
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the second dehydrogenation step in ammonia-borane dehydrocoupling: characterization and reactivity of the key intermediate, B-(cyclotriborazanyl)amine-borane†
Hassan A.
Kalviri
a,
Felix
Gärtner
ab,
Gang
Ye
a,
Ilia
Korobkov
a and
R. Tom
Baker
*a
aDepartment of Chemistry and Centre for Catalysis Research and Innovation (CCRI), University of Ottawa, 10 Marie Curie, Ottawa, Ontario K1N 6N5, Canada. E-mail: rbaker@uottawa.ca; Fax: +1 613 5625613; Tel: +1 613 5625698
bLeibniz-Institut für Katalyse (LIKAT), Albert-Einstein Straβe 29a, 18059 Rostock, Germany
First published on 30th October 2014
Abstract
While thermolysis of ammonia-borane (AB) affords a mixture of aminoborane- and iminoborane oligomers, the most selective metal-based catalysts afford exclusively cyclic iminoborane trimer (borazine) and its B–N cross-linked oligomers (polyborazylene). This catalysed dehydrogenation sequence proceeds through a branched cyclic aminoborane oligomer assigned previously as trimeric B-(cyclodiborazanyl)amine-borane (BCDB). Herein we utilize multinuclear NMR spectroscopy and X-ray crystallography to show instead that this key intermediate is actually tetrameric B-(cyclotriborazanyl)amine-borane (BCTB) and a method is presented for its selective synthesis from AB. The reactivity of BCTB upon thermal treatment as well as catalytic dehydrogenation is studied and discussed with regard to facilitating the second dehydrogenation step in AB dehydrocoupling.
Introduction
The development of clean and efficient technologies for the production, storage and utilization of hydrogen is prerequisite for establishing a hydrogen economy.1 For hydrogen storage,2 chemical hydrides such as methanol,3 formic acid,4 carbazoles,5 hydrazine6 and especially ammonia-borane7 are promising sources that could serve as ‘drop-in’ liquid fuels with the existing fuel delivery infrastructure.Ammonia-borane (AB, NH3BH3) is an especially attractive hydrogen storage material due to its high gravimetric hydrogen content of 194 g H2 per kg (19.6 wt% H2). Since the molecule contains both hydridic as well as protic hydrogens, hydrogen release can be realized under mild conditions by various means such as thermal treatment,8 acid-9 or base10 catalysis, or metal-based catalysis.11,12 A number of AB dehydrocoupling mechanisms have been elucidated8,9,10,11g–k,13 and metal-based catalysis has been shown to offer the best combination of selectivity and H2 release rate. While many of the very active catalysts afford linear polyaminoborane and a single equivalent of hydrogen,11b,d,e,13b,14 the most selective catalysts generate >2 equiv. H2 with concomitant formation of cyclic iminoborane trimer (borazine) and its BN-cross-linked oligomers (polyborazylene; Scheme 1).11c,i,k–m,q,r Mechanistic studies of AB dehydrocoupling in ethereal solutions (kinetics, isotope labelling and intermediate trapping) point to the importance of reactive aminoborane, NH2BH2, shown previously to oligomerize above −150 °C.15 It has been suggested that the very active catalysts can retain aminoborane in the first or second coordination sphere and undergo a coordination polymerization-type mechanism (Scheme 1, path B).13a,16 In contrast, the selective catalysts expel aminoborane into solution where it undergoes oligomerization to cyclotriborazane (1, CTB) and a key branched, cyclic aminoborane intermediate, that we assigned previously as trimeric B-(cyclodiborazanyl)amine-borane (2, BCDB). These intermediates then undergo further catalyzed dehydrogenation to the observed borazine and polyborazylene products (Scheme 1, path A and Fig. 1).
 | ||
| Scheme 1 Proposed mechanisms for the metal catalyzed dehydrogenation of ammonia-borane. | ||
 | ||
| Fig. 1 11B{1H} NMR spectra showing Ni(NHC)2-catalyzed conversion of AB to 3 (*) [(a): after 12 h at 20 °C] and of 3 to borazine (+) and polyborazylene (■) [(b)–(e) after heating at 60 °C in the NMR probe (10 min intervals; NHC = 1,3,4-triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene). Minor resonances in top spectrum are due to CTB (−12 ppm) and NHC-borane (−34 ppm). | ||
In spite of the successful design and development of AB dehydrogenation catalysts, their application is hampered by formation of volatile borazine (bp 51 °C) which contaminates the H2 stream and poisons the fuel cell catalyst.17 More work is thus needed on understanding and optimizing the second and third H2 release steps and in particular on the reactivity of the key cyclic aminoborane intermediate. In this work we describe the first practical synthesis of this intermediate and show unequivocally that it is, in fact, a branched, cyclic aminoborane tetramer, B-(cyclotriborazanyl)amine-borane (3, BCTB). Reactivity studies demonstrate why thermolysis of 3 yields different products than those derived from its catalytic dehydrogenation.
Results
Synthesis and molecular structure of the aminoborane selective oligomerization product
Initial attempts to isolate the key cyclic aminoborane intermediate from AB solution thermolysis at 80 °C, or catalyzed AB dehydrogenation using 10 mol% FeH(CH2PMe2)(PMe3)3 (ref. 18) or Ni N-heterocyclic carbene precursors11c,13a at room temperature over several days invariably produced impure samples in low yields (ESI,† p. 3). In contrast, reaction of AB with 5 mol% of Schwartz' reagent, Cp2ZrHCl (4), at 50 °C afforded much purer samples in fair to good yields (ESI,† p. 4). As reported previously,13a the 11B NMR spectrum of the product consists of doublet, triplet and quartet resonances due to the BH, BH2 and BH3 groups (Fig. S4†). In previous samples the intensity of the BH2 resonance was often seen to be greater than that of the other two, presumed to be due to differing amounts of the aminoborane cyclic trimer, CTB (1). After sublimation (80 °C, 2 × 10−6 torr for 24 h), however, the desired product was contaminated mostly by a small amount of AB. Although the 1H NMR spectrum confirmed just a trace amount of CTB impurity, the BH2 intensity in the 11B NMR spectrum was still approximately twice that of the other two resonances (Fig. S5 and S6†). Turning to single crystal X-ray crystallography, crystals from the sublimed product were grown via two different methods and obtained as a glyme solvate and a crown ether adduct.19 Both structures showed the intermediate to be the branched, cyclic aminoborane tetramer, B-(cyclotriborazanyl)-amine-borane (3, BCTB). (Fig. 2 and S7†). As expected, the bond distances and angles within the 6-membered ring (B3–N3 = 1.572(2) Å, B3–N3–B2 = 116.8(1) ° and N1–B3–N3 = 107.1(1) °) are similar to those in CTB (B–Navg = 1.574(2) Å, B–N–Bavg = 115.6(1) ° and N–B–Navg = 107.0(1) °)20 and the B4–N4 distance (1.588(2) Å) is similar to analogous bonds in other amine-boranes such as propylamine–borane (1.593(3) Å).21 Both structures feature extensive dihydrogen bonding as well as conventional N–H–O hydrogen bonds (Fig. S7 and S8 and Table S6†).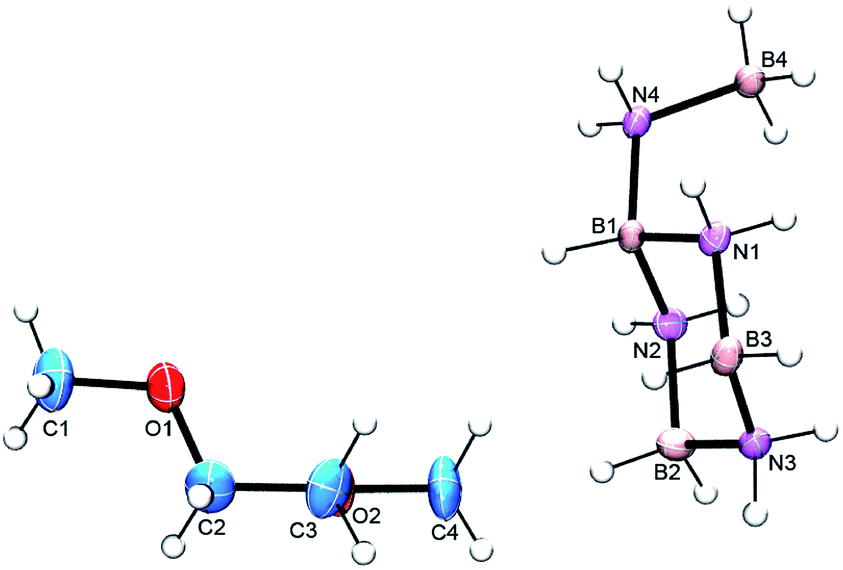 | ||
| Fig. 2 ORTEP view of BCTB (3) co-crystallized with glyme. The thermal ellipsoids are drawn at the 50% probability level. | ||
Proton and nitrogen NMR spectra of BCTB, 3
A series of multinuclear NMR studies confirmed the structure of BCTB in solution. In the 11B decoupled 1H/1H COSY NMR spectrum at −15 °C (Fig. 3), the BH resonance at δ 2.5 ppm is strongly correlated with the NH at δ 3.0 (a in Fig. 3). A weaker correlation is observed with the other NH at δ 3.2 (b in Fig. 3), thus confirming the chair configuration of the BCTB molecule at low temperature (i.e., strong axial–axial correlations). Moreover, two resonances are observed for BH2 groups at δ 2.1 and 2.2 (c in Fig. 3) with integrations close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S6†). These two peaks can be assigned to the axial and equatorial BH2 groups in BCTB. Thus at low temperature separate correlations are observed with axial and equatorial protons of NH2 groups in the ring. On the other hand, the exo-NH2 group (δ 2.7) shows a strong correlation with the BH3 group (δ 1.2) on the side chain (d in Fig. 3). The TOCSY NMR spectrum shows also that the in-ring NH2 resonances are all strongly correlated with each other (e in Fig. 4). As well, the BH2 and BH groups in the ring exhibit strong correlations with the NH2 protons (f in Fig. 4) whereas the BH correlates more weakly with the external NH2 compared with the internal NH2 groups (g in Fig. 4). Variable temperature 1H{14N} NMR spectra of BCTB in THF-d8 show also the sensitivity of the NH chemical shifts to temperature (Fig. S9†), likely due to changes in hydrogen bonding structural motifs. Compared to the literature data for ammonia-borane and CTB,8d we observed similar 15N NMR chemical shifts (Fig. S10†). However, overlapping signals at −362 ppm were assigned to the NH2-groups within the six-membered ring of BCTB (due to correlation with the NH2-protons with an integral of 6) (a in Fig. S10†) and the resonance at −367 ppm was assigned to the exo-cyclic NH2-group (b in Fig. S10†). Finally, the resonance at −377 ppm correlates with the NH3 resonance due to the ammonia-borane impurity in the sample (c in Fig. S10†).
:1 (Fig. S6†). These two peaks can be assigned to the axial and equatorial BH2 groups in BCTB. Thus at low temperature separate correlations are observed with axial and equatorial protons of NH2 groups in the ring. On the other hand, the exo-NH2 group (δ 2.7) shows a strong correlation with the BH3 group (δ 1.2) on the side chain (d in Fig. 3). The TOCSY NMR spectrum shows also that the in-ring NH2 resonances are all strongly correlated with each other (e in Fig. 4). As well, the BH2 and BH groups in the ring exhibit strong correlations with the NH2 protons (f in Fig. 4) whereas the BH correlates more weakly with the external NH2 compared with the internal NH2 groups (g in Fig. 4). Variable temperature 1H{14N} NMR spectra of BCTB in THF-d8 show also the sensitivity of the NH chemical shifts to temperature (Fig. S9†), likely due to changes in hydrogen bonding structural motifs. Compared to the literature data for ammonia-borane and CTB,8d we observed similar 15N NMR chemical shifts (Fig. S10†). However, overlapping signals at −362 ppm were assigned to the NH2-groups within the six-membered ring of BCTB (due to correlation with the NH2-protons with an integral of 6) (a in Fig. S10†) and the resonance at −367 ppm was assigned to the exo-cyclic NH2-group (b in Fig. S10†). Finally, the resonance at −377 ppm correlates with the NH3 resonance due to the ammonia-borane impurity in the sample (c in Fig. S10†).
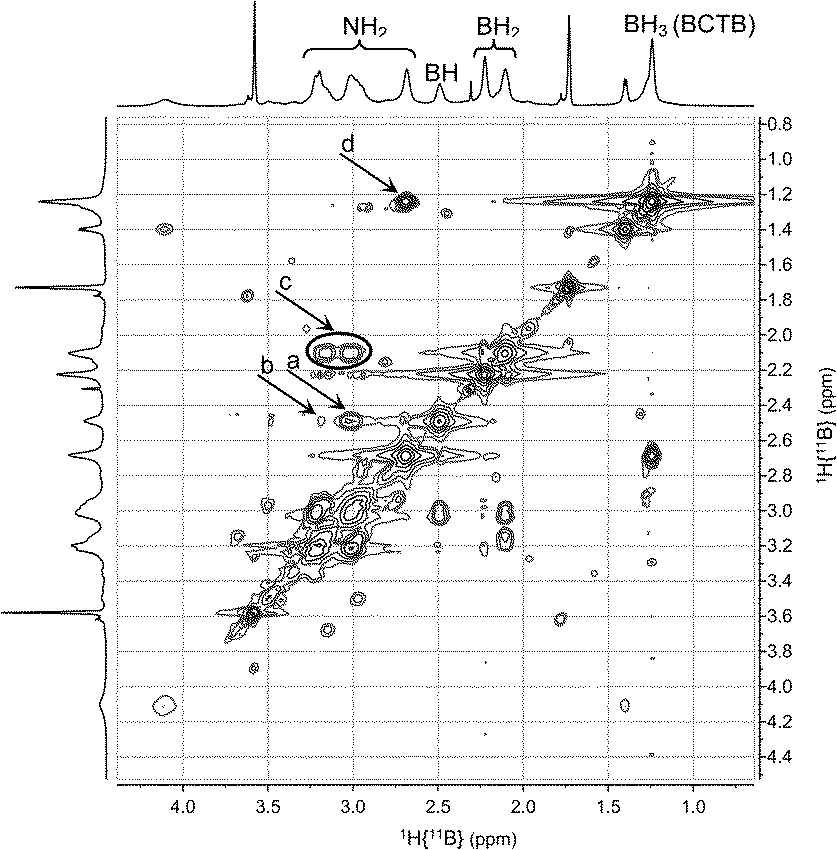 | ||
| Fig. 3 2D-NMR (1H–1H COSY) (THF-d8) of BCTB at −15 °C. | ||
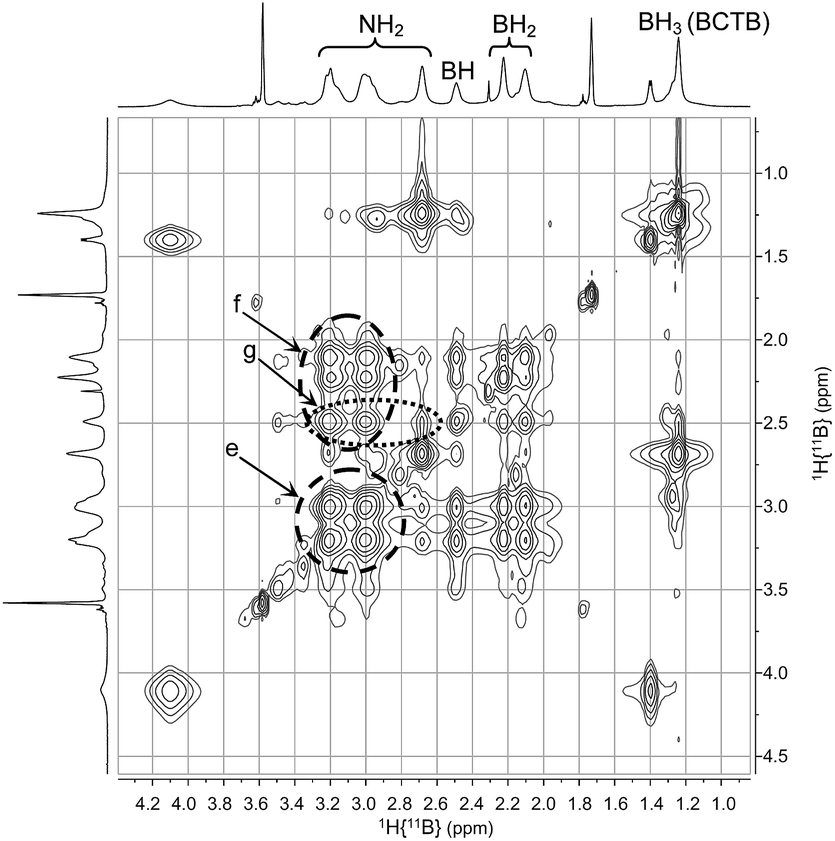 | ||
| Fig. 4 2D-NMR (1H–1H TOCSY) (THF-d8) of BCTB at −15 °C. | ||
Reactivity of BCTB, 3
Reactivity studies demonstrate that thermolysis of 3 yields different products than those derived from its catalytic dehydrogenation. While THF or glyme solutions of AB undergo slow dehydrogenation at 85 °C, thermolysis of CTB does not proceed readily below 125 °C.22 Thermal treatment of BCTB (100 °C, diglyme) led to a mixture of products. After 1 and 3 h CTB, borazine and AB were detected as the main products in a sealed NMR tube experiment (Fig. S11,†Scheme 2A). After 20 h almost all BCTB was converted to borazine (as the main product) and CTB (Fig. S11†). Formation of the latter product suggests a 1,3-hydride transfer from the BH3 to the BH group of BCTB, affording CTB and aminoborane, NH2BH2. Indeed, when cyclohexene was used to trap the latter, a clean reaction ensued with predominate formation of CTB and NH2BCy2 (Fig. S12†). In contrast, heating BCTB at 60 °C in the presence of 7.5 mol% [Rh(μ-Cl)(cod)]2 (5, cod = 1,5-cyclooctadiene) afforded exclusively borazine and polyborazylene (Fig. S18†). | ||
| Scheme 2 Reactivity of BCTB in the presence of complexes 4 to 9 (20 mg BCTB, 15 mol% catalyst, 2.5 mL THF, 60 °C, 16 h). | ||
In order to probe the second dehydrogenation step of AB dehydrocoupling in more detail, we investigated a series of metal complexes as precatalysts for the dehydrogenation of BCTB. The complexes [RuCl2(P^N)2] (6, P^N: 2-(di-tert-butylphosphino)-ethylamine),11d Ni(IMes)2 (7, IMes: N,N′-bis(2,4,6-trimethyl-phenyl)imidazol-2-ylidene),11c,23 and [RuH(PMe3)3(BH4)] (8)24,25 have been applied previously as dehydrogenation catalysts for AB. The iron hydride complex [FeH(PP3)][BPh4] (9, PP3: tris[2-(diphenylphosphino)ethyl]-phosphine)26 has been successfully employed for the catalytic dehydrogenation of formic acid.4c In a typical experiment 20 mg BCTB was heated in THF (60 °C) in the presence of 15 mol% catalyst (with respect to BCTB). The reaction progress was monitored using 11B NMR spectroscopy with samples taken after 1, 3 and 16 h. Based on their reactivity, the catalysts can be divided into three groups (Scheme 2B–D). Complexes 4 and 8 had little effect and the NMR spectra after 16 h were similar to those obtained from the control experiment (Fig. S13–15†). Complexes 7 and 9, however, converted BCTB to a mixture of CTB, borazine and polyborazylene (Fig. S16 and S17†). The inability of complex 7 to dehydrogenate CTB to borazine was confirmed using isolated CTB (no reaction after 24 h at 60 °C). Most interestingly, a third group of catalysts, complexes 5 and 6, effected the dehydrogenation of both BCTB and CTB after the 16 h reaction time (Fig. S18 and S19†).
Discussion
Synthesis and formation of BCTB, 3
Our new synthesis of BCTB takes advantage of the sluggish reactivity of d0 complexes with AB27 and particularly the inability of Cp2ZrHCl to catalyse the dehydrogenation of BCTB to borazine, as shown above. We propose that the AB dehydrogenation proceeds through the amidoborane complex Cp2ZrCl(NH2BH3), 10, characterized previously by Roesler et al.28 β-elimination of the B–H bond then affords the aminoborane monomer, regenerating the catalyst. In the presence of cyclohexene, all the aminoborane was trapped by cyclohexene and no BCTB formation was observed (Fig. S20†). While we did not observe 10 in the 11B NMR spectra of these reactions, resonances were observed at −15.8 (quintet, JB–H = 86 Hz) (minor) and −7.1 ppm (quintet, JB–H = 87 Hz) due to Cp2Zr(BH4)2, 11, and Cp2ZrCl(BH4), 12, respectively (Fig. S3†). As these complexes are likely resting states of the Zr hydride catalysts, we also investigated the reaction of complex 11 with 20 equiv. of AB. Poor BCTB selectivity was observed and borazine was detected as the major product (Fig. S21†).In light of the variety of diverse mechanisms that have been postulated for the thermolytic and catalyzed dehydrogenation of AB, what is the origin of the extremely clean formation of BCTB with selected metal complex catalysts? Substituted primary aminoboranes such as methylaminoborane, NMeH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BH2, are postulated to oligomerize in solution to the cyclic trimer and, with further heating, to N,N′,N′′-trimethylborazine.29 Although the detailed mechanisms of these transformations require additional elucidation, formation of branched oligomers has been confined to reactions of the parent amine-borane, AB. In previous computational studies we13a and others30 showed that this selective oligomerization could proceed via B–H–B bridged intermediates followed by intramolecular attack of NH2 on the three-coordinate boron (Scheme 3). The dimerization thus affords unusual (not yet observed) intermediate I with BH and BH3 groups. Addition of the third aminoborane monomer to I through a similar mechanism then generates intermediate II which was proposed to undergo an additional intramolecular attack of NH2 on three-coordinate boron to give BCDB, 2 selectively. In this work we find instead that intermolecular reaction of II with a fourth equivalent of aminoborane selectively affords the branched cyclic aminoborane tetramer, BCTB, 3.
BH2, are postulated to oligomerize in solution to the cyclic trimer and, with further heating, to N,N′,N′′-trimethylborazine.29 Although the detailed mechanisms of these transformations require additional elucidation, formation of branched oligomers has been confined to reactions of the parent amine-borane, AB. In previous computational studies we13a and others30 showed that this selective oligomerization could proceed via B–H–B bridged intermediates followed by intramolecular attack of NH2 on the three-coordinate boron (Scheme 3). The dimerization thus affords unusual (not yet observed) intermediate I with BH and BH3 groups. Addition of the third aminoborane monomer to I through a similar mechanism then generates intermediate II which was proposed to undergo an additional intramolecular attack of NH2 on three-coordinate boron to give BCDB, 2 selectively. In this work we find instead that intermolecular reaction of II with a fourth equivalent of aminoborane selectively affords the branched cyclic aminoborane tetramer, BCTB, 3.
 | ||
| Scheme 3 Oligomerization of aminoborane through B–H–B bridged intermediates. | ||
On the basis of 11B and 15N NMR spectroscopy, thermolysis of AB in glyme solvent at 80 °C was proposed to generate both cyclodiborazane and cyclotriborazane in addition to a branched aminoborane cyclic oligomer, assigned as BCDB, 2.8d This product distribution results presumably from the slow thermal generation of the aminoborane monomer, likely catalysed by traces of free BH3.31 In the metal-catalysed reaction, however, rapid generation of aminoborane favors selective generation of BCTB, 3, as long as the monomer does not take part in the metal coordination oligomerization pathway to linear polyamino-borane.16 Further work is thus needed to fully characterize the branched aminoborane cyclic oligomer derived from uncatalysed AB themolysis.
Thermal vs. catalysed conversion of BCTB, 3
The branched aminoborane tetramer, BCTB, 3, is considerably more reactive than the cyclic trimer, CTB, 1, which is believed to undergo competing ring-opening and dehydrogenation pathways.22b The formation of some borazine and ammonia-borane from the thermolysis of 3 can be attributed to hydrogen transfer processes (eqn (1)) as studied recently by Manners and co-workers.32 Consistent with this proposal, neither AB nor borazine was observed on thermolysis of 3 in the presence of cyclohexene which effectively traps the aminoborane monomer.33 | (1) |
The catalysed reactions of 3 shed additional light on the second H2 release step from AB. The fact that Ru catalyst precursor 8 is unable to dehydrogenate 3 is consistent with previous studies that showed the sensitivity of 8 to the reaction solvent. While poor selectivity for AB dehydrogenation was observed in THF, formation of insoluble polyaminoborane could be prevented using ionic liquids with nucleophilic anions.25 Similar observations were made using the Ni(NHC)2 catalyst that required a mixed benzene–glyme solvent system to release >2 equiv. of H2.11c In spite of the short lifetime of this catalyst, the small amount of CTB remaining in Fig. 1(d) supports the greater reactivity seen for 3vs. CTB using the similar catalyst 7. In contrast, the [RuCl2(P^N)2]/KOBut catalyst system 6 that was reported to afford a single equivalent of hydrogen from AB readily converted both CTB and 3 to borazine and polyborazylene. This would thus be a good candidate for further solvent studies to avoid the aminoborane coordination polymerization pathway from AB. Finally, the Rh colloids12a,b or clusters34 derived from the chlororhodium diene catalyst precursor 5 are also effective catalysts for conversion of 3 to borazine and polyborazylene although catalyst lifetime experiments have yet to be performed.12b
Conclusions
Utilization of the Cp2ZrHCl catalyst precursor has allowed for the isolation and characterization of the key intermediate in the second step of metal-catalysed AB dehydrogenation. In contrast to previous assignments, the intermediate has now been identified as the BN ethylcyclohexane analog, the branched, cyclic aminoborane tetramer, B-(cyclotriborazanyl)amine-borane (3). Reactivity studies show that higher yields of 3 would likely be obtained if reaction of the aminoborane monomer with CTB to give borazine and AB (eqn (1)) could be controlled. Testing of several catalysts commonly used for dehydrocoupling reactions for this second dehydrogenation step revealed an important difference in selectivity as some catalysts converted 3, but not CTB, into borazine and polyborazylene. With this information in hand we are now poised to address the challenging third step of AB dehydrogenation – identifying an effective catalyst for the BN cross-linking of borazine to polyborazylene.35Acknowledgements
We thank the US Department of Energy for support of this work through the Office of Science Basic Energy Sciences Research Contract #DE-FC36-05GO15051. Thank you also to the Canada Foundation for Innovation, Ontario Ministry of Economic Development and Innovation, Canada Research Chairs and the University of Ottawa for provision of enabling infrastructure and Dr Glenn Facey for help with the NMR measurements. Part of this work was supported by the BMBF (Spitzenforschung und Innovation in den neuen Ländern). F. G. thanks the Fonds der Chemischen Industrie (FCI) for a Kekulé grant.Notes and references
- (a) L. Barreto, A. Makahira and K. Riahi, Int. J. Hydrogen Energy, 2003, 28, 267–284 CrossRef CAS; (b) G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, Phys. Today, 2004, 57, 39–44 CrossRef CAS PubMed; (c) U. Bossel, B. Eliasson and G. Taylor, J. KONES Int. Comb. Eng., 2004, 11, 87–111 Search PubMed.
- (a) U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed; (b) J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- (a) T. C. Johnson, D. J. Morris and M. Wills, Chem. Soc. Rev., 2010, 39, 81 RSC; (b) B. Loges, A. Boddien, F. Gärtner, H. Junge and M. Beller, Top. Catal., 2010, 53, 902–914 CrossRef CAS; (c) A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS PubMed; (d) M. Graseman and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- (a) J. von Wild, T. Friedrich, A. Cooper, B. Toseland, G. Muraro, W. TeGrotenhuis, Y. Wang, P. Humble and A. Karim, WHEC Proc., 2010, 78–4, 189–197 Search PubMed; (b) D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4, 2767–2773 RSC; (c) F. Sotoodeh and K. J. Smith, Can. J. Chem. Eng., 2013, 91, 1477–1490 CrossRef CAS.
- (a) S. K. Singh, X.-B. Zhang and Q. Xu, J. Am. Chem. Soc., 2009, 131, 9894–9895 CrossRef CAS PubMed; (b) S. K. Singh and Q. Xu, J. Am. Chem. Soc., 2009, 131, 18032–18033 CrossRef CAS PubMed.
- (a) F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613–2626 RSC; (b) T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116–8118 CrossRef CAS PubMed; (c) C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC; (d) N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 509–521 CrossRef CAS; (e) A. Staubitz, A. P. M. Roberston and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- (a) F. Baitalow, J. Baumann, G. Wolf, K. Jaenicke-Rossler and G. Leitner, Thermochim. Acta, 2002, 391, 159 CrossRef CAS; (b) G. Wolf, J. Baumann, F. Baitalow and F. P. Hoffmann, Thermochim. Acta, 2000, 343, 19 CrossRef CAS; (c) J. S. Wang and R. A. Geanangel, Inorg. Chim. Acta, 1988, 148, 185–190 CrossRef CAS; (d) W. J. Shaw, J. C. Linehan, N. K. Szymczak, D. J. Heldebrant, C. Yonker, D. M. Camaioni, R. T. Baker and T. Autrey, Angew. Chem., Int. Ed., 2008, 47, 9600–9602 CrossRef PubMed.
- F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., 2007, 119, 760–763 CrossRef.
- (a) D. W. Himmelberger, C. W. Yoon, M. E. Bluhm, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2009, 131, 14101–14110 CrossRef CAS PubMed; (b) W. C. Ewing, A. Marchione, D. W. Himmelberger, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2011, 133, 17093–17099 CrossRef CAS PubMed; (c) W. C. Ewing, P. J. Carroll and L. G. Sneddon, Inorg. Chem., 2013, 52, 10690–10697 CrossRef CAS PubMed.
- Homogeneous Catalysts: (a) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424–9434 CrossRef CAS PubMed; (b) M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048–12049 CrossRef CAS PubMed; (c) R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844–1845 CrossRef CAS PubMed; (d) N. Blaquiere, S. Diallo-Garcia, S. I. Gorelsky, D. A. Black and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 14034–14035 CrossRef CAS PubMed; (e) M. Käβ, A. Friedrich, M. Drees and S. Schneider, Angew. Chem., Int. Ed., 2009, 48, 905–907 CrossRef PubMed; (f) Y. Jiang, O. Blaque, T. Fox, C. M. Frech and H. Berke, Organometallics, 2009, 28, 5493–5504 CrossRef CAS; (g) Y. Kawano, M. Uruichi, M. Shimoi, S. Taki, T. Kawaguchi, T. Kakizawa and H. Ogino, J. Am. Chem. Soc., 2009, 131, 14946–14957 CrossRef CAS PubMed; (h) S.-K. Kim, W.-S. Han, T.-J. Kim, T.-Y. Kim, S. W. Nam, M. Mitoraj, L. Pieko, A. Michalak, S.-J. Wang and S. O. Kang, J. Am. Chem. Soc., 2010, 132, 9954–9955 CrossRef CAS PubMed; (i) C. Boulho and J.-P. Djukic, Dalton Trans., 2010, 39, 8893–8905 RSC; (j) B. L. Conley and T. J. Williams, Chem. Commun., 2010, 46, 4815–4817 RSC; (k) B. L. Conley, D. Guess and T. J. Williams, J. Am. Chem. Soc., 2011, 133, 14212–14215 CrossRef CAS PubMed; (l) M. Vogt, B. de Bruin, H. Berke, M. Trincado and H. Grützmacher, Chem. Sci., 2011, 2, 723–727 RSC; (m) D. F. Schreiber, C. O'Connor, C. Grave, Y. Ortin, H. Müller-Bunz and A. D. Phillips, ACS Catal., 2012, 2, 2505–2511 CrossRef CAS; (n) J. R. Vance, A. Schäfer, A. P. M. Robertson, K. Lee, J. Turner, G. R. Whittell and I. Manners, J. Am. Chem. Soc., 2014, 136, 3048–3064 CrossRef CAS PubMed; (o) M. A. Esteruelas, I. Fernandez, A. M. Lopez, M. Mora and E. Onate, Organometallics, 2014, 33, 1104–1107 CrossRef CAS; (p) H. C. Johnson, E. M. Leitao, G. R. Whittell, I. Manners, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2014, 136, 9078–9093 CrossRef CAS PubMed; (q) P. Bhattacharya, J. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 14212–14215 CrossRef PubMed; (r) J. A. Buss, G. A. Edouard, C. Cheng, J. Shi and T. Agapie, J. Am. Chem. Soc., 2014, 136, 11272–11275 CrossRef CAS PubMed.
- Heterogeneous catalysts: (a) C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 9776–9785 CrossRef CAS PubMed; (b) C. A. Jaska, T. J. Clark, S. B. Clendenning, D. Grozea, A. Turak, Z.-H. Lu and I. Manners, J. Am. Chem. Soc., 2005, 127, 5116–5124 CrossRef CAS PubMed; (c) R. P. Shrestha, H. V. K Diyabalanage, T. A. Semelsberger, K. C. Ott and A. K. Burrell, Int. J. Hydrogen Energy, 2009, 34, 2616–2621 CrossRef CAS PubMed; (d) A. P. M. Robertson, R. Suter, L. Chabanne, G. R. Whittell and I. Manners, Inorg. Chem., 2011, 50, 12680–12691 CrossRef CAS PubMed; (e) M. Zahmakiran, M. Tristany, K. Philippot, K. Fajerwerg, S. Ozkar and B. Chaudret, Chem. Commun., 2010, 46, 2938–2940 RSC; (f) J. F. Sonnenberg and R. H. Morris, ACS Catal., 2013, 3, 1092–1102 CrossRef CAS.
- (a) V. Pons, R. T. Baker, J. Linehan, M. H. Matus and D. A. Dixon, Chem. Commun., 2008, 6597–6599 RSC; (b) A. Staubitz, M. E. Sloan, A. P. M. Robertson, A. Friedrich, S. Schneider, P. J. Gates, J. Schmedt auf der Günne and I. Manners, J. Am. Chem. Soc., 2010, 132, 13332–13345 CrossRef CAS PubMed; (c) Z. Lu, B. L. Conley and T. J. Williams, Organometallics, 2012, 31, 6705–6714 CrossRef CAS PubMed; (d) E. M. Leitao, N. E. Stubbs, A. P. M. Robertson, H. Helten, R. J. Cox, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2012, 134, 16805–16816 CrossRef CAS PubMed; (e) A. N. Marziale, A. Friedrich, I. Klopsch, M. Drees, V. R. Celinski, J. Schmedt auf der Günne and S. Schneider, J. Am. Chem. Soc., 2013, 135, 13342–13355 CrossRef CAS PubMed.
- (a) R. T. Baker, J. G. Gordon, C. W. Hamilton, N. J. Henson, P.-H. Lin and S. Maguire, J. Am. Chem. Soc., 2012, 134, 5598–5609 CrossRef CAS PubMed.
- H. A. McGee Jr and C. T. Kwon, Inorg. Chem., 1970, 9, 2458–2461 CrossRef.
- A. Kumar, H. C. Johnson, T. N. Hooper, A. S. Weller, A. G. Algarra and S. A. Macgregor, Chem. Sci., 2014, 5, 2546–2553 RSC.
- B. Davis, H. Diyabalanage, C. Hamilton, R. Shrestha, T. Baker, A. Burrell, N. Henson, M. Inbody, K. John, T. Semelsberger, F. Stephens, J. Gordon and K. Ott, DOE Annual Merit Review, Project ST6, 2008.
- H. H. Karsch, H.-F. Klein and H. Schmidbauer, Chem. Ber., 1977, 110, 2200–2212 CrossRef CAS.
- AB and several other BN compounds have been structurally characterized previously as 18-crown-6 solvates: (a) H. M. Colquhoun, G. Jones, J. M. Maud, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Dalton Trans., 1984, 63–66 RSC; (b) C. W. Yoon, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2009, 131, 855–864 CrossRef CAS PubMed; (c) X. Chen, J.-C. Zhao and S. G. Shore, J. Am. Chem. Soc., 2010, 132, 10658–10659 CrossRef CAS PubMed.
- (a) P. W. R. Corfield and S. G. Shore, J. Am. Chem. Soc., 1973, 95, 1480–1487 CrossRef CAS; (b) H. K. Lingam, C. Wang, J. C. Galucci, X. Chen and S. G. Shore, Inorg. Chem., 2012, 51, 13430–13436 CrossRef CAS PubMed.
- G. J. Gainsford and M. E. Bowden, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o1395 CAS.
- (a) G. H. Dahl and R. Schaeffer, J. Am. Chem. Soc., 1961, 83, 3032–3034 CrossRef CAS; (b) R. Schellenberg, J. Kriehme and G. Wolf, Thermochim. Acta, 2007, 457, 103–108 CrossRef CAS PubMed.
- A. A. Danopoulos and D. Pugh, Dalton Trans., 2008, 30–31 RSC.
- J. A. Statler, G. Wilkinson, M. Thornton-Pett and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1984, 1731–1738 RSC.
- W. R. H. Wright, E. R. Berkeley, L. R. Alden, R. T. Baker and L. G. Sneddon, Chem. Commun., 2011, 47, 3177–3179 RSC.
- (a) P. Stoppioni, F. Mani and L. Sacconi, Inorg. Chim. Acta, 1974, 11, 227–230 CrossRef CAS; (b) C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS PubMed.
- (a) D. Pun, E. Lobkovsky and P. J. Chirik, Chem. Commun., 2007, 3297–3299 RSC; (b) M. E. Sloan, A. Staubitz, T. J. Clark, C. A. Russell, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2010, 132, 3831–3841 CrossRef CAS PubMed; (c) T. Beweries, S. Hansen, M. Kessler, M. Klahn and U. Rosenthal, Dalton Trans., 2011, 40, 7689–7692 RSC; (d) T. Miyasaki, Y. Tanabe, M. Yuki, Y. Miyake and Y. Nishibayashi, Organometallics, 2011, 30, 2394–2404 CrossRef; (e) H. Helten, B. Dutta, J. R. Vance, M. E. Sloan, M. F. Haddow, S. Sproules, D. Collison, G. R. Whittell, G. C. Lloyd-Jones and I. Manners, Angew. Chem., Int. Ed., 2013, 52, 437–440 CrossRef CAS PubMed.
- T. D. Forster, H. M. Tuonen, M. Parvez and R. Toesler, J. Am. Chem. Soc., 2009, 131, 6689–6691 CrossRef CAS PubMed.
- (a) Y. Yamamoto, K. Miyamoto, J. Umeda, Y. Nakatani, T. Yamamoto and N. Miyaura, J. Organomet. Chem., 2006, 691, 4909–4917 CrossRef CAS PubMed; (b) O. J. Metters, A. M. Chapman, A. P. M. Robertson, C. H. Woodall, P. J. Gates, D. F. Wass and I. Manners, Chem. Commun., 2014, 50, 12146–12149 RSC.
- (a) W. R. Nutt and M. L. Mckee, Inorg. Chem., 2007, 46, 7633–7645 CrossRef CAS PubMed; (b) P. M. Zimmerman, A. Paul, Z. Zhang and C. B. Musgrave, Inorg. Chem., 2009, 48, 1069–1081 CrossRef CAS PubMed; (c) T. Malakar, L. Roy and A. Paul, Chem.–Eur. J., 2013, 19, 5812–5817 CrossRef CAS PubMed.
- M. T. Nguyen, V. S. Nguyen, M. H. Matus, G. Gopakumar and D. A. Dixon, J. Phys. Chem. A, 2007, 111, 679–690 CrossRef CAS PubMed.
- (a) A. P. M. Robertson, E. M. Leitao and I. Manners, J. Am. Chem. Soc., 2011, 133, 19322–19325 CrossRef CAS PubMed; (b) N. E. Stubbs, A. P. M. Robertson, E. M. Leitao and I. Manners, J. Organomet. Chem., 2013, 730, 84–89 CrossRef CAS PubMed; (c) A. P. M. Robertson, E. M. Leitao, T. Jurca, M. F. Haddow, H. Helten, G. C. Lloyd-Jones and I. Manners, J. Am. Chem. Soc., 2013, 135, 12670–12683 CrossRef CAS PubMed.
- Note that the BN analogs of cyclohexene and cyclohexadiene have never been observed and may be unstable with respect to formation of borazine and CTB through intermolecular hydrogen transfer reactions.
- (a) Y. Chen, J. L. Fulton, J. C. Linehan and T. Autrey, J. Am. Chem. Soc., 2005, 127, 3254–3255 CrossRef CAS PubMed; (b) R. Rousseau, G. K. Schenter, J. L. Fulton, J. C. Linehan, M. H. Engelhard and T. Autrey, J. Am. Chem. Soc., 2009, 131, 10516–10524 CrossRef CAS PubMed.
- (a) A. T. Lynch, PhD thesis, 1989, Proquest paper AAI8922559, http://repository.upenn.edu/dissertations/AAI8922559; (b) T. J. Carter and N. K. Szymczak, Angew. Chem., Int. Ed., 2012, 51, 13168–13172 CrossRef CAS PubMed; (c) T. J. Carter, J. Y. Wang and N. K. Szymczak, Organometallics, 2014, 33, 1540–1543 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, X-ray and NMR data. CCDC 1022830 and 1022831. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02710h |
| This journal is © The Royal Society of Chemistry 2015 |