
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioorthogonal prodrug activation driven by a strain-promoted 1,3-dipolar cycloaddition†
Siddharth S.
Matikonda‡
,
Douglas L.
Orsi‡
,
Verena
Staudacher‡
,
Imogen A.
Jenkins
,
Franziska
Fiedler
,
Jiayi
Chen
and
Allan B.
Gamble
*
School of Pharmacy, University of Otago, Dunedin, 9054, New Zealand. E-mail: allan.gamble@otago.ac.nz
First published on 14th November 2014
Abstract
Due to the formation of hydrolysis-susceptible adducts, the 1,3-dipolar cycloaddition between an azide and strained trans-cyclooctene (TCO) has been disregarded in the field of bioorthogonal chemistry. We report a method which uses the instability of the adducts to our advantage in a prodrug activation strategy. The reaction of trans-cyclooctenol (TCO-OH) with a model prodrug resulted in a rapid 1,3-dipolar cycloaddition with second-order rates of 0.017 M−1 s−1 and 0.027 M−1 s−1 for the equatorial and axial isomers, respectively, resulting in release of the active compound. 1H NMR studies showed that activation proceeded via a triazoline and imine, both of which are rapidly hydrolyzed to release the model drug. Cytotoxicity of a doxorubicin prodrug was restored in vitro upon activation with TCO-OH, while with cis-cyclooctenol (CCO-OH) no activation was observed. The data also demonstrates the potential of this reaction in organic synthesis as a mild orthogonal protecting group strategy for amino and hydroxyl groups.
The ability to deliver drugs only at their site of action is highly advantageous in cancer therapy. Unfortunately, the current arsenal of drugs used to treat cancer are generally non-selective, leading to toxic side effects.1–3 To combat toxicity, prodrug activation strategies have been developed.3–5 Activation, via cleavage of a deactivating linker on the prodrug, is usually triggered by an overexpressed enzyme or change in the tumor environment (e.g. pH). However, the changes are often so subtle that selective activation of small molecule prodrugs is low, with many linkers susceptible to non-specific, off-target hydrolysis.3
Antibody–drug conjugates (ADCs) have the potential to avoid some of the problems associated with small molecule prodrugs and have been utilised effectively over the past few decades.6 However, there are still some inherent problems with ADCs including;6 (1) immunogenicity of the antibody, (2) difficulty in maintaining the balance between stability vs. cleavability of the antibody–drug linker, and (3) decreased potency of the drug as the effective concentration is limited by the number of cell surface receptors (∼105 receptors per cell) and number of molecules that can be attached to the antibody. In addition, low target-to-background drug ratios of monoclonal antibodies typically result in low tumor concentrations of the therapeutic agent.7,8 Pre-targeting strategies in which a non-toxic conjugate is allowed to accumulate at the tumor before administration of the prodrug, e.g. antibody-directed enzyme prodrug therapy (ADEPT)9,10 and bioorthogonal chemistry,11–14 could help overcome the low tumor-to-background ratios as well as avoid the requirement of a cleavable (and potentially unstable) linker in the administered prodrug. While ADEPT has shown promise in cancer therapy, anti-enzyme immune responses have hindered its in vivo application.10
Over the past decade, bioorthogonal chemistry has found widespread utility in biological imaging.15 To date, however, there are only a handful of examples using bioorthogonal chemistry for in vitro prodrug activation strategies.11,16–20 The best examples using click chemistry are those of Robillard who utilized the Staudinger reaction,17 and more recently, the trans-cyclooctene (TCO)/tetrazine ligation for prodrug-activation.11 The fast kinetics of the tetrazine ligation provide in vivo potential, however, this is yet to be demonstrated for prodrug activation and we envisage a number of challenges that could be encountered in future studies. These may include; (1) reduced reactivity of the TCO-conjugate when it is present in a sterically demanding environment (e.g. attached to both the antibody and cytotoxic drug), and (2) a low tumor-to-background ADC ratio leading to off-target release of the cytotoxic drug. Attachment of the tetrazine to the antibody would allow for pre-targeting with a non-toxic conjugate, however, the doxorubicin–TCO prodrug which would then need to be administered separately will be exposed to potential isomerization back to the unreactive cis-cyclooctene (CCO), a problem which is avoided when the TCO is attached to the antibody via a suitably designed linker.21
Inspired by the initial bioorthogonal activation strategies and the in vivo challenges that they posed, we set out to develop a new bioorthogonal prodrug activation based on a 1,3-dipolar click reaction between a TCO and an azide. Interestingly, this click reaction has received much less attention than the strain-promoted 1,3-dipolar cycloaddition of cyclooctynes and azides (SPAAC) due to the instability of the formed adducts in aqueous conditions.15a,15c,22,23 In 1992, Shea reported a reaction between a strained TCO 2 and 2,4,6-trinitrophenylazide 1 in CDCl3.24 Triazoline 3 formed from this highly electron-deficient system could not be isolated. Instead a rearrangement in which diatomic nitrogen was released gave three isolated products; aldimine 4, ketimine 5 and aziridine 6 (Scheme 1).
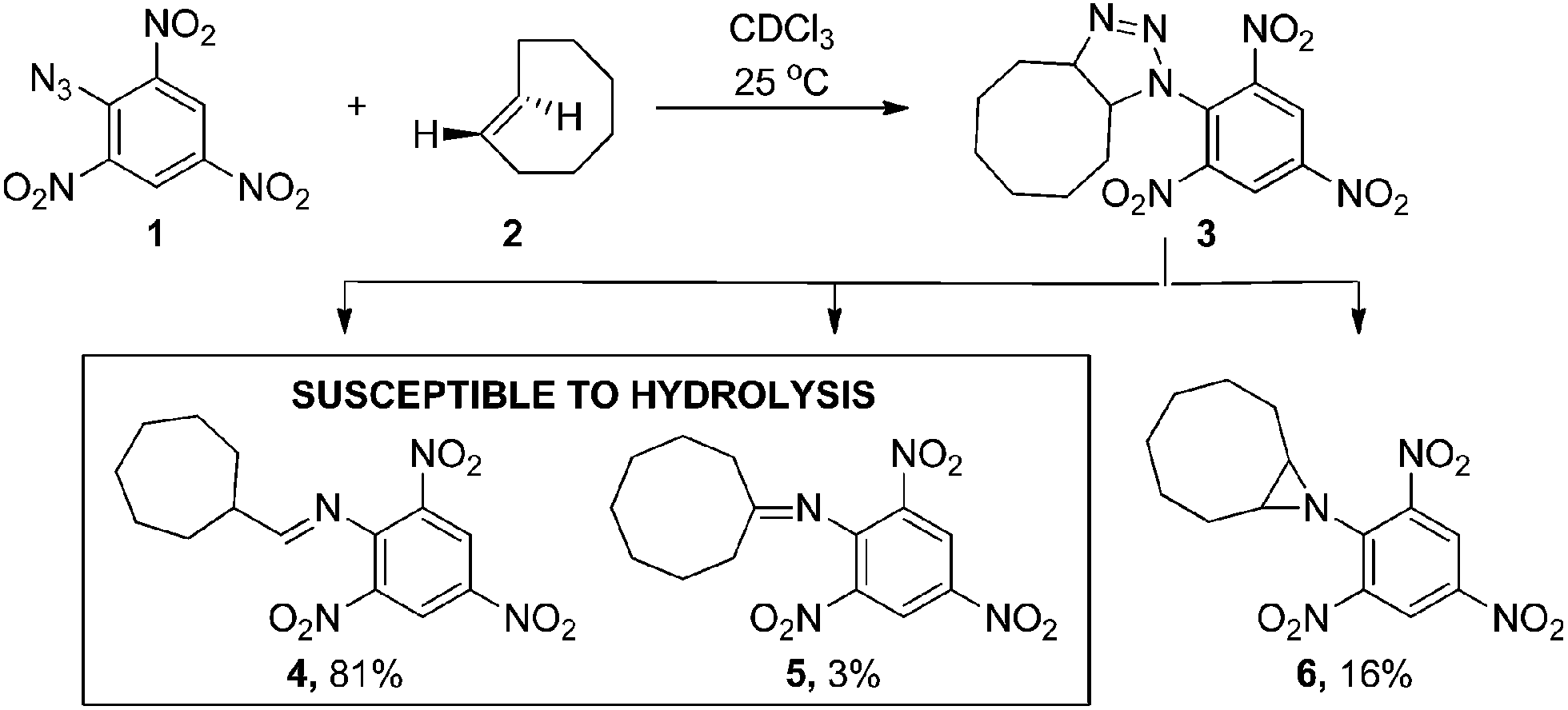 | ||
| Scheme 1 1,3-dipolar cycloaddition reported by Shea.24 | ||
We recognized that analogues of imine 4 or 5, when correctly attached to an electron-deficient self-eliminating linker (e.g. p-aminobenzyloxycarbonyl, PABC),5,25 could have potential in targeted prodrug activation strategies (Fig. 1) or orthogonal protection/deprotection chemistry. If an imine could be generated in situ by a 1,3-dipolar cycloaddition (step 1–3), acid-catalyzed hydrolysis26 in the tumor microenvironment (pH = 6.0–7.4)27,28 would lead to a rapid 1,6-elimination and removal of the PABC protecting group from the prodrug to release the active drug (step 4 and 5).25,26 While simple aryl azides can form stable triazolines in organic solvents,29 we hypothesized that, in an aqueous environment, the reaction of a TCO and an aryl azide bearing an electron-deficient PABC group would favour in situ triazoline rearrangement via alkyl migration towards the desired imine rather than aziridine formation.24
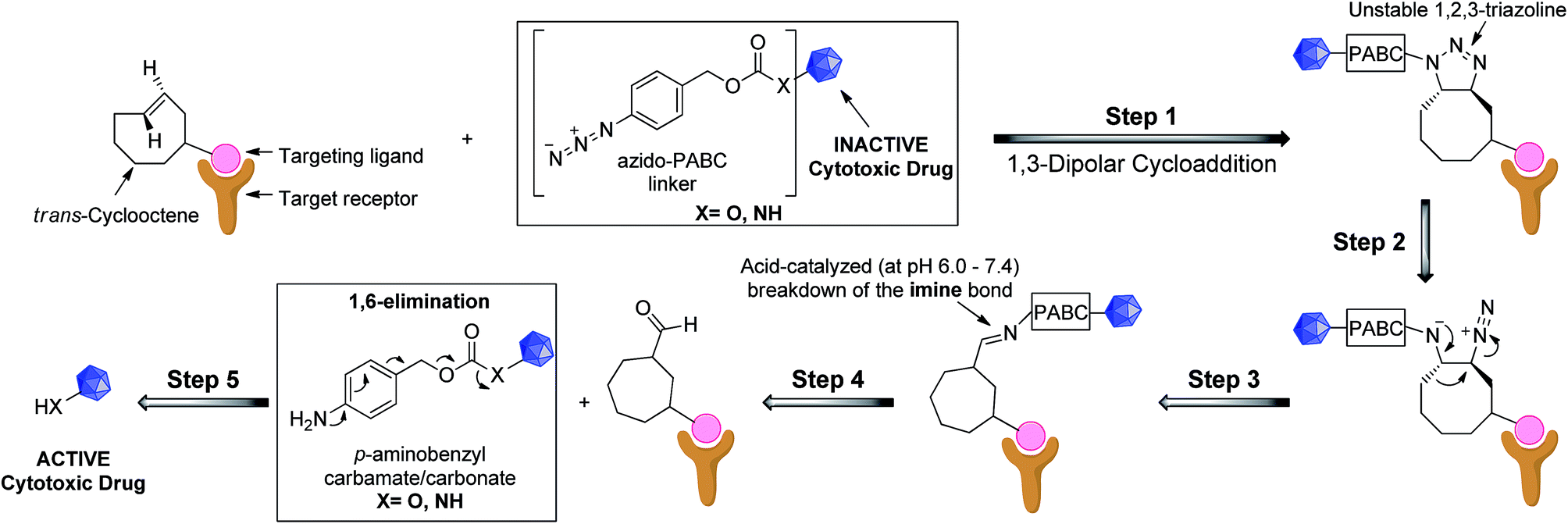 | ||
| Fig. 1 Proposed general strategy for prodrug activation via a 1,3-dipolar cycloaddition. The targeting ligand could be an antibody, peptide or small molecule. | ||
Herein we report the first in vitro example of a bioorthogonal prodrug activation strategy utilizing the 1,3-dipolar cycloaddition of an azide and a strained trans-cyclooctenol (TCO-OH). Our strategy demonstrates greater potential for bioorthogonally activated azide prodrugs in vivo as it holds two key advantages over the use of the Staudinger reaction for prodrug activation;17 (1) activation is 1–2 orders of magnitude faster with potential for future improvements (not possible with the Staudinger reaction),30 and (2) TCO can be modified so that it is more metabolically stable (i.e. no deactivation by isomerization to the cis-isomer)21 than the oxidation prone and serum reactive phosphine.30 Our results also demonstrate the potential use of the cycloaddition in synthetic chemistry as an orthogonal protection/deprotection strategy of –NH2 and –OH functional groups.
In order for our strategy to succeed we first needed to determine if the labile aldimine was favored over the stable aziridine, therefore enabling release of the active drug. For these proof-of-concept experiments two azido-PABC analogues were synthesized (Scheme 2). Coumarin probes 8a and 8b, masking a 7-hydroxycoumarin and 7-amino-4-methylcoumarin, respectively, were selected to investigate the mechanism and rate of the 1,3-dipolar cycloaddition, triazoline/imine degradation, and overall release of the drug from a carbonate and carbamate linked drug. The previously reported doxorubicin prodrug 9 was synthesized17 to examine in vitro bioorthogonal activation, and stability/activation of the aryl azide linker in mouse serum. TCO 2 and trans-cyclooctenol (TCO-OH) 10 were synthesized using a modified procedure of Fox,31 in which the CCO or cis-cyclooctenol (CCO-OH) was irradiated at 254 nm in the presence of methyl benzoate (see ESI†). For TCO-OH 10 a mixture of diastereomers (1.42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was isolated and used in preliminary studies. The major (equatorial) and minor (axial) isomers were subsequently separated for detailed kinetic analyses.
:1) was isolated and used in preliminary studies. The major (equatorial) and minor (axial) isomers were subsequently separated for detailed kinetic analyses.
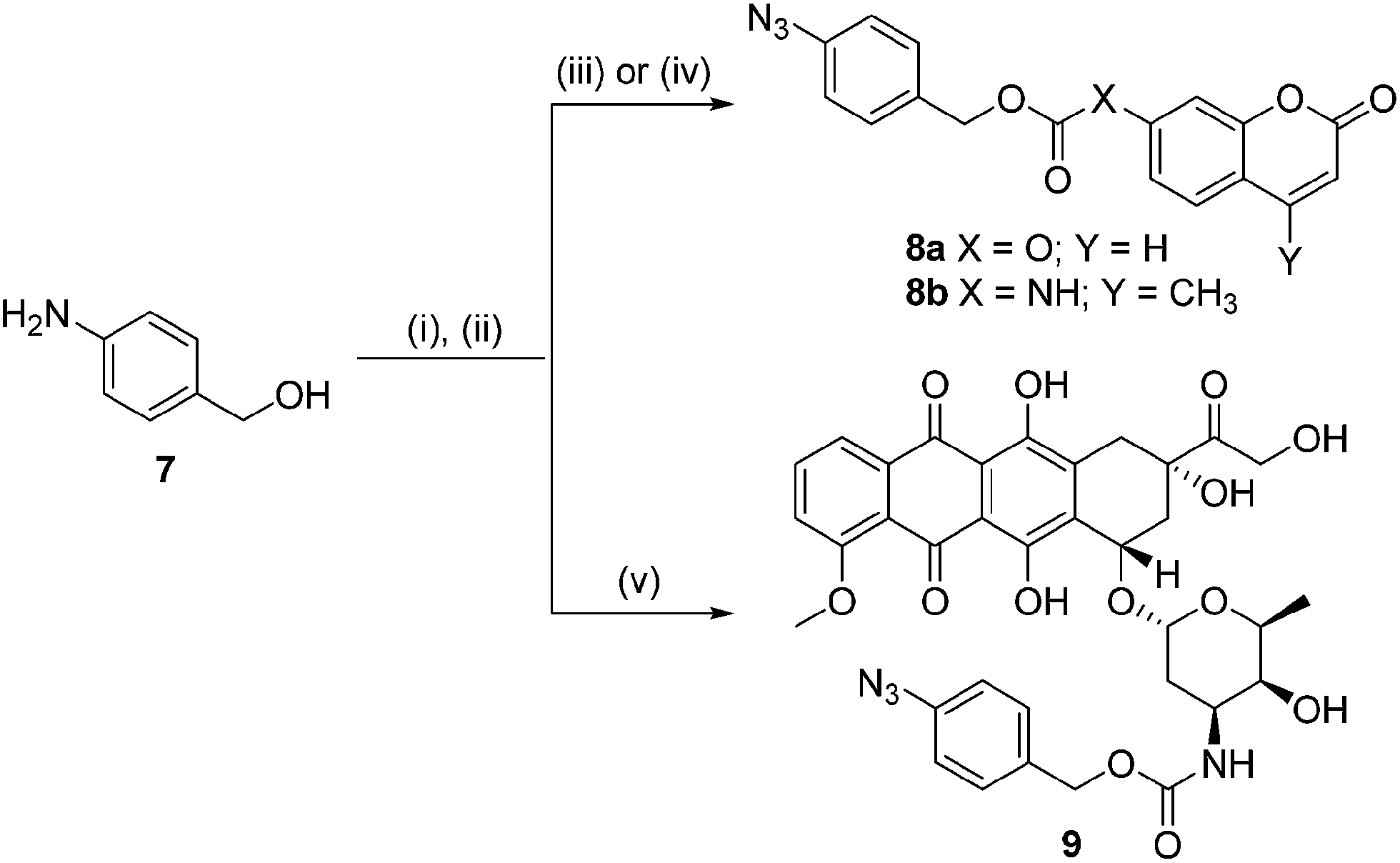 | ||
| Scheme 2 Conditions: (i) NaNO2, 5 M HCl then NaN3, 0 °C, 73%; (ii) 4-nitrophenyl chloroformate, pyridine, DCM, 25 °C, 57%; (iii) 7-hydroxycoumarin, Et3N, DMF, 25 °C, 38%; (iv) 7-amino-4-methylcoumarin, triphosgene, toluene, reflux, then 4-azidobenzyl alcohol, 25 °C, 30%; (v) doxorubicin·HCl, Et3N, 4 Å molecular sieves, DMF, 25 °C, 69%. | ||
Initially the cycloaddition of 8a/8b with TCO-OH 10 (Scheme 3) was investigated using spectrofluorometry (ex 360 nm, em 455 nm). Fluorescence of 7-hydroxycoumarin 13a or 7-amino-4-methylcoumarin 13b was quenched by the azido-PABC linker (probe 8a/8b), which upon addition of TCO-OH 10 (10-fold excess) resulted in activation of 8a/8b (Fig. 2). By 6 h 80–90% of coumarin had been released from both carbonate 8a and carbamate 8b. In the absence of TCO-OH 10, a low level of carbonate hydrolysis in 8a was observed, however, no background hydrolysis was evident for the carbamate analogue 8b. Reaction of 8a with CCO-OH (10-fold excess) had no effect on the release of 13a, with only background hydrolysis measured (10% at 24 h). As amides, esters and ethers of 13a/13b are non-fluorescent (ex 360 nm, em 455 nm),26,32–34 the intermediates 11b and 12b would not exhibit any fluorescence at the measured excitation and emission wavelengths, hence the measured fluorescence is the result of released coumarin 13a/13b. Therefore the results from our spectrofluorometry studies imply that the reaction proceeds via triazoline 11b followed by an imine intermediate such as 12b.
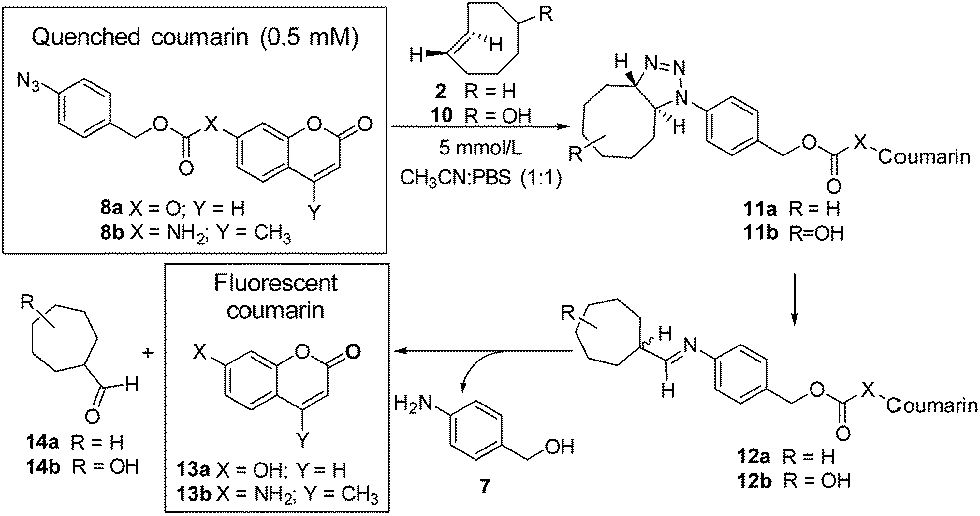 | ||
| Scheme 3 Model prodrug activation strategy. | ||
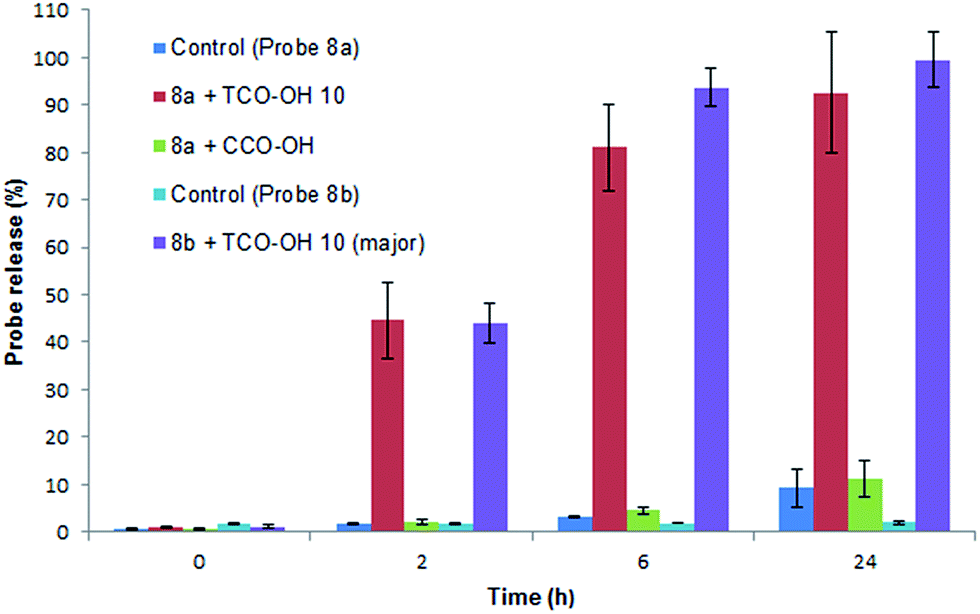 | ||
| Fig. 2 Release of 7-hydroxycoumarin 13a from 8a and 7-amino-4-methylcoumarin 13b from 8b in PBS:MeCN (1:1), measured by fluorescence (ex 360, em 455). Error represented as ±SD (n = 3). | ||
To confirm that the reaction we observed in the initial spectrofluorometry studies proceeds via the triazoline and aldimine, and not some alternate mechanism, a series of 1H NMR experiments in CDCl3 and CD3CN/D2O were performed with 8a (Fig. 3 and Fig. S11–S14†). Reaction of TCO-OH 10 with 8a would have given a complex mixture of regio- and stereo-isomers 11b and 12b, therefore, TCO 2 was used (Scheme 3, conc. 8a: 6.7 mM, TCO 2: 18.7 mM). This allowed us to monitor for the less complex intermediates 11a and 12a in the 1H NMR spectrum. Initially we examined the reaction in CDCl3 and observed that approx. 50% of probe 8a had been consumed within 3 hours and converted to the triazoline 11a. At 24 hours, trace amounts of the imine 12a were observed, and by 5 days 12a was clearly visible in the reaction mixture. The isomers of imine 12a (3:1 ratio) were identified via the imine proton (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) observed as two doublets at δ 7.75 (major, J = 5.2 Hz) and 7.77 (minor, J = 4.4 Hz). Interestingly, no detectable levels of the released coumarin 13a were observed in CDCl3, even after 5 days. This indicates that unlike the example of Shea (no triazoline detected),24 our benzyloxycarbonyl triazoline 11a and imine 12a are stable in organic solvent. If residual acid was not removed from the CDCl3 (dependent on storage time of CDCl3 bottle) by filtration through a short plug of basic alumina, direct conversion of probe 8a to the coumarin 13a occurred, with no evidence of the intermediate imine 12a in the 1H NMR spectrum.
CH) observed as two doublets at δ 7.75 (major, J = 5.2 Hz) and 7.77 (minor, J = 4.4 Hz). Interestingly, no detectable levels of the released coumarin 13a were observed in CDCl3, even after 5 days. This indicates that unlike the example of Shea (no triazoline detected),24 our benzyloxycarbonyl triazoline 11a and imine 12a are stable in organic solvent. If residual acid was not removed from the CDCl3 (dependent on storage time of CDCl3 bottle) by filtration through a short plug of basic alumina, direct conversion of probe 8a to the coumarin 13a occurred, with no evidence of the intermediate imine 12a in the 1H NMR spectrum.
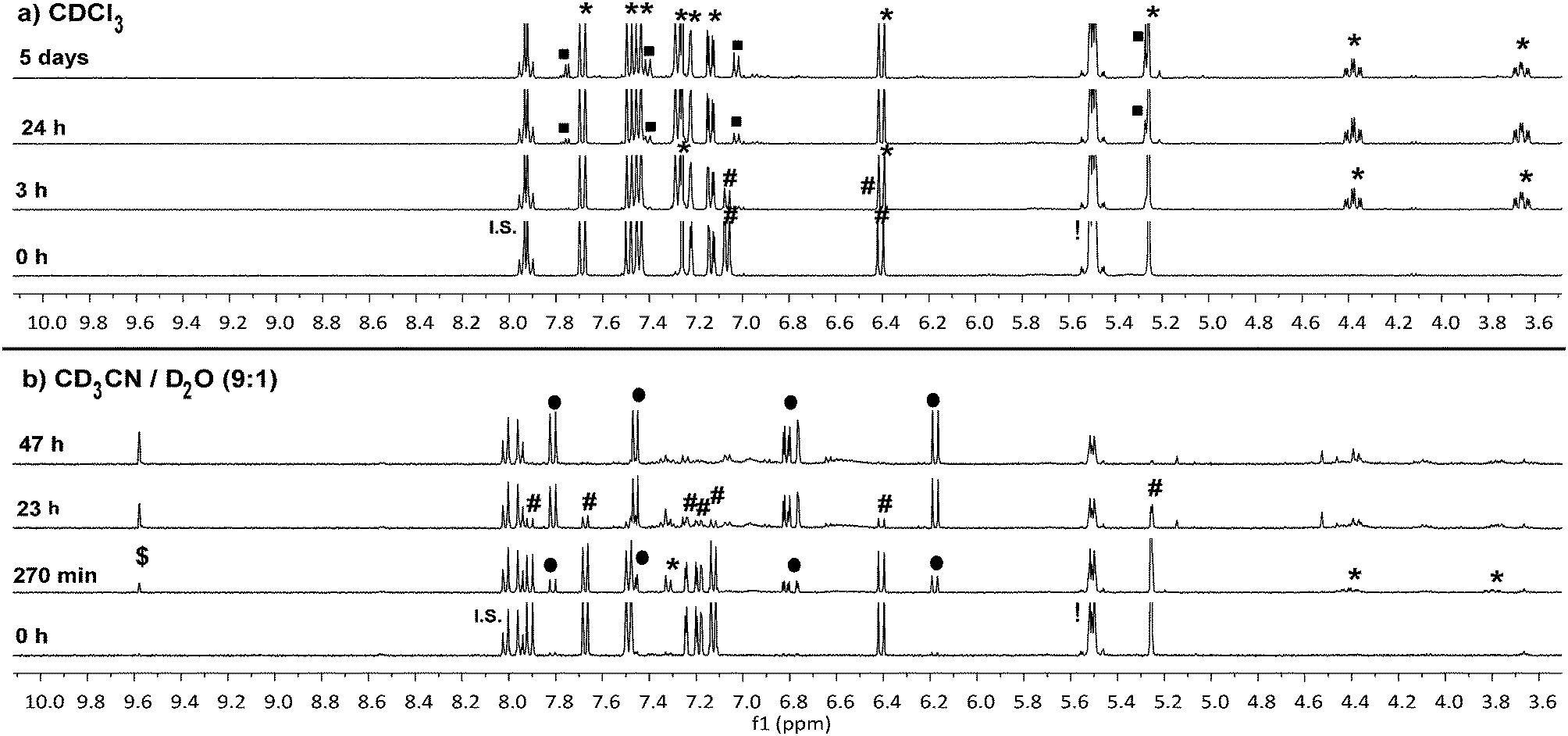 | ||
| Fig. 3
1H NMR experiments in: (a) CDCl3 (filtered through basic alumina) and (b) CD3CN/D2O (9:1); monitoring 1,3-dipolar cycloaddition between TCO 2 and probe 8a. Legend: key structural proton shifts illustrated; #: coumarin probe 8a; *: triazoline 11a; ■: aldimine 12a; ●: 7-hydroxycoumarin 13a; $: exocyclic aldehyde 14a; !: TCO 2; I.S.: Internal Standard, 4-iodonitrobenzene. | ||
In a slightly more protic environment, CD3CN/D2O (9:1), coumarin 13a was rapidly released, with trace amounts immediately observed upon mixing (time = 0 h). At 47 hours none of the initial probe 8a remained, and the major peaks observed were due to the release of coumarin 13a and the imine hydrolysis product, aldehyde 14a. In this solvent system the internal standard masked the area where the major and minor imine protons were expected, but the absence of two doublets corresponding to the aromatic protons of the imine 12a, and the presence of the aldehyde peak at δ 9.58 (d, J = 1.2 Hz) indicated that the imine was rapidly hydrolyzed in the presence of water. The NMR studies indicate that water or acid is essential for protonation and conversion of the triazoline 11a/b and imine 12a/b intermediates to the PABC-analogue. As expected, the PABC-analogue is not observed as it undergoes a rapid 1,6-elimination to release coumarin 13a and aldehyde 14a/b. The ketamine and aziridine intermediates analogous to that reported for Shea's trinitrophenylazide derivatives 5 and 6 (Scheme 1)24 were expected in small quantities, however, apart from possible trace amounts (see aromatic region of 6.5–7.6 ppm in Fig. S14†), they were not observed in the 1H NMR spectra. This indicates that the thermodynamically favored aldimine 12a dominates during the triazoline degradation. Based on our data from the spectrofluorometry and NMR mechanistic studies, the reaction favors the synthesis of the desired and labile aldimine (≥90% conversion), making it a strong candidate for orthogonal protecting group chemistry and bioorthogonal prodrug activation.
The fluorescent release and NMR studies show that TCO 2 and TCO-OH 10 activation of 8a/8b was responsible for the in situ generation of an aldimine with rapid release of coumarin 13a/13b in protic solvents. However, the experiments did not give an idea as to the speed for the initial 1,3-dipolar cycloaddition in protic solvents. Using RP-HPLC we were able to measure the pseudo first-order rate of the 1,3-dipolar cycloaddition in CH3CN:PBS (1:1, 37 °C) via the disappearance of probe 8a at 254 nm and calculate the second-order rate constant (see Fig. S6 and S7†). In the presence of the TCO-OH major-10 (equatorial isomer) or minor-10 (axial isomer), the rate of the 1,3-dipolar cycloaddition was measured as 0.017 M−1 s−1 ± 0.003 and 0.027 M−1 s−1 ± 0.006, respectively. The faster rate for minor-10 can be attributed to the higher energy in the axially substituted ring,31 and will be important for future design of activating molecules. For comparison, the rate of the 1,3-dipolar cycloaddition between probe 8b and TCO major-10 was calculated as 0.020 M−1 s−1 ± 0.0002 (CH3CN:PBS, 1:1), indicating that the presence of a carbamate linker does not affect the rate of cycloaddition (Fig. S8†).
Promisingly, even without modifications to the aromatic ring of the PABC linker or the TCO-OH ring, the rates are comparable to the first generation of SPAAC reactions (100–10−3 M−1 s−1)15a,15d and are an order of magnitude faster than the Staudinger ligation (10−3 M−1 s−1).15a Comparison of our reaction rates to those of Shea24 demonstrate that with the highly electron-deficient tri-nitro substituted aromatic ring, a rate of 0.687 M−1 s−1 in CDCl3 is achieved. While our rate for the cycloaddition is an order of magnitude slower than that of Shea, synthesis of optimized linkers which have been substituted with electron-withdrawing functional groups should enable us to reach or improve on these rates, particularly in an aqueous environment.
While the 1,3-dipolar cycloaddition is considered the key rate-determining step in our approach, the triazoline and imine degradation, and the final 1,6-elimination also contribute to the overall rate of drug/probe release. Therefore, our next goal was to investigate the rate of triazoline and imine degradation. Based on the spectrofluorometry results (Fig. 2) we did not expect the rates of the triazoline rearrangement and subsequent imine hydrolysis to be influenced by the nature of the para-substituted benzyloxy linker (carbonate vs. carbamate drug/probe), thus we selected the more stable carbamate probe 8b to investigate these steps (ESI Section 4†). To enable us to directly measure release of the 7-amino-4-methylcoumarin 13b from the triazoline intermediate, the 1,3-dipolar cycloaddition of 8b with TCO major-10 was monitored by 1H NMR spectroscopy in CD3CN and DMSO-d6 (Fig. S3†). After 19 h, 1H NMR analysis indicated that all of probe 8b had been consumed and converted to the triazoline intermediate 11b (diastereomers), with no imine, aziridine or 7-amino-4-methylcoumarin 13b observed. An aliquot of the NMR sample was then diluted (1000-fold) into PBS and the rate of triazoline and imine degradation was measured by the appearance of 13b on the spectrofluorometer (Fig. S4† ex 360, em 455). By 1 h 88% of 13b had been released, and over the first 40 min, the degradation and release process (three steps in total) appeared to follow pseudo first-order kinetics with a half-life of 19 min (Fig. S5†). While we cannot delineate the rates of the three processes, the kinetics suggest that one of the steps, either triazoline degradation or imine hydrolysis is rate-limiting in a polar protic solvent. The increase in entropy and loss of CO2 provides a significant thermodynamic driving force for the 1,6-elimination,35 thus this is not expected to be rate-determining at a pH of 7.4 (t1/2 = 17 s),26 and is independent on the pKa of the leaving group.36
Our mechanistic studies with probe 8a and 8b demonstrate that there are likely two steps which are rate determining in our prodrug activation strategy, the 1,3-dipolar cycloaddition and either the triazoline or imine degradation. However, as we progress to in vitro and in vivo pre-targeting studies, the rates of triazoline and imine degradation become less significant. Both intermediates would be fixed to the tumor cell surface, unable to diffuse away from the tumor and exhibit off-target effects, thus demonstrating the importance for selective and rapid reactivity in the initial 1,3-dipolar cycloaddition. This will in effect determine how much drug is released at the pre-targeted tumor.
Next we examined the bioorthogonal potential of our strategy via the activation of our doxorubicin-based prodrug 9 with TCO-OH 10 (Scheme 4) in a model murine melanoma cell line (Table 1 and Fig. S16†). TCO-OH 10 was used in place of TCO 2, due to its better solubility, low volatility, and potential to attach a targeting ligand in the future.
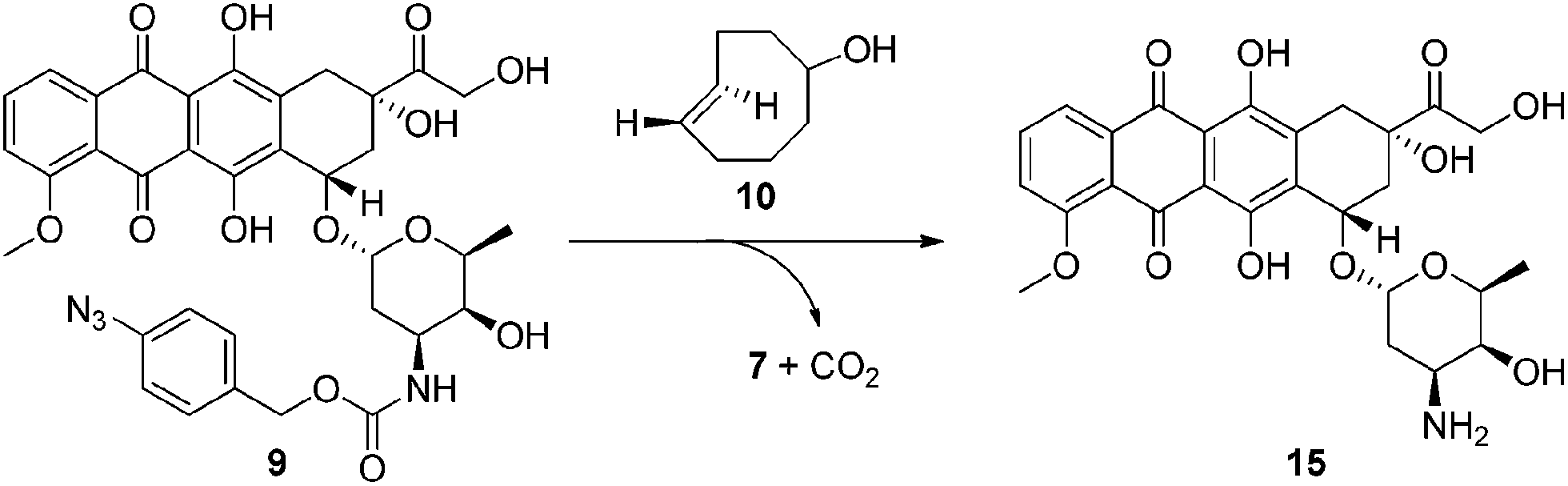 | ||
| Scheme 4 In vitro activation of prodrug 9. | ||
| Compound | IC50a,b,c (μM) |
|---|---|
|
a IC50 = concentration required to kill 50% of cells.
b 95% confidence interval (n ≥ 6) is shown in parenthesis.
c Cell survival at 100 μm of CCO-OH, TCO-OH 10, major-10, minor-10 was 98%, 101%, 97% and 92%, respectively.
d Mixture of major and minor diastereomers (1.42:1).
|
|
| Doxorubicin 15 | 0.71 (0.66–0.77) |
| Dox-prodrug 9 | 49.9 (42.5–58.5) |
| 9 + cis-cyclooctenol (100 μM) | 55.0 (38.0–79.5) |
| 9 + trans-cyclooctenold10 (100 μM) | 0.96 (0.91–1.01) |
| 9 + major-10 (100 μM) | 1.47 (1.36–1.60) |
| 9 + minor-10 (100 μM) | 1.34 (1.27–1.42) |
| 9 + major-10 (10 μM) | 4.98 (4.49–5.52) |
Against the melanoma cell line (Table 1 and Fig. S16†), cytotoxicity of prodrug 9 was low (IC50: 49.9 μM) compared to the parent drug 15 (IC50: 0.71 μM). This indicates that the azido-PABC linker deactivates 15 and is stable in vitro. Combining 9 with TCO-OH 10 (100 μM), itself non-toxic to cells at this concentration (Table 1 and Fig. S15†), restored activity of the prodrug via release of 15 (IC50: 0.96 μM). CCO-OH, also non-toxic at 100 μM, had no effect on release of 15 from 9 (IC50: 55.0 μM), showing that bioorthogonal activation only occurs in the presence of TCO-OH 10. Utilizing major-10 and minor-10 for activation resulted in no significant difference in IC50. Next we examined the cytotoxicity of 9 when activated with a more biologically relevant concentration of TCO-OH major-10 (10 μM). To our excitement, an IC50 of 4.98 μM was obtained. While the IC50 is approximately 5-fold lower than activation with 100 μM TCO-OH 10, we envisage that release of 15 at a targeted and doxorubicin-sensitive tumor (IC50 in low nanomolar range) could still result in tumor cell death when prodrug 9, or analogues thereof, are present at the tumor in the low micromolar range. From the current experiments we do not know where the activation and release occurs, but suspect that it is occurring both inside and outside the cells. However, from the experiments we do know that release of 15 is promoted by the 1,3-dipolar cycloaddition, and that in our proposed in vivo targeting strategy (Fig. 1) release of 15 would be expected to occur outside the cell followed by diffusion of the cytotoxic drug into the closely located tumor cells.
Importantly, with only a 5-fold difference, improving the rate of the cycloaddition (via modifications to the aromatic ring of the azido-PABC linker24,37 and TCO-OH 10 ring) could lead to higher release of 15 at even lower activation concentrations of TCO-OH. Assuming one to six TCOs can be attached to each monoclonal antibody7 which then binds to approximately 105 cell surface receptors,6 we estimate that a concentration of 0.4 to 2.5 μM TCO would be present on the tumor cell surface (assuming cell volume of 400 femto litre).38 While prodrug 9 is significantly less toxic than the parent drug 15 (70-fold) and could be administered in relatively high doses, it will be important to improve the rate of cycloaddition for our future targeted in vivo studies, particularly when the number of activating TCO molecules on targeting ligands and therefore the concentration on the cell surface is limited.7 A faster rate for the cycloaddition will also help to overcome the relatively rapid clearance rates observed in mice (t1/2 = minutes) for small molecules such as doxorubicin and its prodrugs,39 without the requirement for larger and continuous dosing.
With proof-of-concept demonstrated in vitro, we next evaluated the stability and activation of prodrug 9 in mouse serum so as to determine its suitability for in vivo studies (ESI Section 8†). While aryl azides can be prone to thiol reduction,40–42 we expected this to be minimal for prodrug 9 as the most cell-relevant species (GSH and cysteine) require very high levels to carry out azide reduction.40,41 To confirm azide stability we examined the degradation of 9 (100 μM) in 50% mouse serum:PBS and PBS only (Table S1 and S2†). Prodrug 9 (tR = 8.7 min) was relatively stable in mouse serum (at 37 °C), with 95%, 68%, and 56% intact at 4, 24 and 53 h, respectively (Table S1†). The amount of doxorubicin 15 (tR = 4.2 min) released in the absence of TCO-OH 10 was also measured, and at 53 h, only 6% was observed (Table S2†), indicating that most of the prodrug 9 degradation was not due to azide reduction and subsequent 1,6-elimination of 15. Activation of 9 in serum via the addition of TCO-OH major-10 (500 μM) resulted in rapid release of 15 (51% in 4 h, Table S2†). The amount of 15 which had been released following activation decreased over time (4.7% at 48 h, Table S2†), and correlated to an increase in a new peak (tR = 7.6 min, Fig. S23†). The decrease in 15 and appearance of the new peak was not observed in the control (PBS only) activation experiment, with 79% of 15 observed at 48 h (Fig. S20, S24 and Table S2†), indicating that free doxorubicin is slowly metabolized in the serum or binds more strongly than prodrug 9 to serum protein. The peak at 7.6 min was very weak in the serum stability assay, confirming that the product is a metabolite of 15 and not prodrug 9. We are also aware that TCO-OH 10 could undergo slow isomerization back to the unreactive CCO-OH, however, future in vivo studies will use strategically designed linkers attached to targeting ligands (e.g. antibodies) that are known to stabilize the strained TCO-OH in vivo.8,21
Importantly, our serum studies are in contrast to the other potential azide prodrug activation strategy, the Staudinger ligation, in which a byproduct of the phosphine formed in the serum resulted in severely reduced reaction efficiency.30 Examining the in vivo SPAAC reported by Bertozzi14 and Robillard43 indicated that strong covalent and non-covalent interactions to serum proteins could potentially reduce the effective concentration, and therefore reactivity, of our prodrug 9 at the pre-targeted tumor. This could be detrimental to any pre-targeting strategy as the strong serum binding in the case of cyclooctyne resulted in even lower levels of in vivo reactivity than the sluggish Staudinger ligation.14 While both groups reported that their studied cyclooctynes (the reagent added after tumor pre-targeting) suffered from reduced in vivo reaction rates because of strong covalent and non-covalent interactions with serum proteins,14,43 the reactivity of our reagents did not appear to be hindered in the presence of the mouse serum proteins (we observed 51% of doxorubicin 15 being released in 4 h, Table S2†). The rate was slightly faster in serum:PBS than in PBS alone (34% release of doxorubicin 15 in 4 h, Table S2†), providing further evidence that in vivo serum protein binding of prodrug 9 should be minimized (lower binding affinity than the cyclooctynes used in SPAAC).14,43 Promisingly, the second order rate of 1,3-dipolar cycloaddition for prodrug 9 (0.1 mM) with TCO major-10 (10 mM) measured under pseudo first-order reaction conditions in 50% serum:PBS (Fig. S25†) was calculated to be 0.137 ± 0.012 M−1 s−1. This is an order of magnitude faster than that observed for the model probes 8a and 8b in an acetonitrile:PBS mixture (1:1), and two orders of magnitude faster than the Staudinger ligation, demonstrating that we can expect faster 1,3-dipolar cycloadditions under increasingly aqueous conditions.44
Conclusions
To the best of our knowledge there are no examples of in vivo click reactions for chemically triggered release of drugs, making our work reported here and that of Robillard11,45 all the more significant. Although challenges await as we progress to in vivo pre-targeting studies, we have demonstrated, for the first time, a strain promoted 1,3-dipolar cycloaddition with potential use in prodrug activation, imaging applications and orthogonal protecting group strategies. The reaction proceeds with a second-order rate equivalent to the first generation SPAAC and is 1–2 orders of magnitude faster than the Staudinger reaction enabling rapid orthogonal deprotection of azido-PABC protected amino and hydroxyl groups under mild conditions. While azido-PABC carbonate-linked drugs are unlikely to be stable in a biological milieu, the potential of azido-PABC carbamate drugs for bioorthogonal prodrug activation is demonstrated in a melanoma cell line, with the model doxorubicin prodrug 9 remaining deactivated until reaction with TCO-OH 10, upon which it regains the cytotoxicity of the parent drug 15. The prodrug also demonstrates good stability in mouse serum over biologically relevant circulation times for cytotoxic drugs (minutes to hours) and due to the rapid reactivity observed in serum:PBS, the azido-prodrug 9 does not appear to bind strongly to, or react with, serum proteins like the cyclooctynes and triphenylphosphines of the in vivo SPAAC and Staudinger ligation pre-targeting studies.14,30,43We are currently investigating ways to improve the activation rate of our prodrugs for future in vivo studies with modifications to the PABC linkers and TCO molecules (e.g. electron-withdrawing functionalities such as nitro24 and fluorine groups37). Additionally, we are examining other linkers with azide triggers that can be used as stable alternatives to the carbonate linker, expanding the scope of our activation strategy to both alcoholic and phenolic drugs.
Acknowledgements
This research was funded in part by a University of Otago Research Grant (UORG) and a contract from the Health Research Council (HRC) of New Zealand (A.B.G). The authors would also like to thank Sarah Katzemich for her contributions to the synthesis of the 7-hydroxycoumarin probe.Notes and references
- J. R. Trounce, Br. J. Clin. Pharmacol., 1979, 8, 205–207 CrossRef CAS PubMed.
- I. Collins and P. Workman, Nat. Chem. Biol., 2006, 2, 689–700 CrossRef CAS PubMed.
- R. Mahato, W. Tai and K. Cheng, Adv. Drug Delivery Rev., 2011, 63, 659–670 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Jarvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS PubMed.
- A. Warnecke, Drug Delivery in Oncology: From Basic Research to Cancer Therapy, ed. F. Kratz, P. Senter and H. Steinhagen, Wiley-VCH, Weinheim, Germany, 1st edn, 2011, pp. 553–589 Search PubMed.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- N. K. Devaraj, R. Upadhyay, J. B. Haun, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2009, 48, 7013–7016 CrossRef CAS PubMed.
- R. Rossin, S. M. J. van Duijnhoven, T. Lappchen, S. M. van den Bosch and M. S. Robillard, Mol. Pharmaceutics, 2014, 11, 3090–3096 CrossRef CAS PubMed.
- K. D. Bagshawe, Drug Delivery in Oncology: From Basic Research to Cancer Therapy, ed. F. Kratz, P. Senter and H. Steinhagen, Wiley-VCH, Weinheim, Germany, 1st edn, 2011, pp. 169–186 Search PubMed.
- K.-C. Chen, S.-Y. Wu, Y.-L. Leu, Z. M. Prijovich, B.-M. Chen, H.-E. Wang, T.-L. Cheng and S. R. Roffler, Bioconjugate Chem., 2011, 22, 938–948 CrossRef CAS PubMed.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- N. K. Devaraj, R. Upadhyay, J. B. Haun, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2009, 48, 7013–7016 CrossRef CAS PubMed.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS PubMed.
- For selected reviews on bioorthogonal chemistry see: (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed; (b) N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed; (c) M. F. Debets, S. S. van Berkel, J. Dommerholt, J. Dirks, F. P. J. T. Rutjies and F. L. van Delft, Acc. Chem. Res., 2011, 44, 805–815 CrossRef CAS PubMed; (d) C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007–11022 RSC; (e) M. King and A. Wagner, Bioconjugate Chem., 2014, 25, 825–839 CrossRef CAS PubMed; (f) A. Borrmann and J. C. M. van Hest, Chem. Sci., 2014, 5, 2123–2134 RSC; (g) D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605 CrossRef CAS PubMed.
- M. Azoulay, G. Tuffin, W. Sallem and J.-C. Florent, Bioorg. Med. Chem. Lett., 2006, 16, 3147–3149 CrossRef CAS PubMed.
- R. van Brakel, R. C. M. Vulders, R. J. Bokdam, H. Grull and M. S. Robillard, Bioconjugate Chem., 2008, 19, 714–718 CrossRef CAS PubMed.
- K. Gorska, A. Manicardi, S. Barluenga and N. Wissinger, Chem. Commun., 2011, 47, 4364–4366 RSC.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sanchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 Search PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sanchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- R. Rossin, S. M. van den Bosch, W. ten Hoeve, M. Carvelli, R. M. Versteegen, J. Lub and M. S. Robillard, Bioconjugate Chem., 2013, 24, 1210–1217 CrossRef CAS PubMed.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Angew. Chem., Int. Ed., 2012, 51, 920–922 CrossRef CAS PubMed.
- R. H. Smith Jr, B. D. Wladkowski, J. E. Taylor, E. J. Thompson, B. Pruski, J. R. Klose, A. W. Andrews and C. J. Michejda, J. Org. Chem., 1993, 58, 2097–2103 CrossRef.
- K. J. Shea and J.-S. Kim, J. Am. Chem. Soc., 1992, 114, 4846–4855 CrossRef CAS.
- P. L. Carl, P. K. Chakravarty and J. A. Katzenellenbogen, J. Med. Chem., 1981, 24, 479–480 CrossRef CAS.
- I. A. Muller, F. Kratz, M. Jung and A. Warnecke, Tetrahedron Lett., 2010, 51, 4371–4374 CrossRef PubMed.
- R. A. Gatenby and R. J. Gillies, Nat. Rev. Cancer, 2004, 4, 891–899 CrossRef CAS PubMed.
- D. Neri and C. T. Supuran, Nat. Rev. Drug Discovery, 2011, 10, 767–777 CrossRef CAS PubMed.
- A. S. Bailey and J. E. White, J. Chem. Soc. B, 1966, 819–822 RSC.
- D. J. Vugts, A. Vervoort, M. Stigter-van Walsum, G. W. M. Visser, M. S. Robillard, R. M. Versteegen, R. C. M. Vulders, J. D. M. Herscheid and G. A. M. S. van Dongen, Bioconjugate Chem., 2011, 22, 2072–2081 CrossRef CAS PubMed.
- M. Royzen, G. P. A. Yap and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 3760–3761 CrossRef CAS PubMed.
- J.-P. Goddard and J.-L. Reymond, Curr. Opin. Biotechnol., 2004, 15, 314–322 CrossRef CAS PubMed.
- Y. Meyer, J.-A. Richard, M. Massonneau, P.-Y. Renard and A. Romieu, Org. Lett., 2008, 10, 1517–1520 CrossRef CAS PubMed.
- W. Gao, B. Xing, R. Y. Tsien and J. Rao, J. Am. Chem. Soc., 2003, 125, 11146–11147 CrossRef CAS PubMed.
- H. Y. Lee, X. Jiang and D. Lee, Org. Lett., 2009, 11, 2065–2068 CrossRef CAS PubMed.
- B. M. Sykes, M. P. Hay, D. Bohinc-Herceg, N. A. Helsby, C. J. O'Connor and W. A. Denny, J. Chem. Soc., Perkin Trans. 1, 2000, 1601–1608 RSC.
- F. Schoenebeck, D. H. Ess, G. O. Jones and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 8121–8133 CrossRef CAS PubMed.
- E. H. Chapman, A. S. Kurec and F. R. Davey, J. Clin. Pathol., 1981, 34, 1083–1090 CrossRef CAS.
- H. P. Svensson, V. M. Vrudhula, J. E. Emswiler, J. F. MacMaster, W. L. Cosand, P. D. Senter and P. M. Wallace, Cancer Res., 1995, 55, 2357–2365 CAS.
- J. V. Staros, H. Bayley, D. N. Strandring and J. R. Knowles, Biochem. Biophys. Res. Commun., 1978, 80, 568–572 CrossRef CAS.
- S. Chen, Z. Chen, W. Ren and A. Ai, J. Am. Chem. Soc., 2012, 134, 9589–9592 CrossRef CAS PubMed.
- P. K. Sasmal, S. Carregal-Romero, A. A. Han, C. N. Streu, Z. Lin, K. Namikawa, S. L. Elliot, R. W. Koster, W. J. Parak and E. Meggers, ChemBioChem, 2012, 13, 1116–1120 CrossRef CAS PubMed.
- S. M. van den Bosch, R. Rossin, P. Renart Verkerk, W. ten Hoeve, H. M. Janssen, J. Lub and M. S. Robillard, Nucl. Med. Biol., 2013, 40, 415–423 CrossRef CAS PubMed.
- J. W. Wijnen, R. A. Steiner and J. B. F. N. Engberts, Tetrahedron Lett., 1995, 36, 5389–5392 CrossRef CAS.
- R. Rossin and M. S. Robillard, Curr. Opin. Chem. Biol., 2014, 21, 161–169 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc02574a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2015 |