
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective Lewis base catalyzed formal 1,3-dipolar cycloaddition of azomethine imines with mixed anhydrides†
Lena
Hesping
,
Anup
Biswas
,
Constantin G.
Daniliuc
,
Christian
Mück-Lichtenfeld
* and
Armido
Studer
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster Corrensstrasse 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de; Web: http://www.uni-muenster.de/Chemie.oc/studer/ Fax: +49-251-83-36523; Tel: +49-251-83-33291
First published on 19th November 2014
Abstract
Stereoselective synthesis of pyrazolidinones via dipolar cycloaddition of azomethine imines with active esters under Lewis base catalysis is presented. The active esters are readily generated in situ from the corresponding acids. Products, which are obtained with excellent diastereocontrol and high enantioselectivity, contain along with the pyrazolidinone core also the tetrahydroisoquinoline structural motif. Theoretical studies give insight into the mechanism of the formal cycloaddition reaction.
Introduction
Many natural products with interesting biological activity are based on the C1-substituted tetrahydroisoquinoline core 1.1 It is obvious that many synthetic methods have been developed for the stereoselective construction of this important core structure.2 Pyrazolidinones 2 are also interesting compounds which have found wide applications in different fields. This structural motif can be found in dyes, in pharmaceuticals and agriculturally relevant compounds.3 Pyrazolidinones of type 3 combine the structural features of both 1 and 2 and should therefore be valuable compounds which have so far not been intensively investigated (Fig. 1).4 Herein we present a novel straightforward method for the stereoselective synthesis of compounds of type 3via 1,3-dipolar cycloaddition of azomethine imines with mixed anhydrides.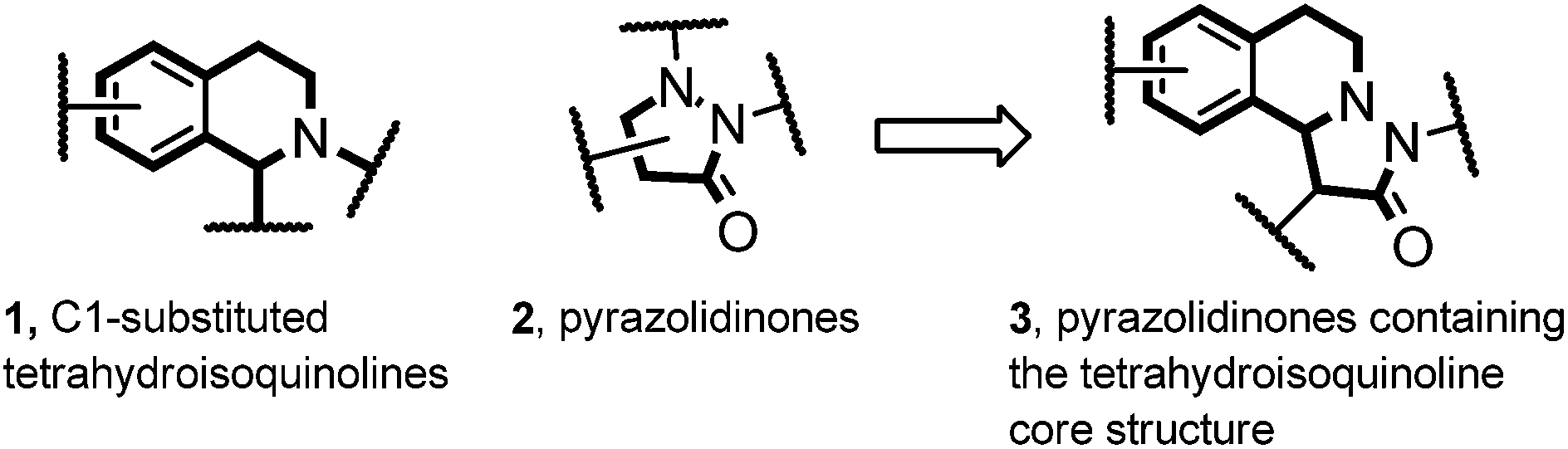 | ||
| Fig. 1 Compounds 3 combining structural features of both 1 and 2. | ||
Results and discussion
Experimental studies
The intermolecular [2 + 3] cycloaddition of azomethine imines with alkynes is known for more than 45 years,5 and an enantioselective version was developed by Fu et al. using Cu-catalysis.6 Chen,7 Sibi,8a Maruoka9 and others8b–d disclosed enantioselective cycloadditions of azomethine imines with electron-poor alkenes and the corresponding enantioselective dipolar cycloaddition with electron-rich alkenes was reported by Leighton10 and Maruoka.11 Very recently, Wang et al. published amine-catalyzed enantioselective 1,3-dipolar cycloadditions of aldehydes to C,N-cyclic azomethine imines, where intermediately generated electron-rich enamines are the actual dipolarophiles.12As a continuation of our studies on oxidative carbene catalysis,13 we decided to investigate the reaction of aliphatic aldehydes with azomethine imines in the presence of a N-heterocyclic carbene (NHC) under oxidative conditions as a novel method for the preparation of compounds of type 3. Disappointingly, we found that the azomethine imine 1a reacted with phenylacetaldehyde in the presence of triazole pre-catalyst and DBU under oxidative conditions to cycloadduct 4 (ref. 14) (Fig. 2). The targeted pyrazolidinone 3aa was not identified.
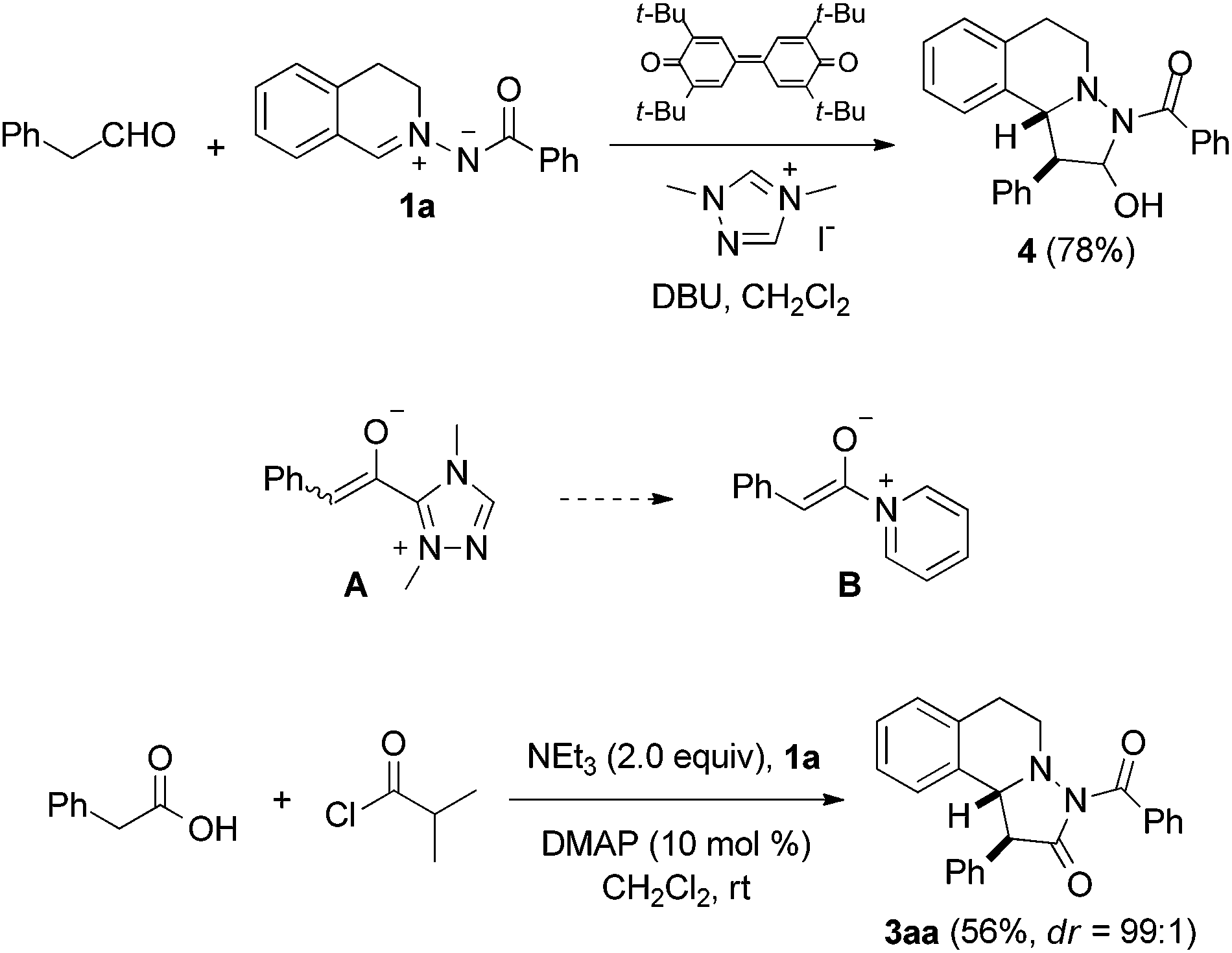 | ||
| Fig. 2 Transformation of 1a to either 4 or 3aa under different conditions. | ||
The reaction of phenylacetaldehyde with the NHC, oxidation to the acylazolium ion and subsequent enolization15 to give an enolate of type A is obviously slower than direct reaction of phenylacetaldehyde with 1a under the applied basic conditions. We therefore switched to enolates of type B as potential dipolarophiles for the [2 + 3] cycloaddition with 1a. It is important to note that stereoselective reactions with enolates formally deriving from acylammonium or acylpyridinium ions have been intensively studied in asymmetric catalysis.16 However, the application of such enolates as dipolarophiles in the reaction with azomethine imines is unknown.
Pleasingly, the mixed anhydride in situ generated from phenylacetic acid and isobutyric acid chloride in the presence of NEt3 reacted with 1a and DMAP (10 mol%) to pyrazolidinone 3aa which was isolated in 56% yield as a 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers, as analyzed by HPLC.17,18 Encouraged by this result, we continued the studies by testing the chiral commercially available Lewis bases C,19D,20 and E,20 which have successfully been used in asymmetric catalysis (Fig. 3).21,22
:1 mixture of diastereoisomers, as analyzed by HPLC.17,18 Encouraged by this result, we continued the studies by testing the chiral commercially available Lewis bases C,19D,20 and E,20 which have successfully been used in asymmetric catalysis (Fig. 3).21,22
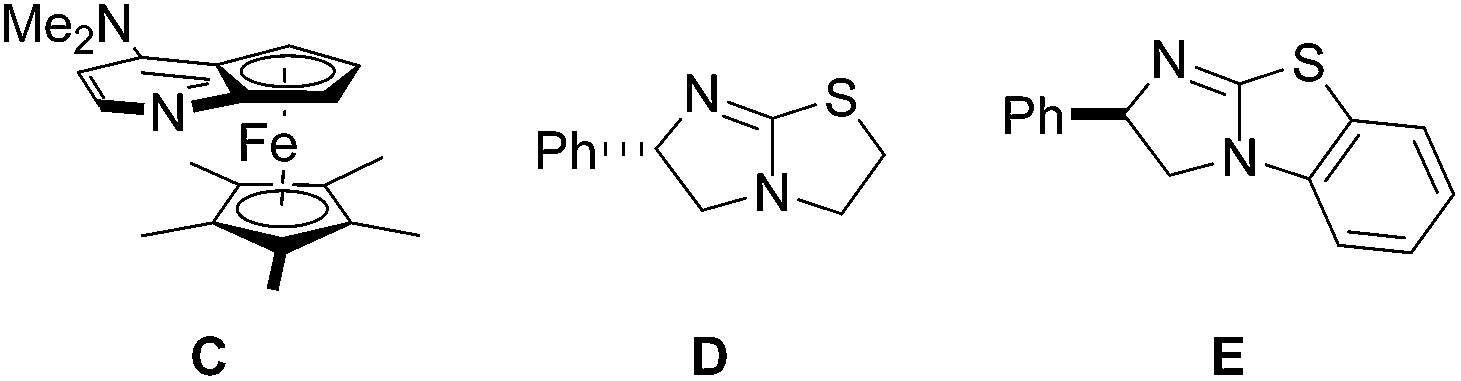 | ||
| Fig. 3 Chiral Lewis bases C, D and E used in this study. | ||
With catalyst C, 3aa was obtained in moderate to good yield with high diastereoselectivity (95:5) but no or very low enantioselectivity by using i-Pr2NEt or Et3N as base (Table 1, no. 1, 2). Pleasingly, tetramisole D (10 mol%) provided 3aa with significant ee (56%) but low diastereoselectivity (67:33) which was improved to 91:9 upon increasing the catalyst loading to 20 mol% (Table 1, no. 3, 4). Likely, back ground reaction, which is cycloaddition of the free ketene with the azomethine imine, competes at lower catalyst loading. This assumption is supported by the fact that the diastereoselectivity at lower catalyst loading was significantly lower (67:33 versus 91:9) and accordingly also the ee was lower. Enantioselectivity was determined by HPLC analysis (see ESI†). With phenylacetyl chloride as substrate, ee and dr were further improved; however, yield was very low in that case (Table 1, no. 5). As compared to D, catalyst E provided slightly improved selectivities (Table 1, no. 6, 7). From the experiment run with the acid chloride as a substrate it became obvious that the leaving group at the activated acid derivatives may play an important role on the selectivity. Therefore, we tested the in situ generated mixed anhydride derived from 2,4,6-trimethylbenzoyl chloride. Disappointingly, both diastereoselectivity and enantioselectivity decreased (Table 1, no. 8). However, the mixed anhydride formed from benzoyl chloride gave 3aa with excellent ee (99%) and high diastereoselectivity (94:6) in good yield (Table 1, no. 9). We found that reaction works far more efficiently at room temperature without diminishing selectivity to a large extent and pyrazolidinone 3aa was obtained in 95% yield, 94:6 diastereoselectivity with 98% ee (Table 1, no. 10). The absolute and relative configuration of 3aa were unambiguously assigned by X-ray analysis (Fig. 4).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| No. | R | Cat. (mol%) | Base | Temp (°C)/Time (h) | dr | ee (%) | Yield (%) |
| a With 2.1 equiv. base. b With 1.1 equiv. base. c Other enantiomer formed as major isomer. d Phenylacetyl chloride used as substrate. | |||||||
| 1 | i-Pr | C (10) | Et3Na | rt/16 | 95:5 |
6 | 46 |
| 2 | i-Pr | C (10) | i-Pr2NEta | rt/14 | 95:5 |
rac | 71 |
| 3 | i-Pr | D (10) | i-Pr2NEtb | 0/23 | 67:33 |
56c | 90 |
| 4 | i-Pr | D (20) | i-Pr2NEta | 0/23 | 91:9 |
76c | 74 |
| 5 | —d | D (10) | i-Pr2NEtb | 0/24 | 99:1 |
92c | 12 |
| 6 | i-Pr | E (10) | i-Pr2NEta | 0/43 | 95:5 |
64 | 64 |
| 7 | i-Pr | E (10) | i-Pr2NEtb | rt/43 | 94:6 |
84 | 86 |
| 8 | Mes | E (10) | i-Pr2NEtb | 5/24 | 83:17 |
32 | 33 |
| 9 | Ph | E (10) | i-Pr2NEtb | 5/24 | 94:6 |
99 | 68 |
| 10 | Ph | E (10) | i-Pr2NEtb | rt/16 | 94:6 |
98 | 95 |
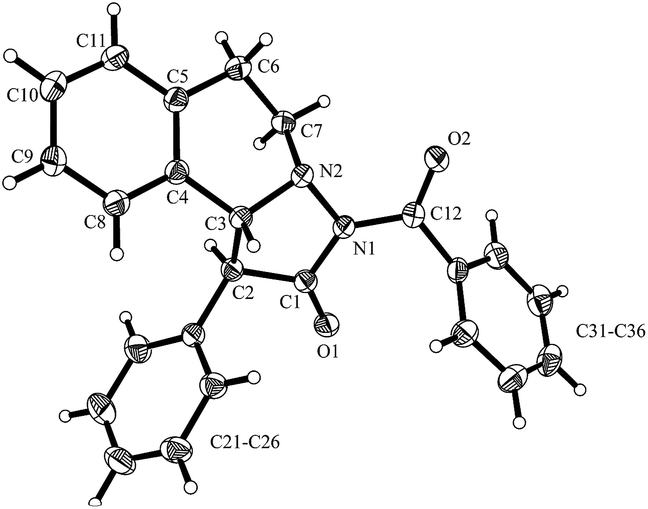 | ||
| Fig. 4 X-ray structure of pyrazolidinone 3aa. Thermal ellipsoids are shown with 30% probability. | ||
With optimized conditions in hand, we tested scope and limitation of the stereoselective cycloaddition by varying the acid component and also the azomethine imine (Fig. 5 and 6). All azomethine imines used in this study were prepared according to literature procedures (see ESI†). Phenylacetic acids with both electron-donating and electron-withdrawing groups were tolerated to afford cycloaddition products 3ac–3ag in good to high yields and high enantio- and diastereoselectivity. Furthermore, the sterically demanding o-tolylacetic acid could be utilized furnishing 3ab with high ee and dr. Excellent selectivity and good yield were achieved with 2-naphthylacetic acid as the substrate (product 3ah, >99% ee, dr = 98:2, 82% yield). Additionally, the heteroaromatic substrate thiopheneacetic acid worked well providing cycloadduct 3ai in good yield and excellent diastereoselectivity, albeit with slightly lower ee.
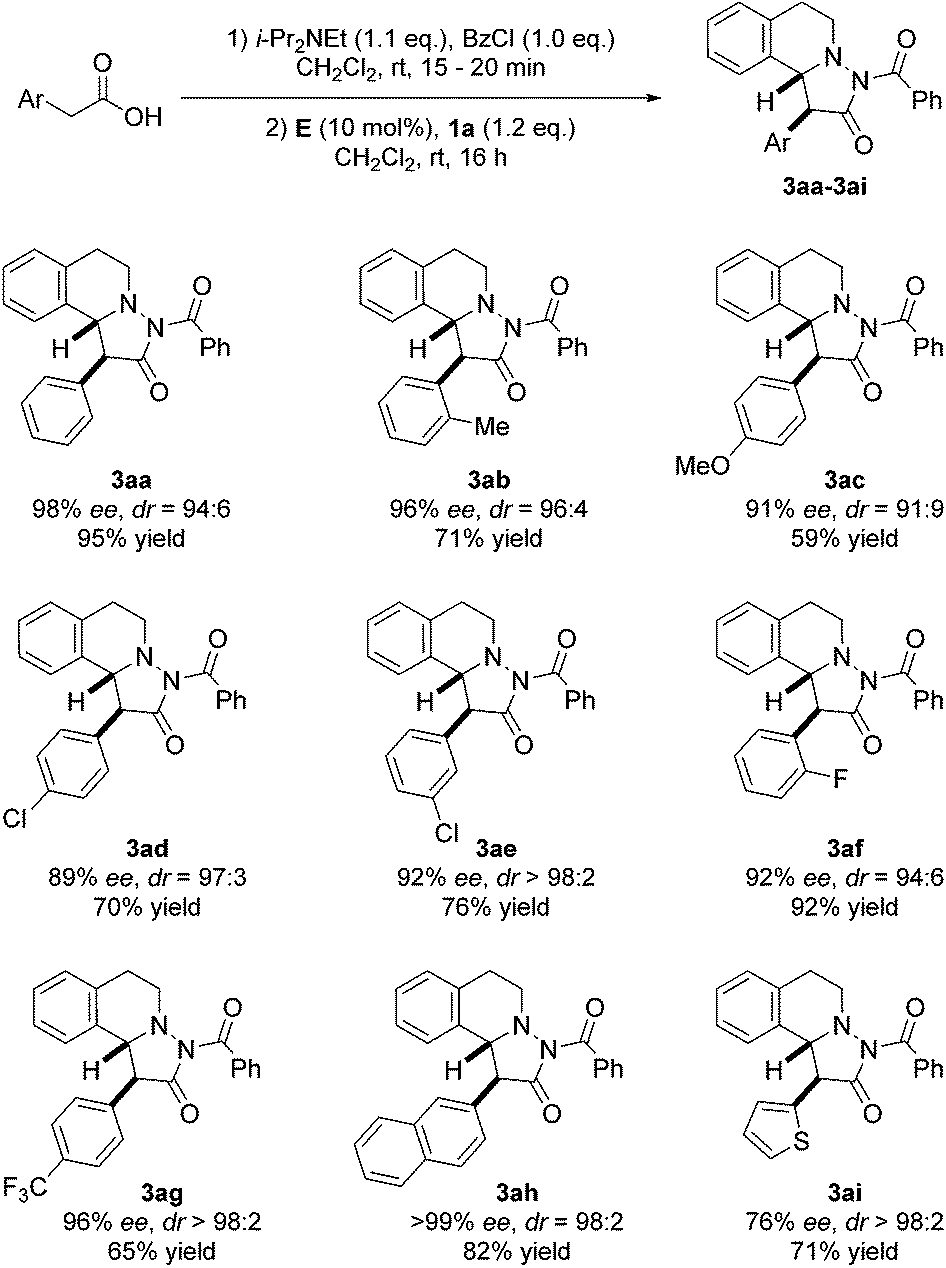 | ||
| Fig. 5 Variation of the acetic acid component. | ||
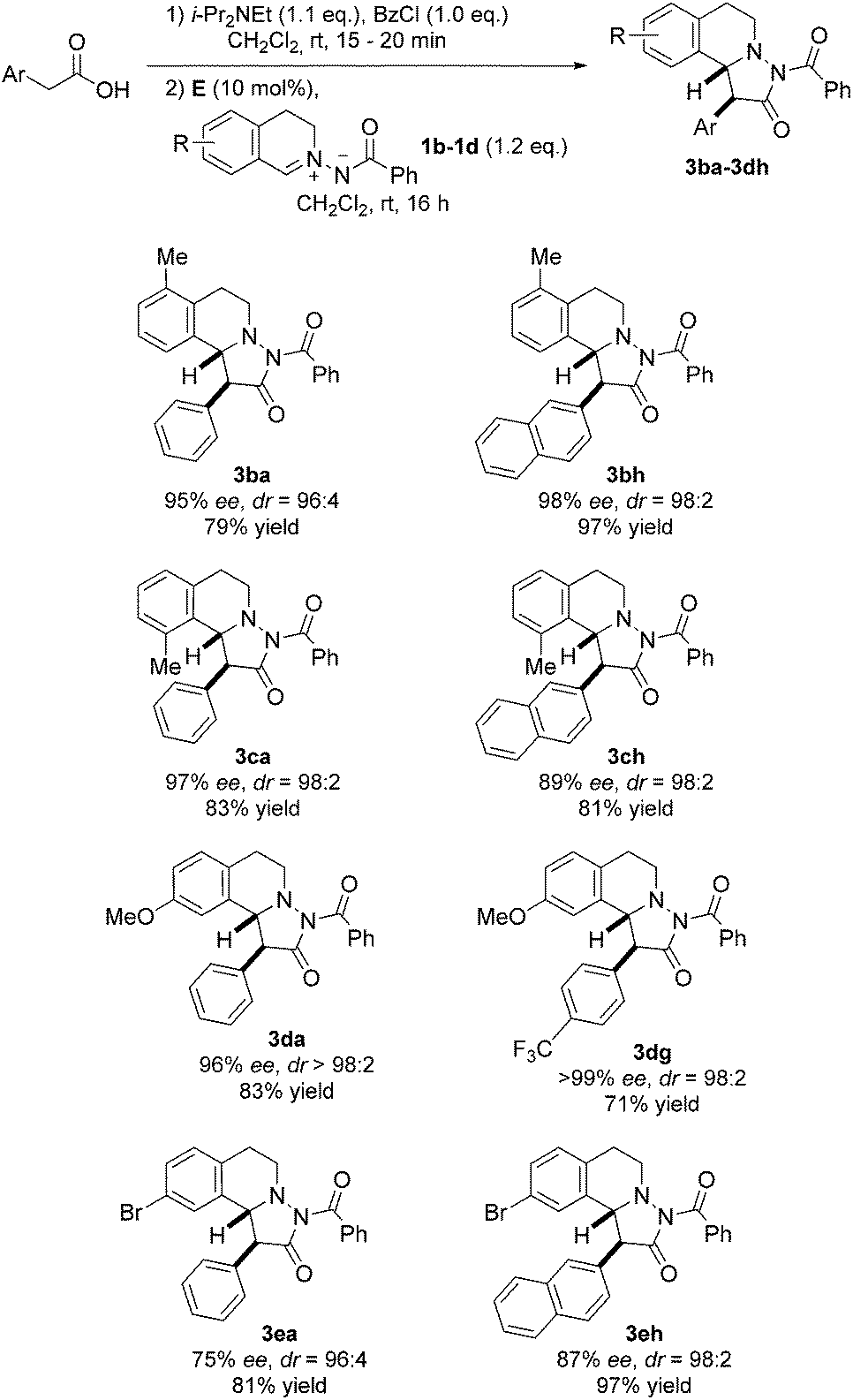 | ||
| Fig. 6 Variation of the azomethine imine and the acetic acid component. | ||
Next, differently substituted azomethine imines were tested in the reaction with various acetic acid derivatives (Fig. 6). Good to high yields were obtained by using 7-bromo and 7-methoxy-substituted azomethine imines 1d and 1e (see 3da–3eh). The electron-rich azomethine imine 1d provided significantly higher ee as compared to the ee obtained with the Br-derivative 1e. Along these lines, 5-methyl-substituted azomethine imine 1b afforded pyrazolidinones 3ba and 3bh in high selectivities and good to excellent yields. Moreover, the sterically hindered 8-methyl-substituted azomethine imine 1c was a good substrate and cycloadducts 3ca and 3ch were obtained in good to excellent selectivities with good yields.
All attempts to conduct the cycloaddition of the chiral ammonium enolate derived from phenylacetic acid with in situ generated C,N-cyclic azomethine imines according to the elegant work of Maruoka et al.9 failed. Moreover, the N,N-cyclic azomethine imine 2-benzylidene-5-oxopyrazolidin-2-ium-1-ide, often used in dipolar cycloaddtions,6–8 did not react with the in situ generated ammonium enolate under the tested conditions.
To show the value of pyrazolidinones as building blocks in synthesis, we tested a first follow-up reaction. To this end, N-benzoyl deprotection in pyrazolidinones 3aa and 3da was achieved with DBU/LiBr23 in MeOH to give the corresponding NH-pyrazolidinones. N–N bond cleavage by reduction with Raney-Ni/H2 eventually provided stereospecifically the tetrahydroisoquinolines 5aa and 5da in good yields (Fig. 7). These β-amino acid derivatives might be valuable for preparation of β-peptides.24
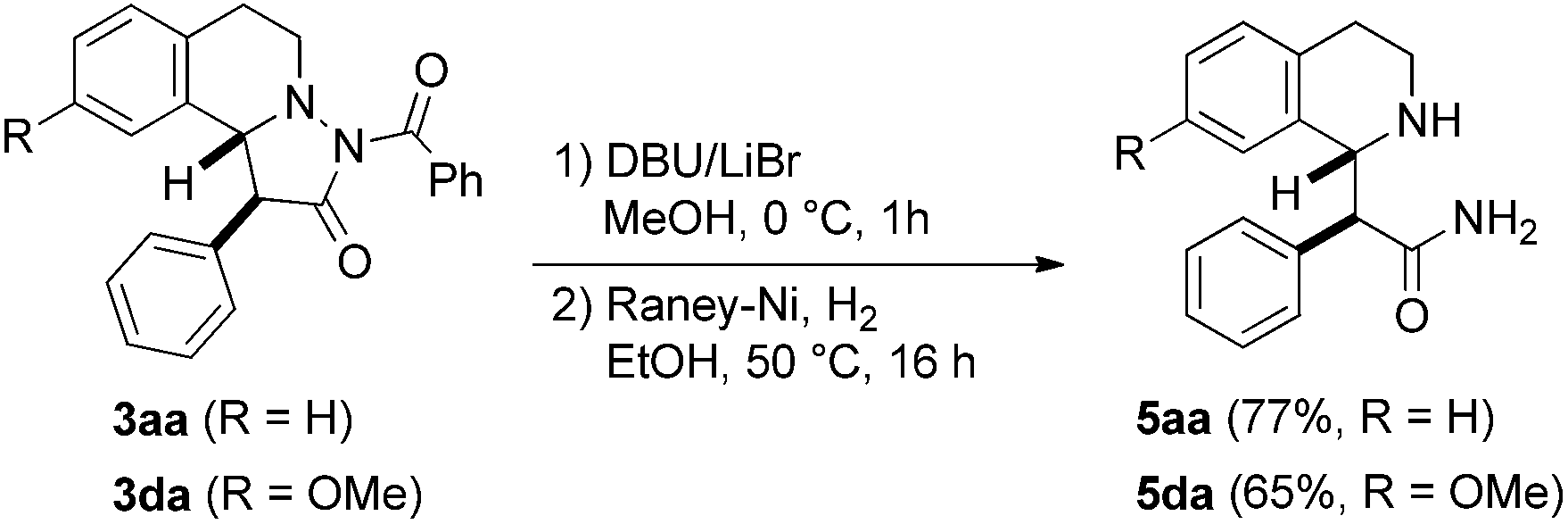 | ||
| Fig. 7 Reductive cleavage of the N–N bond for preparation of β-amino acid derivatives. | ||
Theoretical studies of the mechanism
In our DFT study of the mechanism of the cycloaddition reaction, we used TPSS-D3/def2-TZVP and an implicit solvation model (COSMO), for details, see ESI.† We chose the reaction of intermediate F deriving from phenylacetic acid and catalyst E with azomethine imine 1a as our model system. The free energies, including solvation energies in CH2Cl2 and thermodynamic corrections at 298 K starting from the enolate F and 1a are given in Fig. 8.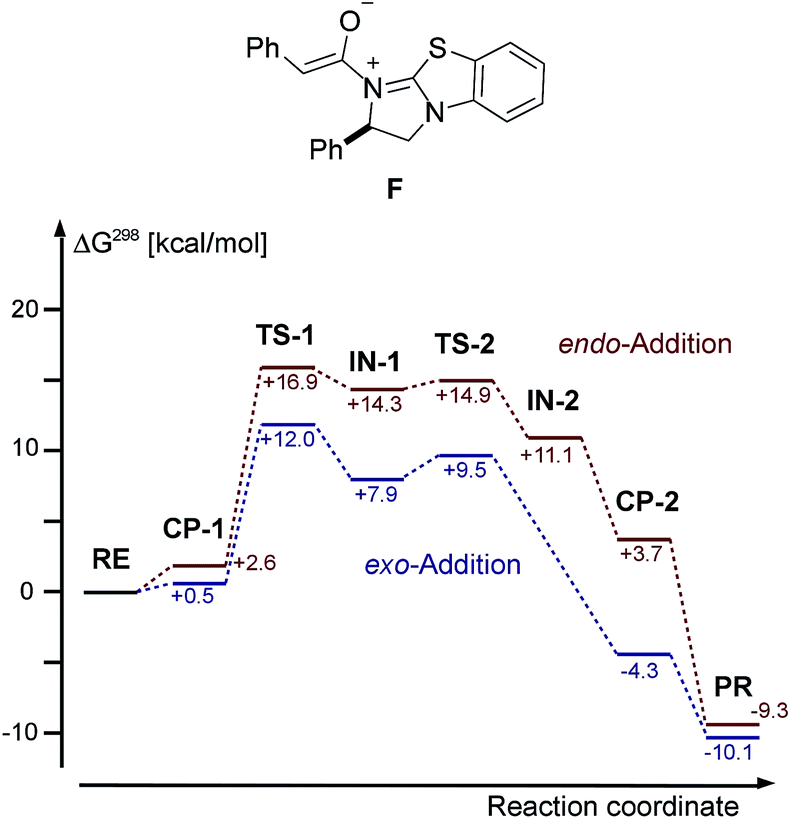 | ||
| Fig. 8 Chemical structure of enolate F and DFT calculated (TPSS-D3/def2-TZVP + COSMO (CH2Cl2) free energies of the cycloaddition of F and 1a. RE = 1a/F, PR = 3aa/E. | ||
We were not able to locate a transition structure of synchronous formation of both bonds (C–C and C–N) of the product (3aa). The mechanism proceeds stepwise with C–C bond formation as the first step (TS-1), leading to an intermediate (IN-1), which subsequently forms the second C–N bond with a very low barrier. Thus, the first step is determining the rate and the stereochemical outcome of the process. The preferred orientation of the two reactants in the pre-reactive complex CP-1 and for TS-1 is exo (see Fig. 9), in accordance with the observed diastereoselectivity of the reaction. Moreover, the absolute stereochemistry obtained in the calculations agreed with the stereochemistry observed in the experiment. In the exo reaction, we could not identify a tetrahedral intermediate IN-2 as for endo, the catalyst E is released instantaneously upon formation of the C–N bond of 3aa. The breakup of the product complex CP-2 releases product 3aa, of which the trans-diastereoisomer is also thermodynamically the more stable one.
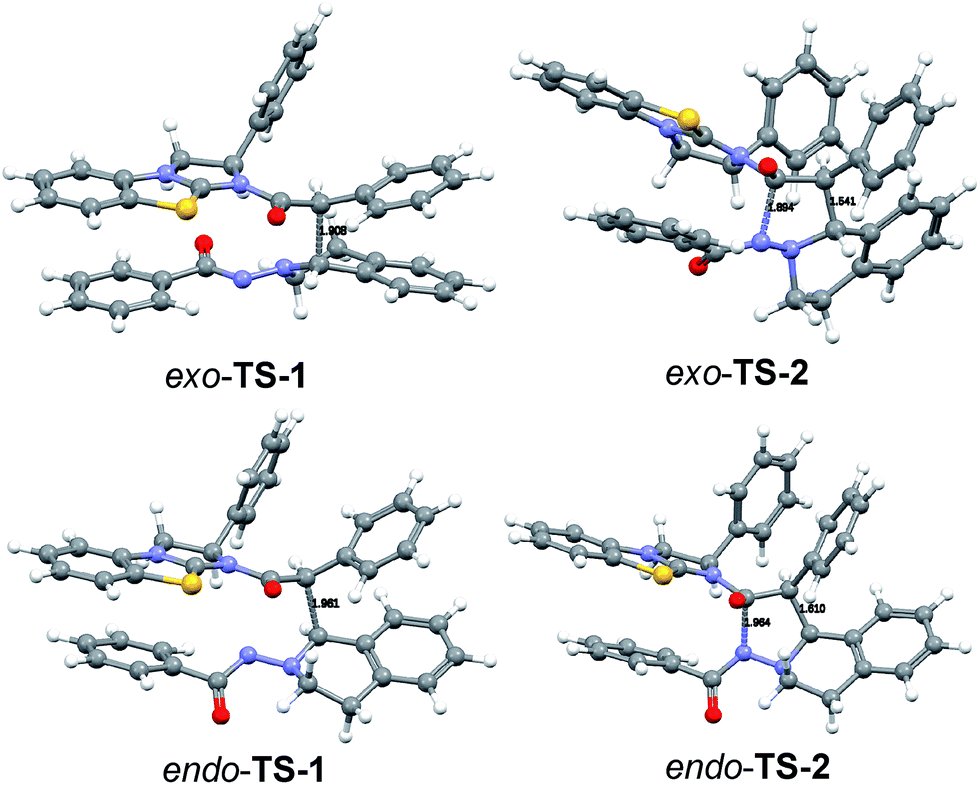 | ||
| Fig. 9 DFT calculated transition structures in the cycloaddition of F and 1a. | ||
Conclusion
In summary, the chiral Lewis base benzotetramisole (E) catalyzes the highly enantio- and diastereoselective formation of complex pyrazilidinones with a tetrahydroisoquinoline core by 1,3-dipolar cycloaddition of C,N-cyclic azomethine imines and activated arylacetic acid derivatives. Reactions proceed in high yields with good to excellent diastereo- and enantioselectivity. Reductive N–N bond cleavage and imide hydrolysis provide β-aminoamides. DFT studies reveal a stepwise mechanism with the formation of the C–C bond as the first step which determines the rate and stereochemical outcome of the formal dipolar cycloaddition. The following C–N bond formation occurs with a low barrier.Acknowledgements
We thank the University of Münster and the Deutsche Forschungsgemeinschaft (DFG) within the SFB 858 (project Z1) for supporting our research. We also thank Ulrich Schreiber for the preparation of some cycloadducts.Notes and references
- (a) G. J. Aune, T. Furuta and Y. Pommier, Anti-Cancer Drugs, 2002, 13, 545–555 CrossRef CAS PubMed; (b) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669–1730 CrossRef CAS PubMed; (c) D. Mujahidin and S. Doye, Eur. J. Org. Chem., 2005, 2005, 2689–2693 CrossRef; (d) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444–463 RSC; (e) F. Werner, N. Blank and T. Opatz, Eur. J. Org. Chem., 2007, 2007, 3911–3915 CrossRef; (f) R. J. Reddy, N. Kawai and J. i. Uenishi, J. Org. Chem., 2012, 77, 11101–11108 CrossRef CAS PubMed.
- (a) I. M. P. Huber and D. Seebach, Helv. Chim. Acta, 1987, 70, 1944–1954 CrossRef CAS; (b) N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916–4917 CrossRef CAS; (c) D. L. Comins, P. M. Thakker, M. F. Baevsky and M. M. Badawi, Tetrahedron, 1997, 53, 16327–16340 CrossRef CAS; (d) M. Chrzanowska and M. D. Rozwadowska, Chem. Rev., 2004, 104, 3341–3370 CrossRef CAS PubMed; (e) Z. Li and C.-J. Li, Org. Lett., 2004, 6, 4997–4999 CrossRef CAS PubMed; (f) M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Angew. Chem., 2005, 117, 6858–6862 CrossRef; (g) N. Sasamoto, C. Dubs, Y. Hamashima and M. Sodeoka, J. Am. Chem. Soc., 2006, 128, 14010–14011 CrossRef CAS PubMed; (h) K. Umetsu and N. Asao, Tetrahedron Lett., 2008, 49, 2722–2725 CrossRef CAS PubMed; (i) M. Chang, W. Li and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 10679–10681 CrossRef CAS PubMed.
- (a) G. J. Blackwell and R. J. Flower, Br. J. Pharmacol., 1978, 63, 360P CrossRef CAS PubMed; (b) H. L. White, J. L. Howard, B. R. Cooper, F. E. Soroko, J. D. McDermed, K. J. Ingold and R. A. Maxwell, J. Neurochem., 1982, 39, 271–273 CrossRef CAS PubMed; (c) L. N. Jungheim, S. K. Sigmund and J. W. Fisher, Tetrahedron Lett., 1987, 28, 285–288 CrossRef CAS; (d) J. M. Indelicato and C. E. Pasini, J. Med. Chem., 1988, 31, 1227–1230 CrossRef CAS; (e) C. Turk, J. Svete, B. Stanovnik, L. Golič, S. Golič-Grdadolnik, A. Golobič and L. Selič, Helv. Chim. Acta, 2001, 84, 146–156 CrossRef CAS; (f) K. Yoshimura, T. Oishi, K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2011, 17, 3827–3831 CrossRef CAS PubMed.
- (a) R. Huisgen, R. Grashey, Germany Pat., DE1203793, 1965; (b) S. Andreae, E. Schmitz, H. Sonnenschein, G. Dörnyei, C. Szántay and J. Tamás, J. Prakt. Chem., 1985, 327, 445–454 CrossRef CAS.
- H. Dorn and A. Otto, Chem. Ber., 1968, 101, 3287–3301 CrossRef CAS.
- R. Shintani and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 10778–10779 CrossRef CAS PubMed.
- W. Chen, X.-H. Yuan, R. Li, W. Du, Y. Wu, L.-S. Ding and Y.-C. Chen, Adv. Synth. Catal., 2006, 348, 1818–1822 CrossRef CAS.
- (a) M. P. Sibi, D. Rane, L. M. Stanley and T. Soeta, Org. Lett., 2008, 10, 2971–2974 CrossRef CAS PubMed; (b) H. Suga, A. Funyu and A. Kakehi, Org. Lett., 2007, 9, 97–100 CrossRef CAS PubMed; (c) W. Chen, W. Du, Y.-Z. Duan, Y. Wu, S.-Y. Yang and Y.-C. Chen, Angew. Chem., Int. Ed., 2007, 46, 7667–7670 CrossRef PubMed; (d) K. Tanaka, T. Kato, Y. Ukaji and K. Inomata, Heterocycles, 2010, 80, 887–893 CrossRef CAS PubMed.
- T. Hashimoto, Y. Maeda, M. Omote, H. Nakatsu and K. Maruoka, J. Am. Chem. Soc., 2010, 132, 4076–4077 CrossRef CAS PubMed.
- S. Shirakawa, P. J. Lombardi and J. L. Leighton, J. Am. Chem. Soc., 2005, 127, 9974–9975 CrossRef CAS PubMed.
- T. Hashimoto, M. Omote and K. Maruoka, Angew. Chem., Int. Ed., 2011, 50, 3489–3492 CrossRef PubMed.
- W. Li, Q. Jia, Z. Du, K. Zhang and J. Wang, Chem.–Eur. J., 2014, 20, 4559–4562 CrossRef CAS PubMed.
- Reviews: (a) C. E. I. Knappke, A. Imami and A. J. Von Wangelin, ChemCatChem, 2012, 4, 937–941 CrossRef; (b) S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem.–Eur. J., 2013, 19, 4664–4678 CrossRef CAS PubMed.
- Compound 4 was formed as a single diastereoisomer. The relative configuration at the acetal stereocenter could not be unambiguously assigned. The relative configuration of the other two stereocenters could be assigned as trans by oxidation of 4 to 3aa. The structure of 3aa was assigned by X-ray analysis, see below.
- (a) M. Wang, Z. Huang, J. Xu and Y. R. Chi, J. Am. Chem. Soc., 2014, 136, 1214–1217 CrossRef PubMed See also ; (b) X. Zhao, K. E. Ruhl and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 12330–12333 CrossRef CAS PubMed; (c) J. Mo, R. Yang, X. Chen, B. Tiwari and Y. R. Chi, Org. Lett., 2013, 15, 50–53 CrossRef CAS PubMed.
- Review on ammonium enolate chemistry, see: M. J. Gaunt and C. C. C. Johansson, Chem. Rev., 2007, 107, 5596–5605 CrossRef CAS PubMed Review on enolates derived from ketenes, see: D. H. Paull, A. Weatherwax and T. Lectka, Tetrahedron, 2009, 65, 6771–6803 CrossRef PubMed.
- In situ generation of ammonium enolates from acid derivatives, see: (a) G. S. Cortez, R. L. Tennyson and D. Romo, J. Am. Chem. Soc., 2001, 123, 7945–7946 CrossRef CAS; (b) G. S. Cortez, S. H. Oh and D. Romo, Synthesis, 2001, 1731–1736 CrossRef CAS PubMed; (c) S. H. Oh, G. S. Cortez and D. Romo, J. Org. Chem., 2005, 70, 2835–2838 CrossRef CAS PubMed; (d) H. Nguyen, G. Ma, T. Gladysheva, T. Fremgen and D. Romo, J. Org. Chem., 2011, 76, 2–12 CrossRef CAS PubMed and references cited thein. From acid fluorides: E. Bappert, P. Müller and G. C. Fu, Chem. Commun., 2006, 2604–2606 Search PubMed.
- In situ generation of ammonium enolates and usage of catalytic amounts of a chiral Lewis base, see: (a) T. Bekele, M. H. Shah, J. Wolfer, C. J. Abraham, A. Weatherwax and T. Lectka, J. Am. Chem. Soc., 2006, 128, 1810–1811 CrossRef CAS PubMed; (b) J. Wolfer, T. Bekele, C. J. Abraham, C. Dogo-Isonagie and T. Lectka, Angew. Chem., Int. Ed., 2006, 45, 7398–7400 CrossRef CAS PubMed; (c) X. Xu, K. Wang and S. G. Nelson, J. Am. Chem. Soc., 2007, 129, 11690–11691 CrossRef CAS PubMed; (d) L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC.
- G. C. Fu, Acc. Chem. Res., 2000, 33, 412–420 CrossRef CAS PubMed.
- (a) V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351–1354 CrossRef CAS PubMed . Application as catalyst for kinetic resolution of secondary alcohols, see: X. Yang, G. Lu and V. B. Birman, Org. Lett., 2010, 12, 892–895 Search PubMed.
- Review on Lewis base catalysis, see: S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS PubMed . Review on the use of isothioureas as catalysts, see: J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC.
- In situ generation of ammonium enolates and usage of catalytic amounts of tetramisole type Lewis bases in C–C-bond forming reactions, see: (a) C. A. Leverett, V. C. Purohit and D. Romo, Angew. Chem., Int. Ed., 2010, 49, 9479–9483 CrossRef CAS PubMed; (b) D. Belmessieri, L. C. Morrill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714–2720 CrossRef CAS PubMed; (c) L. C. Morrill, T. Lebl, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2012, 3, 2088–2093 RSC; (d) C. Simal, T. Lebl, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2012, 51, 3653–3657 CrossRef PubMed; (e) G. Liu, M. E. Shirley, K. N. Van, R. L. McFarlin and D. Romo, Nat. Chem., 2013, 5, 1049–1057 CrossRef CAS PubMed.
- A. Studer, T. Hintermann and D. Seebach, Helv. Chim. Acta, 1995, 78, 1185–1206 CrossRef CAS.
- (a) D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef PubMed; (b) D. Seebach and G. Lelais, Biopolymers, 2004, 76, 206–243 CrossRef PubMed; (c) D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 4, 1366–1375 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, and spectral data for all compounds, including scanned images of 1H and 13C NMR spectra. CCDC 1019046. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02612h |
| This journal is © The Royal Society of Chemistry 2015 |