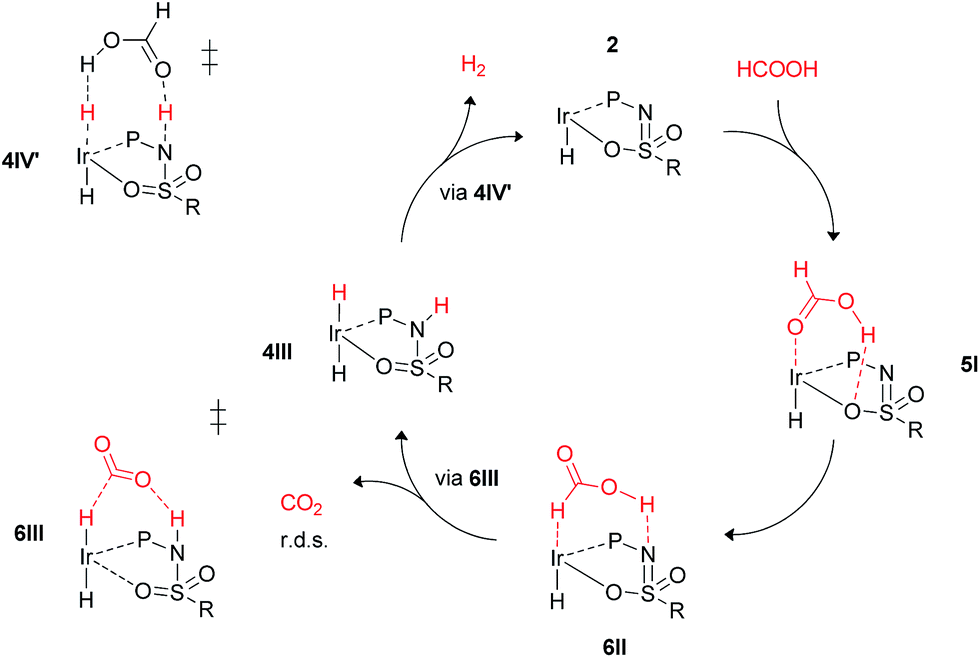
These calculations suggest that HCOOH dehydrogenation using bisMETAMORPhos-derived complexes 2a–c follows an outer-sphere direct hydride-transfer mechanism at the axial vacant site of these monohydride species. Starting from formic acid adduct 5I, the energetically most favored pathway requires rearrangement of 5I to 6I. This is likely a facile process due to the high proton-mobility within the ligand scaffold (from N–H to O–H), as was also inferred from the rapid proton-shuttling in complexes 1a–c. This results in an overall activation barrier of 23.3 kcal mol−1, taking 5I as the resting state. This is in agreement with the variable temperature 1H NMR observations, as the change in chemical shift upon addition of HCOOH to 2a indicates the formation of a 2a–HCOOH adduct akin to structure 5I. Protonation of the ligand takes place in transition state structure 6III. The hydride ligand already present in complexes 2a–c, trans to the axial coordination site of HCOOH, acts purely as a spectator ligand. The lower activity of complex 2c, bearing electron-withdrawing CF3 groups, can therefore potentially be explained by the decreased basicity of the nitrogen atom in the ligand. The electronic effect was also investigated theoretically by comparing p-CF3 with p-CH3 substituents. Indeed, a higher activation barrier was found for p-CF3 (24 kcal mol−1) compared to p-CH3 (22.5 kcal mol−1), see ESI† for energy profiles. We propose the following overall dual role for the bisMETAMORPhos ligand in the mechanism of formic acid dehydrogenation (Scheme 3). Species 2 coordinates HCOOH at the vacant axial site, aided by hydrogen-bonding with the coordinated sulfon–oxygen atom to give structure 5I. Reorientation of the formate group gives a pre-activated HCOOH unit that participates in hydrogen-bonding with the deprotonated nitrogen of the ligand arm (6II). Release of CO2 is achieved via transition state 6III (rate determining step) wherein the ligand deprotonates HCOOH and stabilizes the direct hydride transfer by a hydrogen bonding interaction. This gives trans-dihydride 4III that releases H2 aided by an additional equivalent of HCOOH, which protonates the Ir–H and in turn is reprotonated by the ligand, thereby regenerating starting complex 2. The reversibility of this catalytic system is currently under investigation.
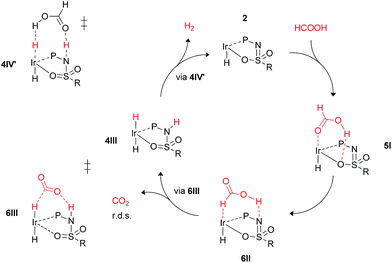 |
| | Scheme 3 Schematic proposed catalytic cycle of the ligand-assisted dehydrogenation of HCOOH by bisMETAMORPhos complexes 2a–c. | |
Conclusions
We report several bisMETAMORPhos ligands (La–Lc) based on the xanthene backbone bearing two sulfonamide-phosphine units. The prototropic nature of these PN(H)SO2R fragments results in different ratios for the PIII and PV tautomers, depending on the electronic and steric nature of the sulfonamide substituents. Formation of IrI complexes 1a–c was achieved by coordination of La–Lc to Ir(acac)(cod). Subsequent formal oxidative addition of the remaining N–H group resulted in the formation of the corresponding IrIII-monohydride complexes IrIII(H)(L) (2a–c). These three complexes are active catalysts for the base-free dehydrogenation of formic acid, with TOFs of 3090, 877 and 1791 h−1 for 2a–c, respectively. These data reflect the influence of subtle electronic and steric changes in the ligand architecture. The role of the ligand during catalysis was investigated by variable temperature 1H and 31P NMR spectroscopic measurements and DFT calculations. Variable temperature NMR data point to the formation of a 2a–HCOOH adduct. DFT calculations indicate that the hydrogen-bonding abilities of the ligand play an important role in the mechanism, resulting in an uncommon direct hydride-transfer mechanism instead of the more commonly proposed β-hydride elimination for HCOOH dehydrogenation. This was also found to be the rate-determining step (23.3 kcal mol−1), which confirms experimental observations using DCOOH and HCOOD.4f A similar mechanism has been proposed for Al- and Fe-based systems reported by Berben and Milstein, respectively.6c,8c It was found that the combined hydrogen-bonding and proton-responsive properties of the bisMETAMORPhos ligands are essential for the reactivity of complexes 2a–c. These interactions facilitate the pre-assembly of the HCOOH substrate and the stabilization of catalytically relevant intermediates and transition states, allowing an otherwise inaccessible reaction pathway. We thus show that a single coordination site is effective for the dehydrogenation reaction to occur when using a functional ligand scaffold. The proton-responsive and hydrogen-bonding features of ligands (La–Lc) are currently being explored for other reactions. The mechanistic findings presented herein potentially play a role in other HCOOH dehydrogenative catalysts bearing hydrogen-bonding functionalities in their ligand systems.
Acknowledgements
We kindly acknowledge NWO for financial support, Frédéric G. Terrade and Däniel L. J. Broere for fruitful discussions and Ed Zuidinga for mass analysis.
Notes and references
-
(a) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC;
(b) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC;
(c) T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC.
-
(a) B. Breit and W. Seiche, J. Am. Chem. Soc., 2003, 125, 6608–6609 CrossRef CAS PubMed;
(b) S. Zhu, J. V. Ruppel, H. Lu, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2008, 130, 5042–5043 CrossRef CAS PubMed;
(c) J. Meeuwissen, M. Kuil, A. M. van der Burg, A. J. Sandee and J. N. H. Reek, Chem.–Eur. J., 2009, 15, 10272–10279 CrossRef CAS PubMed;
(d) L. Diab, T. Šmejkal, J. Geier and B. Breit, Angew. Chem., Int. Ed., 2009, 48, 8022–8026 CrossRef CAS PubMed ; for reviews see: J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 Search PubMed;
(e) A. J. Sandee and J. N. H. Reek, Dalton Trans., 2006, 3385–3391 RSC;
(f) S. Carboni, C. Gennari, L. Pignataro and U. Piarulli, Dalton Trans., 2011, 40, 4355–4373 RSC.
-
(a) S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941–1943 CrossRef CAS PubMed;
(b) L. Du, P. Cao, J. Xing, Y. Lou, L. Jiang, L. Li and J. Liao, Angew. Chem., Int. Ed., 2013, 52, 4207–4211 CrossRef CAS PubMed;
(c) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC;
(d) D. B. Grotjahn, Chem.–Eur. J., 2005, 11, 7146–7153 CrossRef CAS PubMed;
(e) T. Šmejkal and B. Breit, Angew. Chem., Int. Ed., 2008, 47, 311–315 CrossRef PubMed;
(f) P.-A. R. Breuil, F. W. Patureau and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 2162–2165 CrossRef CAS PubMed;
(g) P. Dydio, W. I. Dzik, M. Lutz, B. de Bruin and J. N. H. Reek, Angew. Chem., 2011, 123, 416–420 CrossRef;
(h) P. Dydio, C. Rubay, T. Gadzikwa, M. Lutz and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 17176–17179 CrossRef CAS PubMed;
(i) P. Dydio and J. N. H. Reek, Angew. Chem., Int. Ed., 2013, 52, 3878–3882 CrossRef CAS PubMed;
(j) Y. Gumrukcu, B. de Bruin and J. N. H. Reek, ChemSusChem, 2014, 7, 890–896 CrossRef CAS PubMed;
(k) W. I. Dzik, X. Xu, X. P. Zhang, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2010, 132, 10891–10902 CrossRef CAS PubMed;
(l) V. Lyaskovskyy, A. I. O. Suarez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264–12273 CrossRef CAS PubMed;
(m) A. I. O. Suarez, H. Jiang, X. P. Zhang and B. de Bruin, Dalton Trans., 2011, 40, 5697–5705 RSC;
(n) Y. Chen, K. B. Fields and X. P. Zhang, J. Am. Chem. Soc., 2004, 126, 14718–14719 CrossRef CAS PubMed.
-
(a) F. W. Patureau, M. Kuil, A. J. Sandee and J. N. H. Reek, Angew. Chem., Int. Ed., 2008, 47, 3180–3183 CrossRef CAS PubMed;
(b) F. W. Patureau, S. de Boer, M. Kuil, J. Meeuwissen, P.-A. R. Breuil, M. A. Siegler, A. L. Spek, A. J. Sandee, B. de Bruin and J. N. H. Reek, J. Am. Chem. Soc., 2009, 131, 6683–6685 CrossRef CAS PubMed;
(c) T. León, M. Parera, A. Roglans, A. Riera and X. Verdaguer, Angew. Chem., Int. Ed., 2012, 51, 6951–6955 CrossRef PubMed;
(d) F. W. Patureau, M. A. Siegler, A. L. Spek, A. J. Sandee, S. Jugé, S. Aziz, A. Berkessel and J. N. H. Reek, Eur. J. Inorg. Chem., 2012, 496–503 CrossRef CAS;
(e) F. G. Terrade, M. Lutz, J. I. van der Vlugt and J. N. H. Reek, Eur. J. Inorg. Chem., 2014, 1826–1835 CrossRef CAS;
(f) S. Oldenhof, B. de Bruin, M. Lutz, M. A. Siegler, F. W. Patureau, J. I. van der Vlugt and J. N. H. Reek, Chem.–Eur. J., 2013, 19, 11507–11511 CrossRef CAS PubMed.
-
(a) R. Hille, Chem. Rev., 1996, 96, 2757–2816 CrossRef CAS PubMed;
(b) C. M. P. T. Reda, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654–10658 CrossRef PubMed;
(c) M. J. Romao, Dalton Trans., 2009, 4053–4068 RSC;
(d) C. Mota, M. Rivas, C. Brondino, I. Moura, J. G. Moura, P. González and N. F. S. A. Cerqueira, J. Biol. Inorg. Chem., 2011, 16, 1255–1268 CrossRef CAS PubMed;
(e) M. Tiberti, E. Papaleo, N. Russo, L. De Gioia and G. Zampella, Inorg. Chem., 2012, 51, 8331–8339 CrossRef CAS PubMed;
(f) K. Schuchmann and V. Müller, Science, 2013, 342, 1382–1385 CrossRef CAS PubMed.
-
(a) A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed;
(b) J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed;
(c) T. Zell, B. Butschke, Y. Ben-David and D. Milstein, Chem.–Eur. J., 2013, 19, 8068–8072 CrossRef CAS PubMed;
(d) E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed;
(e) Y. Manaka, W.-H. Wang, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, Catal. Sci. Technol., 2014, 4, 34–37 RSC;
(f) W.-H. Wang, S. Xu, Y. Manaka, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, ChemSusChem, 2014, 7, 1976–1983 CrossRef CAS PubMed.
-
(a) S. Kuwata and T. Ikariya, Chem.–Eur. J., 2011, 17, 3542–3556 CrossRef CAS PubMed;
(b) S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 412–429 CrossRef CAS;
(c) J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 363–375 CrossRef CAS;
(d) B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744–4788 CrossRef CAS PubMed;
(e) G. A. Filonenko, M. P. Conley, C. Copéret, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2013, 3, 2522–2526 CrossRef CAS;
(f) D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed;
(g) C. M. Moore and N. K. Szymczak, Chem. Commun., 2013, 49, 400–402 RSC;
(h) G. Zhang, K. V. Vasudevan, B. L. Scott and S. K. Hanson, J. Am. Chem. Soc., 2013, 135, 8668–8681 CrossRef CAS PubMed.
-
(a) G. A. Filonenko, E. J. M. Hensen and E. A. Pidko, Catal. Sci. Technol., 2014, 4(10), 3474–3485 RSC;
(b) T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed;
(c) T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC.
-
(a) T. Koike and T. Ikariya, Adv. Synth. Catal., 2004, 346, 37–41 CrossRef CAS;
(b) P. Š. J. Václavík, B. Vilhanová, J. Pecháček, M. Kuzma and P. Kačer, Molecules, 2013, 18, 6804–6828 CrossRef PubMed;
(c) A. Nova, D. J. Taylor, A. J. Blacker, S. B. Duckett, R. N. Perutz and O. Eisenstein, Organometallics, 2014, 33, 3433–3442 CAS.
-
(a) R. Sánchez-de-Armas, L. Xue and M. S. G. Ahlquist, Chem.–Eur. J., 2013, 19, 11869–11873 CrossRef PubMed;
(b) X. Yang, Dalton Trans., 2013, 42, 11987–11991 RSC.
-
(a) H. Bauer, U. Nagel and W. Beck, J. Organomet. Chem., 1985, 290, 219–229 CrossRef CAS;
(b) M. Feller, A. Karton, G. Leitus, J. M. L. Martin and D. Milstein, J. Am. Chem. Soc., 2006, 128, 12400–12401 CrossRef CAS PubMed;
(c) J. J. Adams, A. Lau, N. Arulsamy and D. M. Roddick, Organometallics, 2011, 30, 689–696 CrossRef CAS.
-
(a) C. Yin, Z. Xu, S.-Y. Yang, S. M. Ng, K. Y. Wong, Z. Lin and C. P. Lau, Organometallics, 2001, 20, 1216–1222 CrossRef CAS;
(b) M. S. G. Ahlquist, J. Mol. Catal. A: Chem., 2010, 324, 3–8 CrossRef CAS PubMed;
(c) X. Yang, ACS Catal., 2011, 1, 849–854 CrossRef CAS;
(d) R. Tanaka, M. Yamashita, L. W. Chung, K. Morokuma and K. Nozaki, Organometallics, 2011, 30, 6742–6750 CrossRef CAS;
(e) A. Pavlova and E. J. Meijer, ChemPhysChem, 2012, 13, 3492–3496 CrossRef PubMed.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- See ESI.†.
- A. J. C. Walters, J. N. H. Reek and B. de Bruin, ACS Catal., 2014, 4, 1376–1389 CrossRef CAS.
-
(a) D. H. Wertz, J. Am. Chem. Soc., 1980, 102, 5316–5322 CrossRef CAS;
(b) N. Schneider, M. Finger, C. Haferkemper, S. Bellemin-Laponnaz, P. Hofmann and L. Gade, Angew. Chem., Int. Ed., 2009, 48, 1609–1613 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1020151. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02555e |
| ‡ Dehydrogenation studies were performed with diastereomeric mixtures of complexes 2a–c (upon anionic coordination of the sulfon group these become chiral and give rise to diastereomeric mixtures of the complexes 2), see ESI† and ref. 4f for more information. |
| § VT-NMR studies were performed with a diastereomerically pure form of 2a; see ref. 4f for additional information. |
| ¶ The 31P NMR chemical shifts of 2a are significantly affected by the temperature, but these changes are completely reversible, see ESI.† |
| || All calculations were performed with R = phenyl on the sulfon group for computational simplicity using the diastereomeric form that was utilized in VT NMR studies and obtained as a crystal structure. |
| ** Note that these are gas-phase calculations for which the translational entropy contributions are much larger than in solution. By calculation of the partition function of the molecules in the gas phase, the entropy of dissociation or coordination for reactions in solution is overestimated. Therefore, for reactions in solution, the Gibbs free energies for all steps involving a change in the number of species should be corrected. Several methods have been proposed for the correction of gas phase to solution phase data. The minimal correction term is a correction for the condensed phase (CP) reference volume (1 L mol−1) compared to the gas phase (GP) reference volume (24.5 L mol−1). This leads to an entropy correction term (SCP = SGP + Rln{1/24.5}) for all species, lowering the relative free energies (298 K) of all associative steps by 2.5 kcal mol−1.3k,15 According to some authors, this correction term is too small, and larger correction terms up to 6.0 kcal mol−1 have been suggested.16 Which correction term is best remains somewhat debatable. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a