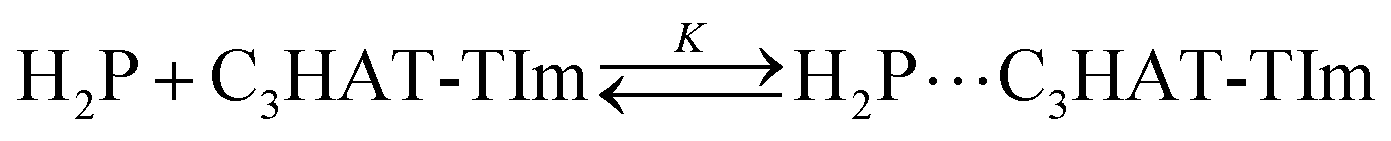DOI:
10.1039/C4SC02787F
(Edge Article)
Chem. Sci., 2015,
6, 1498-1509
Ultrafast photoinduced electron transfer in face-to-face charge-transfer π-complexes of planar porphyrins and hexaazatriphenylene derivatives†
Received
10th September 2014
, Accepted 28th November 2014
First published on 28th November 2014
Abstract
Charge-transfer (CT) π-complexes are formed between planar porphyrins and 1,4,5,8,9,12-hexaazatriphenylene (HAT) derivatives with large formation constants (e.g., 104 M−1), exhibiting broad CT absorption bands. The unusually large formation constants result from close face-to-face contact between two planar π-planes of porphyrins and HAT derivatives. The redox potentials of porphyrins and HAT derivatives measured by cyclic voltammetry indicate that porphyrins and HAT derivatives act as electron donors and acceptors, respectively. The formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CT complexes between porphyrins and HAT derivatives was examined by UV-vis, fluorescence and 1H NMR measurements in nonpolar solvents. The occurrence of unprecedented ultrafast photoinduced electron transfer from the porphyrin unit to the HAT unit in the CT π-complex was observed by femtosecond laser flash photolysis measurements. A highly linear aggregate composed of a planar porphyrin and an HAT derivative was observed by transmission electron microscopy (TEM) and atomic force microscopy (AFM).
:1 CT complexes between porphyrins and HAT derivatives was examined by UV-vis, fluorescence and 1H NMR measurements in nonpolar solvents. The occurrence of unprecedented ultrafast photoinduced electron transfer from the porphyrin unit to the HAT unit in the CT π-complex was observed by femtosecond laser flash photolysis measurements. A highly linear aggregate composed of a planar porphyrin and an HAT derivative was observed by transmission electron microscopy (TEM) and atomic force microscopy (AFM).
Introduction
Extensive efforts have been devoted to the construction of covalently linked electron donor–acceptor (D–A) ensembles to mimic the efficient photoinduced electron-transfer processes in the photosynthetic reaction center, which is essential to realize artificial photosynthesis.1–9 Non-covalent interactions have also been utilized to construct more sophisticated D–A ensembles with highly ordered nanoarchitectures.10–19 The use of non-covalent interactions has great advantage over the step-by-step synthesis of many covalent bonds due to the self-assembling features of supramolecules with non-covalent interactions. However, supramolecular D–A ensembles have a disadvantage in terms of weak interactions between electron donors and acceptors, which prohibit ultrafast photoinduced electron transfer through space.10–19
In order to surmount this disadvantage of supramolecular D–A ensembles, the non-covalent interactions should be strengthened by the close contact of two large planar π-planes of electron donors and acceptors. Porphyrins which have a large planar π-plane have been used as good electron donors as well as chromophores absorbing visible light in D–A ensembles (e.g., electrostatic host–guest assemblies with C60).2–5,14–20 On the other hand, there are many examples of supramolecular assemblies of porphyrins, in which efficient energy transfer occurred.20–22 Thus, porphyrins act as electron donors or energy acceptors but not electron acceptors due to the low one-electron reduction potentials. Although a variety of supramolecular D–A ensembles have been reported so far, there has been no example of a supramolecular D–A ensemble composed of porphyrins utilizing the simple close face-to-face contact for the occurrence of ultrafast photoinduced electron transfer.
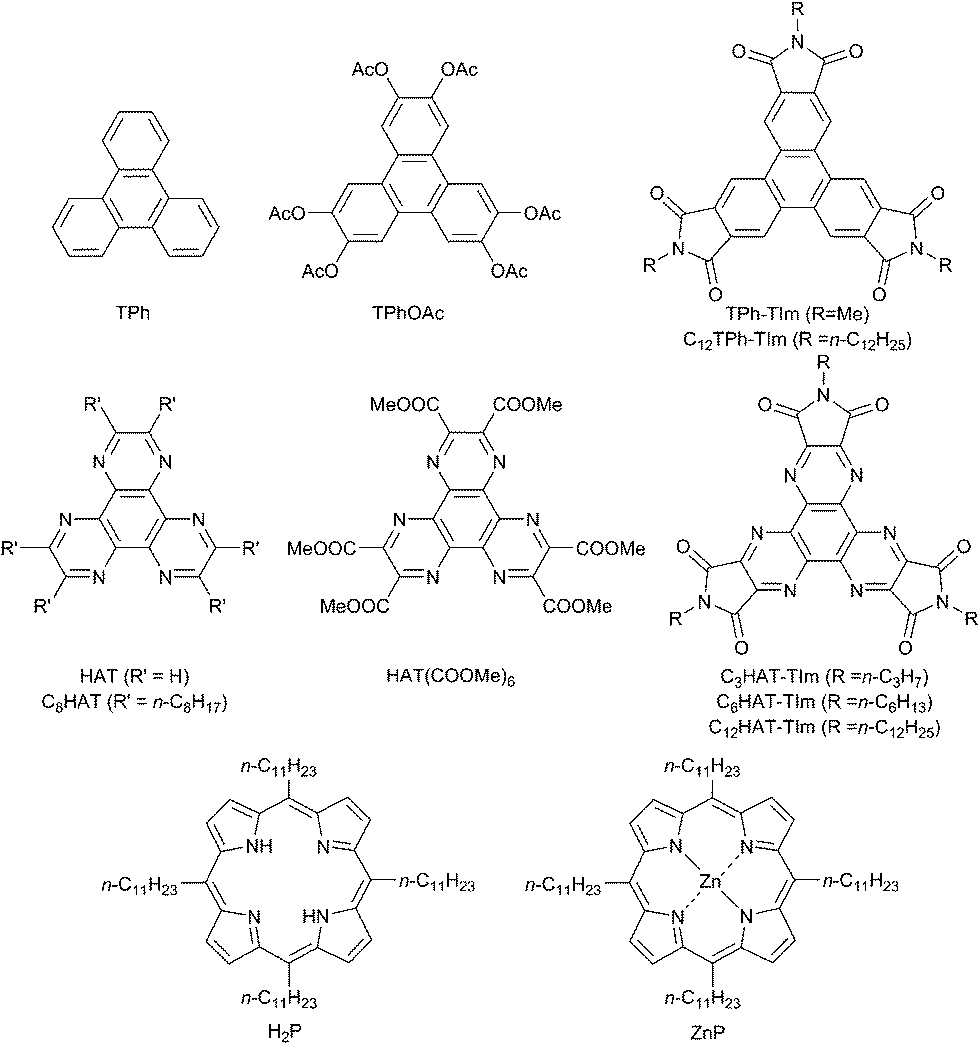
Disc-like polycyclic aromatic hydrocarbon (PAH) derivatives such as triphenylene (TPh) functionalized with alkyl groups, which self-assemble into supramolecular columnar structures with hexagonal and nematic phases because of the stacking π–π interactions,23–26 may be good candidates for use as electron acceptors to construct face-to-face complexes with planar porphyrins. In this context, 1,4,5,8,9,12-hexaazatriphenylene (HAT) derivatives have merited special attention, because these molecules possess electron deficient pyrazine units, acting as good electron acceptors.27,28 Moreover, hexaazatriphenylene hexacarbonitrile (HAT-CN) and hexaazatriphenylene-hexacarboxy triimide (HAT-TIm) have a quite low-lying energy of the lowest unoccupied molecular orbital (LUMO) compared to the pristine HAT because of the introduction of strong electron withdrawing groups such as nitrile and imide groups. The reported LUMO level of HAT-CN (−4.4 eV)29 and the first reduction potential of HAT-TIm (−0.35 V vs. SCE)30 indicate efficient electron accepting properties, which are even better than C60 (−0.44 V vs. SCE).31 Disc shaped charge-transfer (CT) complexes of HAT-TIm with triphenylene (TPh: electron donors) have been reported recently.32 Thus, a combination of HAT-TIm and porphyrin seems to be ideal for fulfilling an enhanced light-harvesting efficiency of chromophores throughout the solar spectrum and efficient photoinduced electron transfer. However, the photoinduced electron transfer of D–A supramolecular complexes with close contact of two large planar π-planes has yet to be examined.
We report herein ultrafast photoinduced electron transfer in supramolecular CT π-complexes formed between alkyl-substituted porphyrins and N-alkyl-substituted HAT-TIm with face-to-face close contact of two planar π-planes. First we examine and compare the electrochemical and photophysical properties of HAT and TPh derivatives (Chart 1). Then, the formation of strong HAT-TIm–porphyrin complexes and the ultrafast photoinduced electron transfer in the complexes including alkyl chain dependencies are discussed in detail.
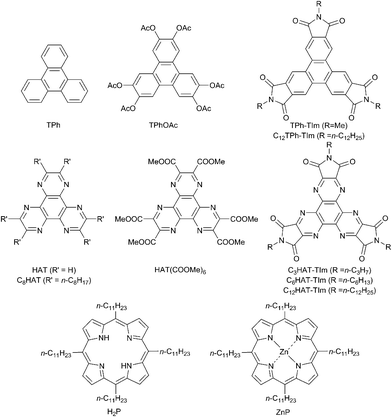 |
| | Chart 1 Chemical structures of TPh, HAT and porphyrin derivatives used in this study. | |
Results and discussion
Synthesis
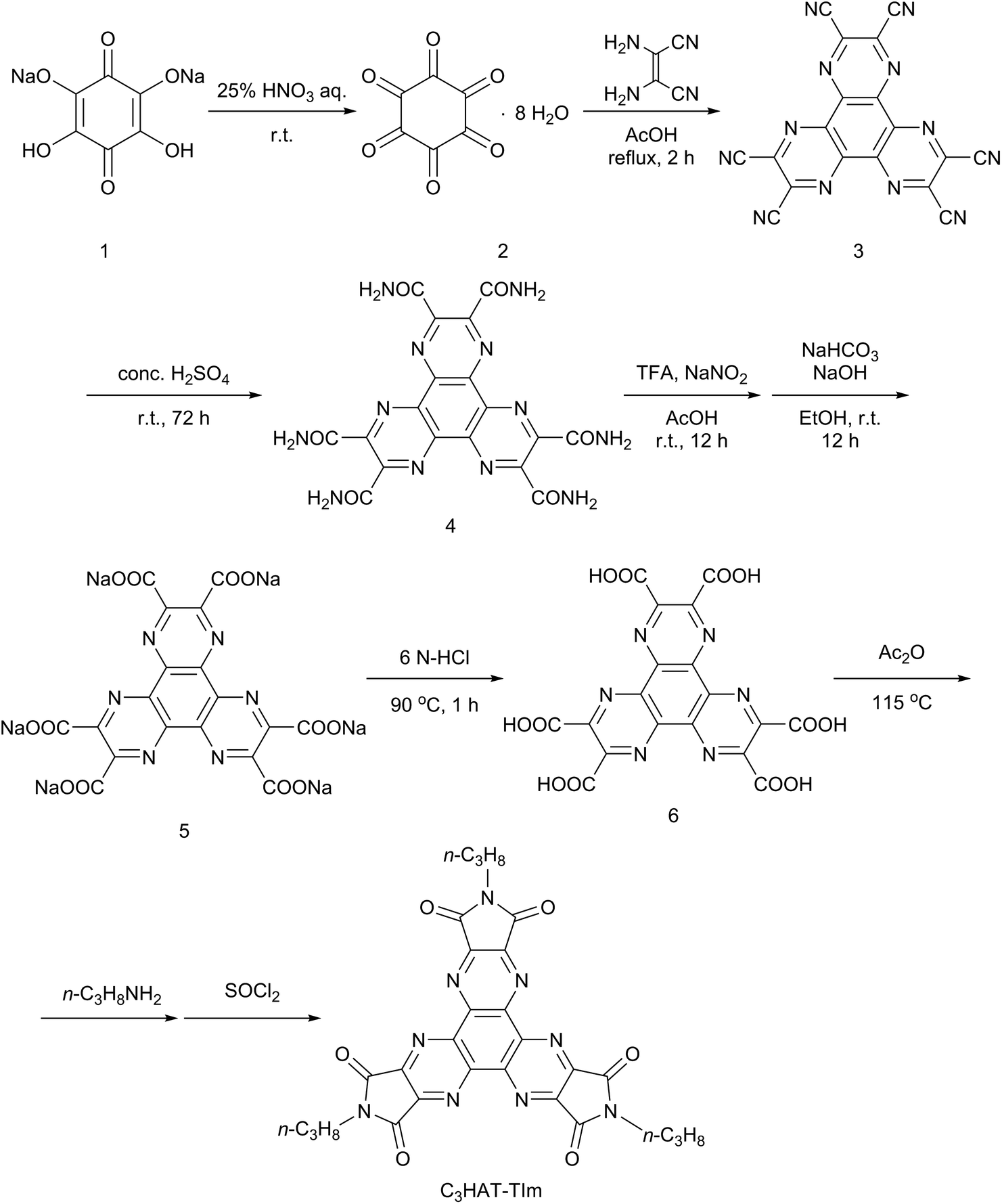
The HAT-TIm derivatives were synthesized according to the reported method by Kanakarajan and coworkers (Scheme 1).33,34 First, compound 2 was synthesized from compound 1 under acidic conditions. Next, the dehydration condensation of compound 2 and 2,3-diaminomaleonitrile was carried out to prepare the HAT skeleton. Then, compound 4 was synthesized by acidic hydrolysis of compound 3. To synthesize compound 5, an HAT derivative 4 was reacted with TFA, NaNO2 and AcOH, which was followed by the reaction under basic conditions. Then, compound 6 was obtained by the reaction of compound 5 with 6 N HCl at 90 °C. Compound 6 was then reacted with Ac2O at 115 °C, which was followed by reaction with each alkyl amine. Finally, the HAT-triimide derivatives were obtained by a reaction of the crude material and thionyl chloride. HAT(COOMe)6 was obtained by a reaction of compound 6 with MeOH and H2SO4 according to the reported method.34 The details of C3HAT-TIm (1H, 13C NMR and MALDI-TOF mass spectra) are shown in Fig. S1–S3 (ESI†). H2P was synthesized by the reported literature35 as shown in Scheme S1 (ESI†). Then, the synthesis of ZnP was achieved through the insertion of zinc into the porphyrin center.
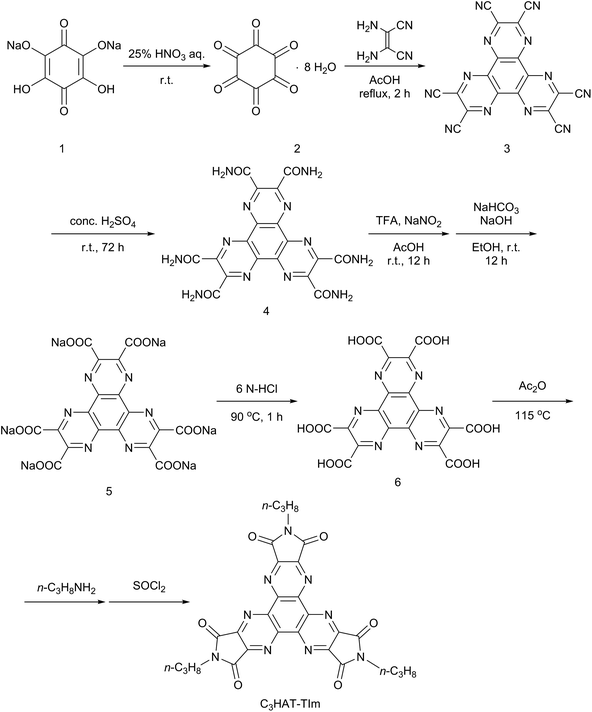 |
| | Scheme 1 Synthetic schemes of C3HAT-TIm. | |
Steady-state spectroscopic measurements
Absorption spectra were measured to evaluate the electronic structures of the HAT and TPh derivatives. Fig. 1 shows the absorption spectra of TPh (spectrum a), TPhOAc (spectrum b), HAT(COOMe)6 (spectrum c) and C3HAT-TIm (spectrum d) in CH2Cl2. The spectrum of C8HAT (spectrum e) is also shown for comparison although the ε value was not calculated.36 The spectra of TPh and TPhOAc have characteristic strong peaks at around 250–270 nm, whereas the peaks of HAT(COOMe)6, C3HAT-TIm and C8HAT have become broadened and red-shifted. According to the electrochemical measurements and DFT calculations (Table 2), the red-shift trend may result from the relatively low levels of the LUMO states by introducing electron-withdrawing groups compared to those of the HOMO states, which leads to a decrease of the HOMO–LUMO gap.37,38
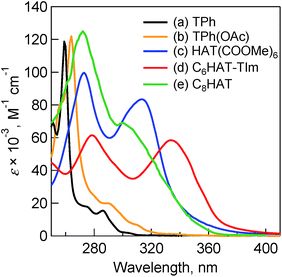 |
| | Fig. 1 Absorption spectra of (a) TPh (black), (b) TPhOAc (yellow), (c) HAT(COOMe)6 (blue) and (d) C6HAT-TIm (red) in 10 μM CH2Cl2. (e) Normalized absorption spectrum of C8HAT (green) for comparison.36 | |
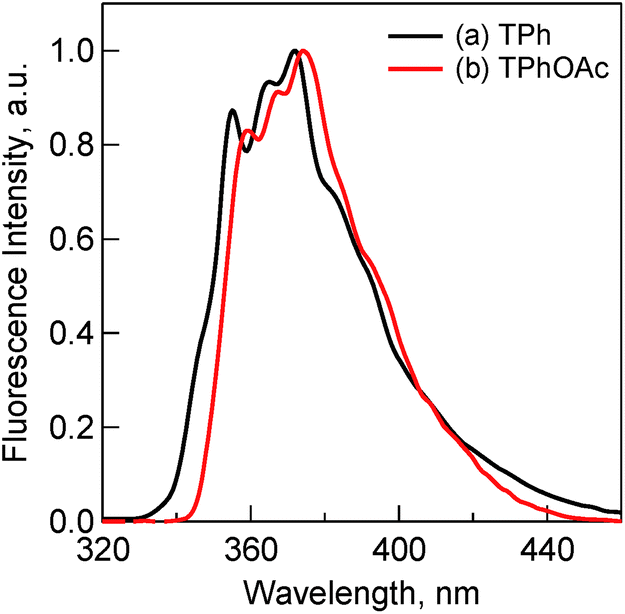
The fluorescence spectra of TPh and TPhOAc (Fig. 2) show a fluorescence peak at ca. 355 nm, whereas the spectra of HAT(COOMe)6 and C3HAT-TIm exhibit extremely small intensities. Consequently, we observed the corresponding phosphorescence spectra at 77 K as shown in Fig. 3. The energies of the triplet excited states of HAT(COOMe)6 and C3HAT-TIm were determined from the phosphorescence maxima, 474 nm and 492 nm, to be 2.61 eV and 2.52 eV, respectively. Additionally, the phosphorescence lifetimes of HAT(COOMe)6 and C3HAT-TIm (77 K) were determined to be 320 ms and 280 ms, respectively (Table 1). This suggests that intersystem crossing occurs efficiently in both compounds. The absorption and emission spectra of C6HAT-TIm and C12HAT-TIm are also very similar to those of C3HAT-TIm.
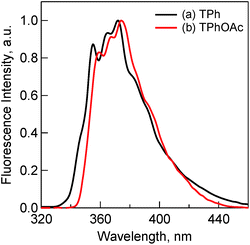 |
| | Fig. 2 Fluorescence spectra of (a) TPh (1.0 μM) (black) and (b) TPhOAc (1.0 μM) (red) in CH2Cl2. Excitation wavelengths are (a) 275 nm and (b) 300 nm, respectively. | |
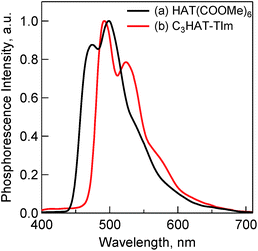 |
| | Fig. 3 Phosphorescence spectra of (a) HAT(COOMe)6 (10 μM) and (b) C3HAT-TIm (50 μM) in MeCN. The excitation wavelength is 330 nm. The measurements were performed at 77 K. | |
Table 1 Emission parameters of TPh and HAT derivativesa
| Compound |
Φ
F
|
Φ
Other
|
τ
FL, ns |
τ
PL, ms |
k
F × 10−6, s−1 |
k
Other × 10−7, s−1 |
|
τ
FL: fluorescence lifetime. τPL: phosphorescence lifetime (77 K). ΦF: fluorescence emission quantum yield. ΦOther: nonradiative quantum yield; ΦOther = 1 − ΦF, kF = ΦFτFL−1, kOther = ΦOtherτFL−1.
Reported values.39
|
| TPh |
0.07b |
0.93 |
37b |
— |
1.8 |
2.5 |
| TPhOAc |
0.13 |
0.87 |
9.4 |
— |
14 |
9.3 |
| HAT(COOMe)6 |
<0.01 |
>0.99 |
— |
320 |
— |
— |
| C3HAT-TIm |
<0.01 |
>0.99 |
— |
280 |
— |
— |
Fluorescence quantum yields
To evaluate the detailed light-emitting properties, we measured the absolute fluorescence quantum yields (ΦF) of these derivatives and the ΦF values are listed in Table 1. The ΦF value of TPhOAc (ΦF = 0.13) is slightly larger than that of TPh (ΦF = 0.07), whereas the ΦF values of HAT(COOMe)6 and C3HAT-TIm are extremely low (ΦF ∼ 0). The low ΦF values of HAT(COOMe)6 and C3HAT-TIm are consistent with the intensities of the phosphorescence spectra in Fig. 3. This is attributable to the enhancement of the intersystem crossing based on spin–orbit coupling because of the introduction of carbonyl groups.38 The low ΦF values and the phosphorescence spectra of C6HAT-TIm and C12HAT-TIm were also similarly observed.
To further investigate and compare the fluorescence properties of TPh and TPhOAc, fluorescence lifetime measurements of these derivatives were performed. The fluorescence decays were examined in CH2Cl2 using a pulsed 404 nm laser light, which excited these moieties. The fluorescence lifetimes (τFL) were evaluated from a monoexponential fitting for the respective compounds and the τFL values are listed in Table 1. The τFL value of TPhOAc (9.4 ns) is much shorter than that of TPh (37 ns). To discuss the excited dynamics carefully, the net rate constants of the above two processes, fluorescence emission (kF) and other processes (kOther), were determined, as shown in Table 2. The kF value of TPhOAc is greater than that of TPh by one order of magnitude. Additionally, in both freebase and zinc porphyrins (e.g., tetraphenylporphyrin), the quantum yields of intersystem crossing (ΦISC:ca. 0.8–0.9) are much larger than those of the fluorescence pathways (ΦF:ca. 0.05–0.10).39b Based on these results, we can conclude that the introduction of substituents successfully contributes to the improvement of the light-emitting property of TPh derivatives.
Table 2 Redox potentials and HOMO–LUMO energies of TPh and HAT derivatives
| Compound |
E
red1
|
E
red2
|
E
red3
|
E
ox
|
HOMOg, eV |
LUMOg, eV |
gap, eV |
|
V vs. SCE in CH2Cl2.
Reported value in dimethylamine/THF.40
Reported value.41
Determined by differential pulse voltammetry (DPV) in THF.
Reported value in CH2Cl2.42
Reported value in MeCN.43
Calculated by B3LYP/6-31+G(d) level.
|
| TPh |
−2.42b |
— |
— |
1.64c |
−5.89 |
−0.92 |
4.97 |
| TPhOAc |
−2.19d |
— |
— |
1.63e |
−6.20 |
−1.54 |
4.66 |
| C12TPh-TIm |
−1.21e |
— |
— |
— |
−7.31 |
−3.25 |
4.06 |
| HAT |
−1.42f |
−1.72f |
— |
— |
−6.89 |
−2.16 |
4.73 |
| HAT(COOMe)6 |
−0.59 |
−1.08 |
— |
— |
−7.58 |
−3.43 |
4.15 |
| C6HAT-TIm |
−0.39 |
−0.85 |
−1.21 |
— |
−7.54 |
−3.63 |
3.91 |
Electrochemical studies and DFT calculations
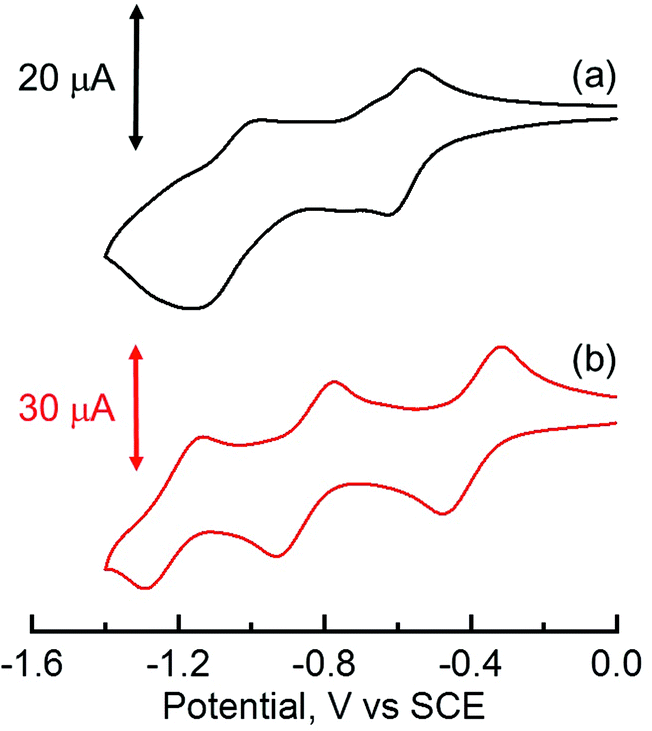
The electrochemical behaviors of TPh and HAT derivatives were investigated by cyclic voltammetry to examine the substituent effects on the reduction and oxidation potentials. The representative voltammograms of HAT(COOMe)6 and C6HAT-TIm in DMF or CH2Cl2 containing 0.10 M tetra-n-butylammonium hexafluorophosphate (n-Bu4NPF6) are shown in Fig. 4. The measured half-wave potentials of these compounds together with reference TPh and HAT are listed in Table 1. The first reduction (Ered1) and oxidation (Eox) potentials of TPh were reported to be −2.42 V and +1.64 V against a saturated calomel electrode (SCE).40,41 Similarly, we determined the first reduction potentials of TPhOAc (Ered1 = −2.19 V), C12TPh-TIm (Ered1 = −1.21 V),42 HAT (Ered1 = −1.42 V),43 HAT(COOMe)6 (Ered1 = −0.59 V) and C6HAT-TIm (Ered1 = −0.39 V). However, no corresponding oxidation potentials of the HAT derivatives could be determined because of the higher oxidation potentials than the solvent. The Ered1 value of C6HAT-TIm is quite similar to those of C3HAT-TIm (Ered1 = −0.39 V) and C12HAT-TIm (Ered1 = −0.40 V) as shown in the ESI (Fig. S4†). With increasing the number of electron-withdrawing groups such as pyrazine, COOMe, OAc and imide units, successive positive shifts of the reduction potentials were observed. In particular, the Ered1 value of HAT-TIm is quite comparable to that of C60 (−0.44 V vs. SCE),31 which indicates that HAT-TIm is a good electron acceptor.
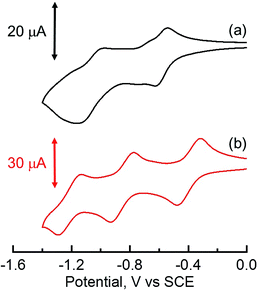 |
| | Fig. 4 Cyclic voltammograms of (a) HAT(COOMe)6 in DMF and (b) C6HAT-TIm in CH2Cl2 with 0.10 M n-Bu4NPF6 as the supporting electrolyte. Reference electrode: standard calomel electrode. Scan rate: 0.10 V s−1. | |
Moreover, the cyclic voltammogram of C6HAT-TIm exhibits the further reduction up to trianions, C6HAT-TIm3−. The HOMO and LUMO levels calculated by the DFT method (Fig. S5 and S6†) also support the above trends observed in the electrochemical data in Table 2. Namely, these shifts of the HOMO and LUMO levels are largely dependent on the electron-withdrawing nature and the number of substituents. Additionally, the LUMO of HAT-TIm is energetically low lying and doubly degenerate, and thus capable of accepting three electrons upon reduction (Fig. 4).
With regard to the porphyrin derivatives such as H2P and ZnP, the first oxidation potentials of H2P and ZnP were determined to be +0.78 V and +0.68 V, respectively (Fig. S7 in the ESI†). The energy level of the charge-separated state of the HAT-TIm and porphyrin (e.g., ZnP) composites (i.e., the HAT-TIm radical anion and the ZnP radical cation) was determined from the difference between the Eox of ZnP and the Ered1 of HAT-TIm (−0.39 V vs. SCE) to be 1.07 eV. This value is smaller than the excited energies of each chromophore: ∼2.1 eV and ∼1.5 eV for the singlet and triplet excited states of ZnP,44 respectively, and ∼3.7 eV and 2.52 eV for the singlet and triplet excited states of HAT-TIm, respectively. Thus, the photoinduced electron transfers from the excited state of ZnP to HAT-TIm and/or ZnP to the excited state of HAT-TIm are energetically favorable because the free energy changes of photoinduced electron transfer are always negative. In such a case, the combination of porphyrin and HAT-TIm units is expected to perform efficient photoinduced electron transfer to form the charge-separated state (vide infra).
Spectroscopic characterization of the reduced C3HAT-TIm
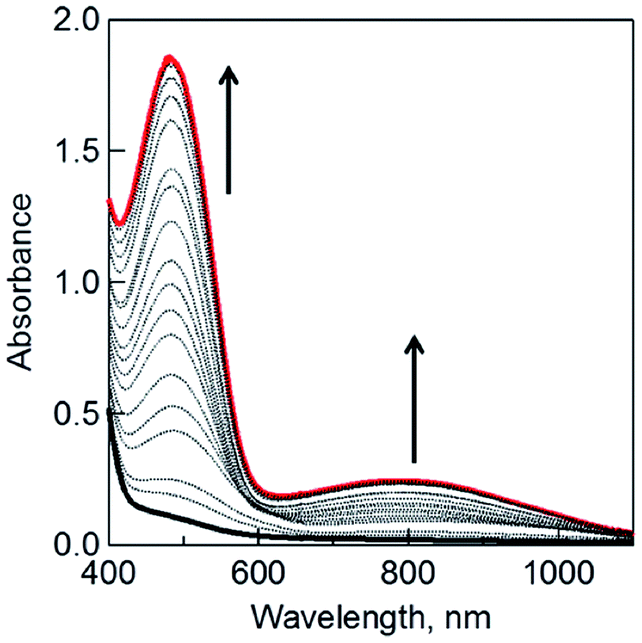
When the dimeric 1-benzyl-1,4-dihydronicotinamide [(BNA)2] was used as an electron donor,45,46 irradiation of a CH2Cl2 solution containing (BNA)2 and C3HAT-TIm with visible light resulted in the photoinduced one-electron reduction of C3HAT-TIm to produce C3HAT-TIm˙−. Fig. 5 shows the absorption spectral change in the photoinduced electron-transfer reduction of C3HAT-TIm to C3HAT-TIm˙−. In this case, a new absorption band at λmax = 482 nm with a broad near-IR absorption band at 800 nm appeared, corresponding to the radical anion species. The molar absorption coefficient of C3HAT-TIm˙− was determined to be 6.2 × 103 M−1 cm−1 at 482 nm.
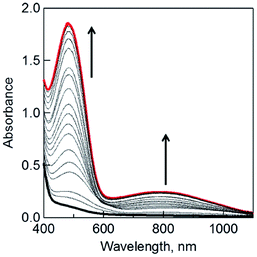 |
| | Fig. 5 The UV-vis spectral changes observed in the photoinduced electron-transfer reduction of C3HAT-TIm (300 μM) with (BNA)2 (150 μM) in deaerated CH2Cl2 at 298 K under photoirradiation with a xenon lamp. | |
Formation constants of CT complexes between HAT-TIm and porphyrins
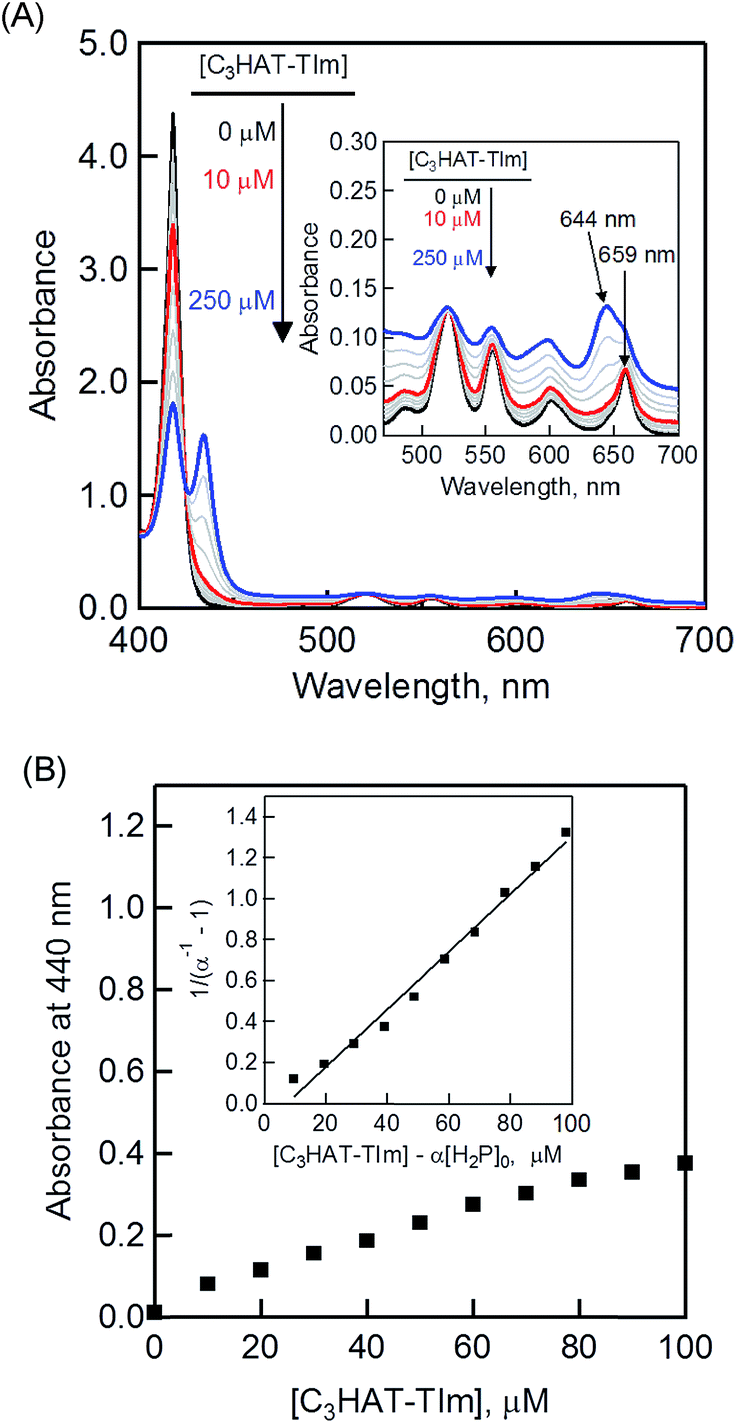
The CT complex formation between HAT-TIm and H2P was examined by the absorption spectral changes (Fig. 6). The absorption of the original Soret band of H2P at 418 nm decreased and a strong absorption peak newly appeared at around 440 nm due to the complexation of C3HAT-TIm with H2P in CH2Cl2 as shown in Fig. 6A, where the inserted figure clearly indicates formation of the typical CT π-complex between H2P and C3HAT-TIm. The CT complex absorption extends up to ∼800 nm (ESI Fig. S8†). The Job's plot in Fig. 7 exhibits a triangle-like shape with a maximum value of 0.48, which indicates that a 1:1 complex (theoretical maximum: 0.50) is formed between H2P and C3HAT-TIm (eqn (1)).| | 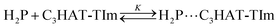 | (1) |
| | | (α−1 − 1)−1 = K([C3HAT-TIm] − α[H2P]0) | (2) |
| | | α = (A − A0)/(A∞ − A0) | (3) |
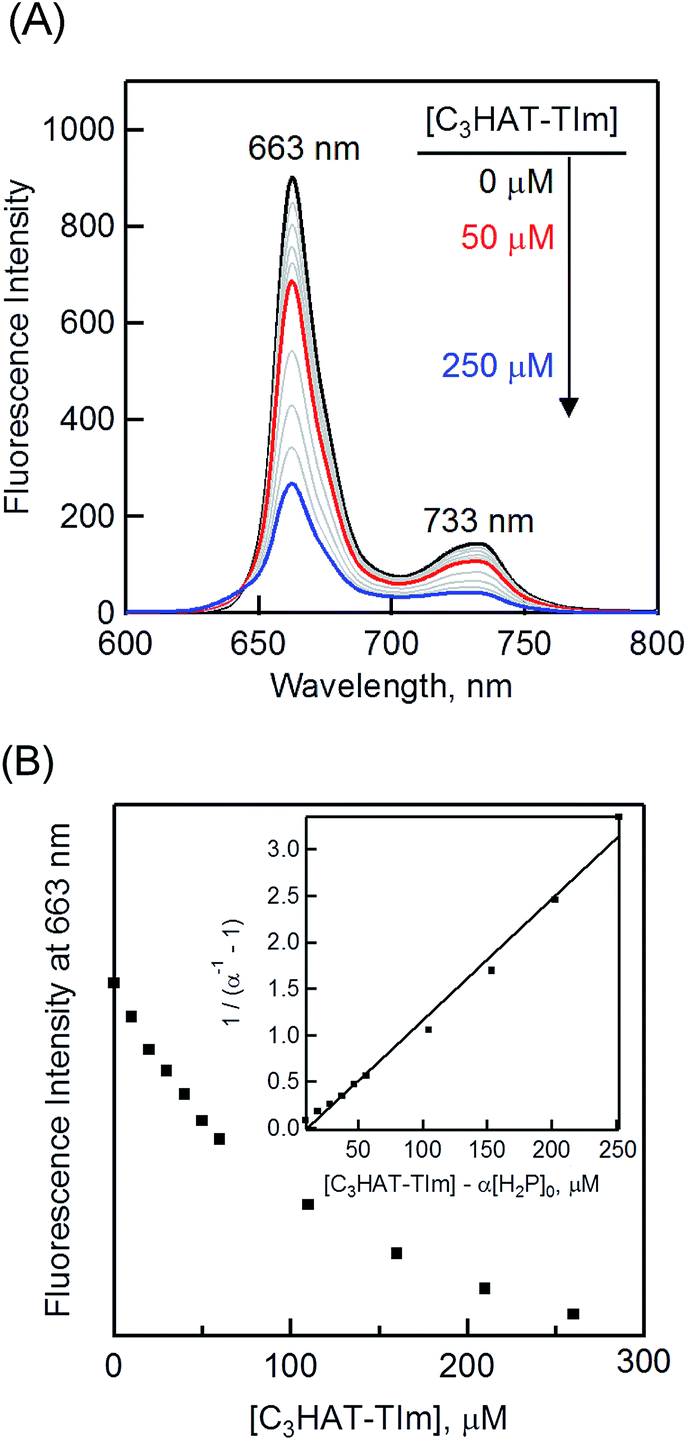
The formation constant (K) was determined by a linear correlation between (α−1 − 1)−1 and ([C3HAT-TIm] − α[H2P]0) in eqn (2) and (3), where A0 and A are the absorbance of H2P at 424 nm in the absence and presence of C3HAT-TIm, and [H2P]0 is the initial concentration of H2P. From the linear plot in the inset of Fig. 6B, the formation constant (K) of the H2P–C3HAT-TIm complex was determined to be 1.4 × 104 M−1 in CH2Cl2 (Fig. 6B). As H2P forms the CT complex with HAT-TIm, the fluorescence emission of H2P was quenched by intramolecular electron transfer from the singlet excited state of H2P to C3HAT-TIm in the complex (Fig. 8A).
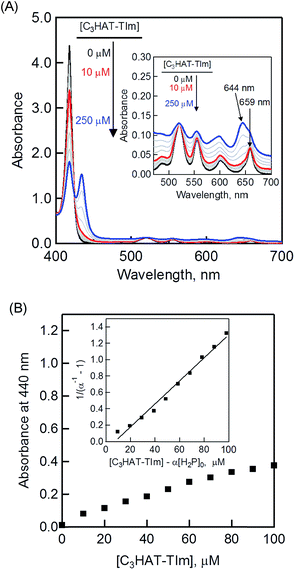 |
| | Fig. 6 (A) Absorption spectral changes of H2P ([H2P] = 10 μM) upon addition of C3HAT-TIm (0–100 μM) in CH2Cl2. The inserted expanded figure indicates a broad CT absorption of the H2P–C3HAT-TIm complex. (B) Absorption profile at 440 nm. Inset: plot of (α−1 − 1) −1vs. [C3HAT-TIm] − α[H2P]0. | |
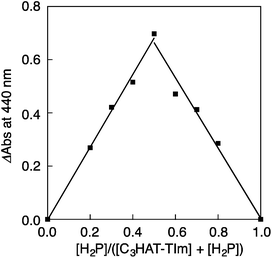 |
| | Fig. 7 The Job's plot obtained by the absorption change at 440 nm for the complex formation between H2P and C3HAT-TIm. The symmetric plot with maxima at 0.5 mole fraction indicates formation of the 1:1 complex in the present system. | |
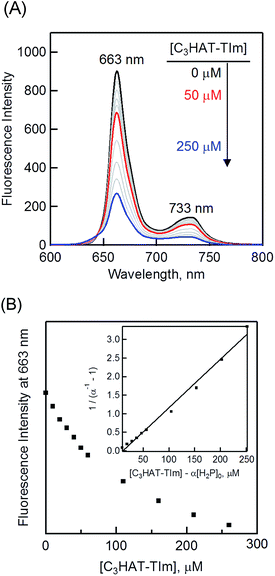 |
| | Fig. 8 (A) The fluorescence spectral changes of H2P ([H2P] = 10 μM) upon addition of increasing equivalents of C3HAT-TIm (0–250 μM) in CH2Cl2. Excitation wavelength: 550 nm. (B) Plot of the fluorescence intensity vs. [C3HAT-TIm] at 663 nm. Inset: plot of (α−1 − 1)−1vs. [C3HAT-TIm] − α[H2P]0 according to eqn (2) and (3). | |
The formation constant K of the H2P–C3HAT-TIm complex was determined from the changes in the fluorescence intensities at 663 nm (Fig. 8B) to be 1.3 × 104 M−1, which agrees with the value determined from the absorption spectral changes in Fig. 6B.47 The formation constants K between the porphyrins (i.e., H2P and ZnP) and the HAT-TIm derivatives are summarized in Table 3. The largest value of formation constant K was obtained for H2P–C6HAT-TIm as 2.1 × 104 M−1. The longer alkyl chain unit may enhance the CT π-complex formation because of the additional van der Waals interaction. This is similar to that obtained for H2P–C12HAT (Fig. S9 and S10 in ESI†). When H2P was replaced by ZnP, the formation constants became smaller (Table 3 and Fig. S11–S13 in ESI†).
Table 3 Formation constants determined by fluorescence titration of H2P/ZnP and the HAT-TIm derivatives in CH2Cl2
| HAT-TIm |
K (H2P), M−1 |
K (ZnP), M−1 |
| C3HAT-TIm |
1.3 × 104 |
3.0 × 103 |
| C6HAT-TIm |
2.1 × 104 |
6.5 × 103 |
| C12HAT-TIm |
1.8 × 104 |
5.2 × 103 |
1H NMR titration
The 1H NMR signals of H2P exhibit upfield shifts of β protons upon complexation with HAT-TIm as shown in Fig. 9. This is ascribed to the influence of the large porphyrin aromatic ring current. This result indicates that the two-dimensional π-structure of HAT-TIm interacts with that of H2P.
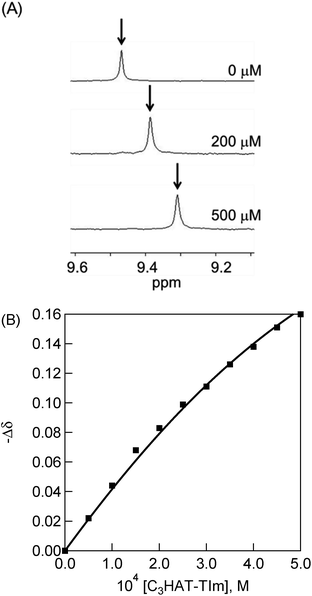 |
| | Fig. 9 (A) 1H NMR titration of H2P ([H2P] = 500 μM) upon addition of C3HAT-TIm (0–500 μM) in CDCl3. (B) 1H NMR titration curve obtained from the chemical shift changes of the β proton of H2P by adding C3HAT-TIm. | |
The signals of the free H2P and the complexed H2P always coalesce into a single signal. This indicates that the complexation and exchange occur at a faster rate than the NMR time scale. The formation constant between H2P and HAT-TIm was determined from the peak shifts of the β protons in the H2P moiety using the following eqn (4):48
| | 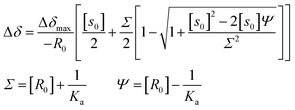 | (4) |
where Δ
δ is the observed change in chemical shift, Δ
δmax is the saturation value, and
S0 and
R0 are the total concentrations of guest and receptor, respectively.
A sample titration curve can be seen in the case of C3HAT-TIm as the guest (Fig. 9B), and the formation constant was determined to be 5.1 × 103 M−1. Similarly, the formation constant of H2P–C6HAT-TIm was determined to be 8.9 × 103 M−1 (Fig. S14 in the ESI†). The formation constants between the porphyrins and the HAT-TIm derivatives in CDCl3 increase with increasing alkyl chain lengths as observed in the spectroscopic measurements in CH2Cl2. The K values in CDCl3 are somewhat smaller than those in CH2Cl2 due to the less stabilization of CT complexes in the less polar solvent (CDCl3).
DFT computational studies of supramolecular CT π-complexes
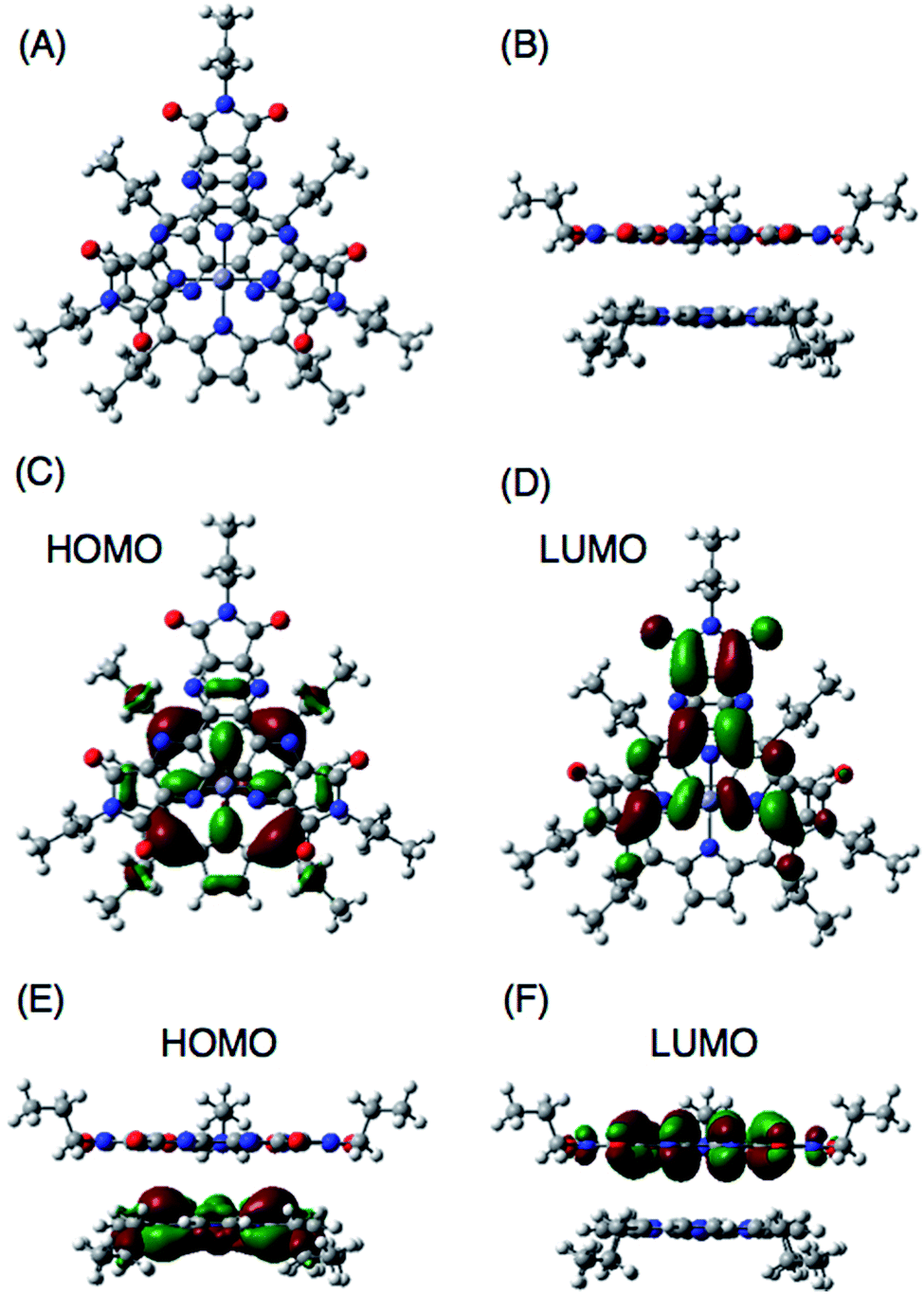
DFT calculations also support the CT π-complex formation between the porphyrin and HAT-TIm units. Fig. 10A and B show the face-to-face planar structure of the CT complex composed of ZnP and C3HAT-TIm. The distance between the porphyrin and HAT units is found to be 3.8 Å. The calculated HOMO and LUMO orbitals of the CT π-complex are localized on the porphyrin and HAT-TIm units, respectively (Fig. 10C–F).49 A similar trend was observed in the case of ZnP and C3HAT-TIm. A TD-DFT calculation for ZnP–C3HAT-TIm was carried out using the TD-B3LYP/6-31G(d)//B3LYP/6-31G(d) basis set to assign the absorption band at the NIR region (λmax = 800 nm, Fig. 6A and Fig. S8†).50 The calculated absorption band was obtained at 815 nm with an oscillator strength of f = 0.172, ascribable to charge transfer from the ZnP moiety to the C3HAT-TIm moiety. The calculated results are shown in S15 (ESI†). We also carried out TD-DFT calculations for ZnP and C3HAT-TIm as references, indicating that no absorption band was obtained at the NIR region (S15–S17 in the ESI†).
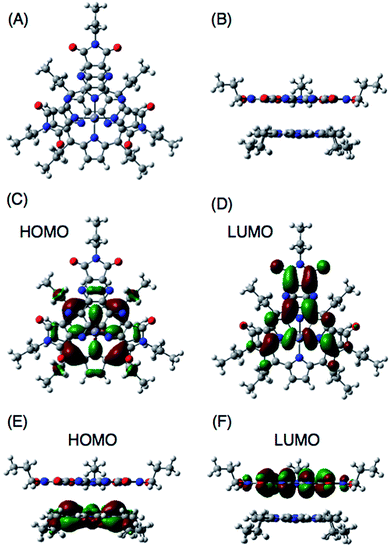 |
| | Fig. 10 B3LYP/6-31G(d) optimized structure of ZnP and C3HAT-TIm (A and B). The HOMO and LUMO of the π-complex are shown in top view (C and D) and side view (E and F), respectively. In ZnP, the 11-carbon alkyl chains were replaced with 3-carbon chains to save computing time. | |
Ultrafast photoinduced electron transfer in CT π-complexes between porphyrins and HAT-TIm
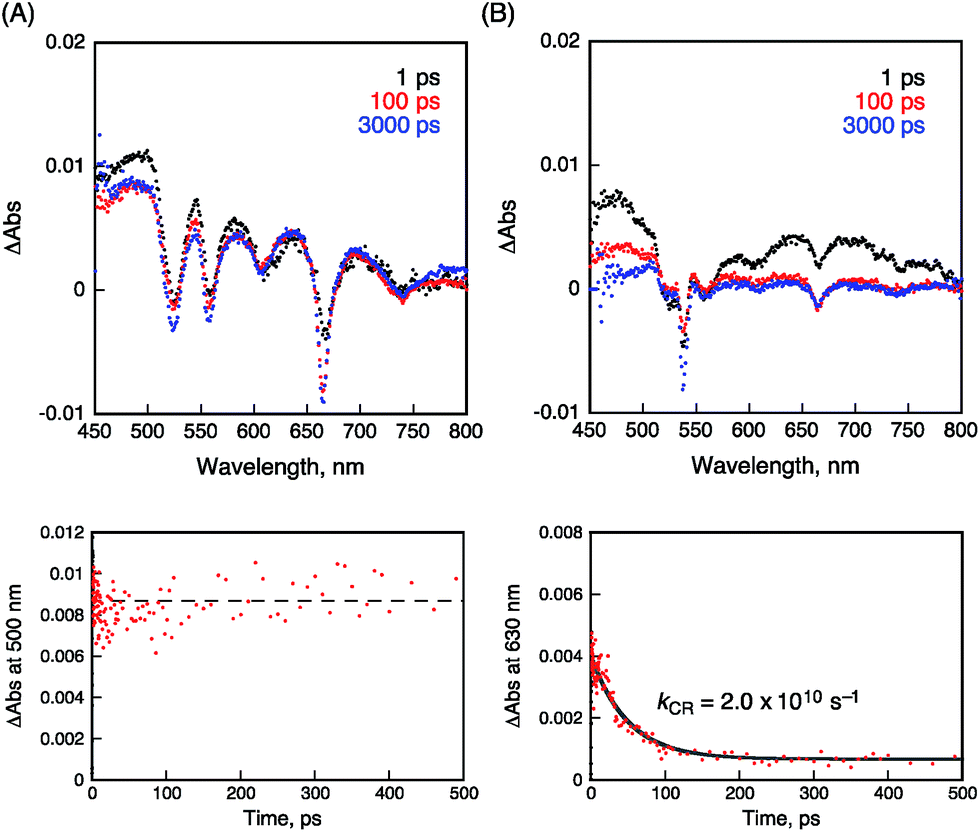
The occurrence of ultrafast electron transfer from the singlet excited states of the porphyrins to HAT-TIm in the complexes was further confirmed by femtosecond laser-induced transient absorption measurements. The transient absorption spectra of pristine H2P in toluene using a 430 nm laser pulse, which selectively excited only the porphyrin units, show the singlet–singlet transient absorption and fluorescence bleaching bands (ca. 660 nm) of H2P (Fig. 11A). In the case of H2P–C3HAT-TIm, we employed a large excess concentration of C3HAT-TIm (8 mM) relative to that of H2P to make sure that all of the H2P molecules formed the CT complex with C3HAT-TIm (>99%) in Fig. 11B. The transient absorption spectra of H2P–C3HAT-TIm exhibit a broad absorption in the ca. 600–700 nm region within 1 ps after laser pulse excitation due to H2P˙+,44,51 without fluorescence bleaching at ca. 660 nm (Fig. 11B). The radical anion species of C3HAT-TIm can also be seen at around 500 nm by comparing the absorption spectrum of the reduced C3HAT-TIm (Fig. 5).
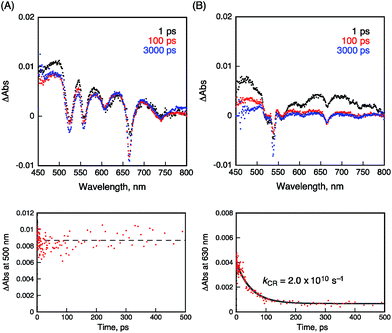 |
| | Fig. 11 Femtosecond laser-induced transient absorption spectra and corresponding time profiles of (A) H2P and (B) H2P–C3HAT-TIm obtained at 1.0 ps (black), 100 ps (red) and 3000 ps (blue) after laser pulse in toluene. The time profiles were detected at 500 and 630 nm, respectively. The concentrations of H2P and C3HAT-TIm were 10 μM and 8.0 μM, respectively. Excitation wavelength was 430 nm. | |
This indicates that photoinduced charge separation occurs immediately within 1 ps upon photoexcitation of the charge-transfer without showing the singlet–singlet transient absorption due to 1H2P*. Such ultrafast charge separation is unprecedented for supramolecular electron donor–acceptor complexes,12,17–22,52–54 resulting from a strong interaction between H2P and a HAT-TIm derivative in the face-to-face planar structure in the CT π-complex (Fig. 10B).
The charge-recombination dynamics were monitored from the decay of the transient absorption at 630 nm due to H2P˙+ as shown in Fig. 11B. The rate constant of the charge recombination (kCR) was determined to be 2.0 × 1010 s−1. A similar kCR value (2.2 × 1010 s−1) was obtained for the H2P–C6HAT-TIm complex as shown in Fig. S18 (ESI†). Additionally, the transient spectra of ZnP and ZnP–C3HAT-TIm are shown in the ESI Fig. S19.† However, photoinduced charge separation of ZnP–C3HAT-TIm was not confirmed under our experimental set-up. This is probably attributable to the smaller formation constant (K = 3.0 × 103 M−1) than that of H2P–C3HAT-TIm (K = 1.3 × 104 M−1) as shown in Table 3.
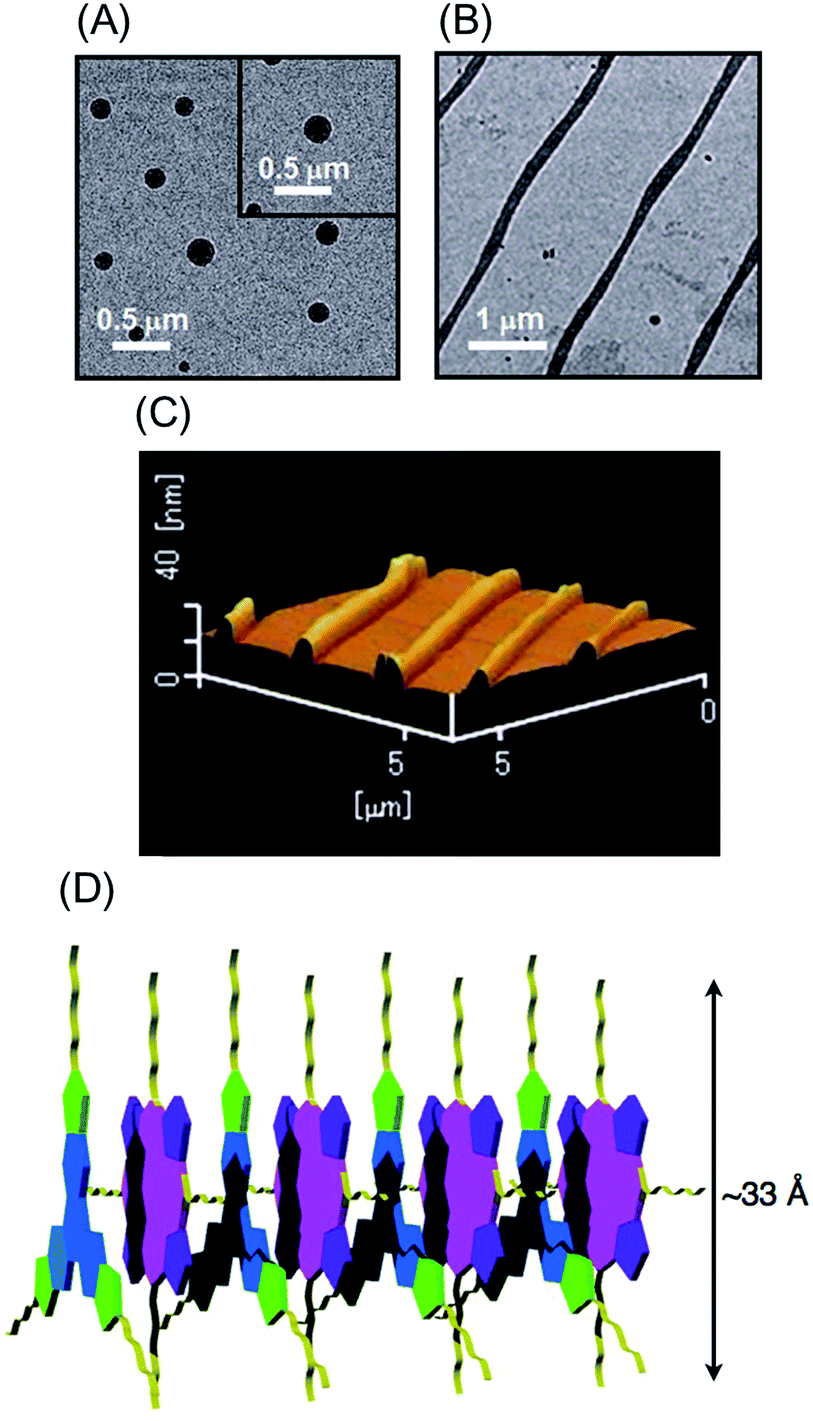
High-order organization of the H2P–C3HAT-TIm complex observed by TEM and AFM
Evaporation of the solvent from a CH2Cl2 solution of the H2P–C3HAT-TIm complex resulted in high-order supramolecular organization, which was observed by TEM and AFM. The supramolecular organization patterns composed of C3HAT-TIm and H2P were obtained by the following method.55 First, we optimized the experimental conditions (10 μM CH2Cl2 solution of C3HAT-TIm and H2P) by examining the concentration effect (5–50 μM) on the aggregate structures. Then, the solution (the optimized concentration: 10 μM) was simply cast onto the carbon-coated copper film (TEM grid) and dried in air. In the solvent evaporation process, the linear molecular pattern was effectively formed. The assemblies of the C3HAT-TIm reference system were also prepared in the same manner. TEM measurements of the C3HAT-TIm reference system without porphyrins showed many spherical assemblies (Fig. 12A), whereas aligned fibrous patterns (linear aggregates) were observed for the H2P–C3HAT-TIm complex (Fig. 12B). The approximate average width of the H2P–C3HAT-TIm assemblies was estimated to be ∼200 nm. AFM measurements including the cross-sectional height information were also performed as shown in Fig. 12C. In the AFM image, we could see the surface patterning, which is very similar to the corresponding TEM images (Fig. 12B). The cross-sectional data showed the average height: 10.1 nm. Considering the chemical structure of H2P (approximate molecular size: ∼33 Å estimated by DFT) in Fig. 12D, the average height approximately corresponds to a few layers of the H2P–C3HAT-TIm composite units.
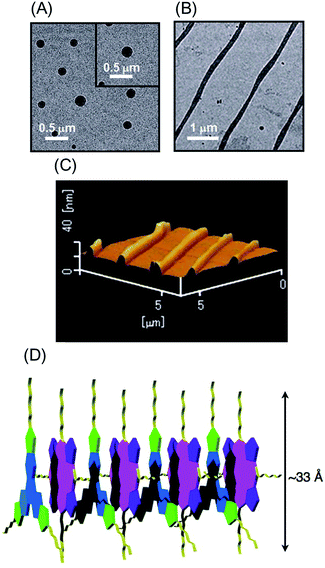 |
| | Fig. 12 TEM images of (A) the C3HAT-TIm assembly and (B) the H2P–C3HAT-TIm assembly. The images were taken by drop cast of the CH2Cl2 solution of the components onto the grid. (C) AFM image of the H2P–C3HAT-TIm composites. (D) Schematic illustration of the proposed supramolecular structures between H2P (pink/purple) and C3HAT-TIm (blue/green). | |
Conclusions
The present study has demonstrated the formation of face-to-face 1:1 CT π-complexes between 1,4,5,8,9,12-hexaazatriphenylene (HAT) derivatives and porphyrins, which undergo ultrafast photoinduced electron transfer in which HAT derivatives with electron-withdrawing groups act as good electron acceptors. Unprecedented ultrafast charge-separated states were successfully formed by CT π-complexes. The CT π-complexes also contribute to the highly ordered patterning on the solid-state film. This simple method for molecular organization provides a new perspective for the construction and development of efficient molecular electronic and energy conversion systems.
Experimental section
General information
Triphenylene and hexaacetoxytriphenylene were purchased from Tokyo Chemical Industry (TCI). They were used after reprecipitation from dichloromethane and hexane for the spectroscopic and electrochemical measurements. All solvents and reagents of the best grade available were purchased from commercial suppliers and were used without further purification. Column flash chromatography was performed on silica gel (Kanto Chemical Silica gel 60N, 40–50 μm or 100–210 μm). We used an LC-9204 apparatus equipped with a pump (JAI PI-60, flow rate 2.5 mL min−1), a UV detector (JAI UV-3740) and two columns (JAIGEL 2H and 1H, 40 × 600 mm for each). All experiments except single crystal X-ray diffraction measurements were performed at room temperature. 1H NMR and 13C NMR spectra were recorded on a 400 MHz spectrometer JEOL JNM-A400, JNM-Al400, or JNM-ECX 400, using the solvent peak as the reference standard, with chemical shifts given in parts per million. CDCl3 was used as a solvent for NMR measurements. MALDI-TOF mass spectra were recorded on a Bruker Ultraflex.
Electrochemical measurements
Cyclic voltammograms were recorded on an Iviumstat 20 V/2.5 A potentiostat using a three electrode system. A platinum electrode was used as the working electrode. A platinum wire served as the counter electrode, and a saturated calomel electrode was used as the reference electrode. A ferrocene/ferrocenium redox couple was used as the internal standard. All the solutions were purged using nitrogen gas prior to the electrochemical and spectral measurements.
Spectroscopic measurements
UV/Vis absorption spectra were recorded on a PerkinElmer (Lambda 750) UV-VIS-NIR spectrophotometer. Fluorescence and phosphorescence emission spectra were recorded on a PerkinElmer (LS-55) spectrofluorophotometer. Fluorescence lifetimes were measured on a HORIBA Scientific time-correlated single-photon counting system (FluoroCube) with the laser light (DeltaDiode, laser diode head, 404 nm, pulse width: 100 ps) as the excitation source. Phosphorescence lifetimes were measured on a JASCO FP-8500. The absolute fluorescence quantum yields were determined by a Hamamatsu Photonics C9920-02 system equipped with an integrating sphere and a red-sensitive multichannel photodetector (PMA-12): excitation wavelength = 300 nm.
Laser flash photolysis measurements
Femtosecond laser-induced transient absorption measurements were conducted using an ultrafast source: Integra-C (Quantronix Corp.) and a commercially available optical detection system: Helios provided by Ultrafast Systems LLC. The detailed instrumentations are given in the ESI.†
Synthesis of C3HAT-TIm
HAT 6 (0.52 mol, 0.26 g) was dissolved in acetic anhydride (15 mL), and the solution was stirred for 15 min at 115 °C. After evaporation of the solvent, the resulting solid was dissolved in acetonitrile (20 mL). Then, n-propylamine (12.2 mol, 1 mL) was injected into the mixture solution with a syringe and precipitation immediately arose. The resulting solid was collected by filtration. The solid was dissolved in thionyl chloride (10 mL), and the solution was stirred for 12 h at room temperature. After the evaporation of thionyl chloride, flash column chromatography on silica gel with chloroform/methanol (1:1 v/v) as the eluent afforded C3HAT-TIm. Yield: 0.23 g (77.4%). 1H NMR (400 MHz, CDCl3): δ = 3.83 (t, J = 7.1 Hz, 2H, NCH2), 1.78 (dd, J = 7.3 Hz, 7.6 Hz, 2H, CH2), 1.02 (t, J = 7.6 Hz, 3H, CH3); 13C NMR (98.5 MHz, CDCl3): δ = 164.1 (CAr), 149.0 (CAr), 143.8 (CAr), 40.1 (CAl), 21.3 (CAl), 11.4 (CAl); MALDI-TOF MS: calcd for C27H21N9O6: 567.16, found 567.18 [M]. 1H NMR, 13C NMR and MALDI-TOF mass spectra are shown in the ESI Fig. S1–S3.†
Theoretical calculations
Density functional theory (DFT) calculations of ZnP–C3HAT-TIm, ZnP and C3HAT-TIm were performed with Gaussian 09 (Revision A.02, Gaussian, Inc.). The calculations were performed on a 32-processor QuantumCube™ at the B3LYP/6-31G(d) level of theory.56 Graphical outputs of the computational results were generated with the GaussView software program (ver. 3.09) developed by Semichem, Inc.57 Electronic excitation energies and intensities were computed by the time-dependent (TD)-DFT calculation at the B3LYP/6-31G(d) level. The size of the integration grid used for all calculations was 4. In each case, 30 excited states were calculated by including all one-electron excitations within an energy window of ±3 hartrees with respect to the HOMO–LUMO energies.
Acknowledgements
This work was partially supported by Grant-in-Aid for Scientific Research (no. 26286017 & 26620159 to T.H., no. 26620154 & 26288037 to K.O., no. 26102012 to T.T.) and the Science Research Promotion Fund from the Promotion and Mutual Aid Corporation for Private Schools from MEXT, Japan, and the Mitsubishi Foundation.
Notes and references
-
(a) S. Kirner, M. Sekita and D. M. Guldi, Adv. Mater., 2014, 26, 1482–1493 CrossRef CAS PubMed
 ;
(b) J. Malig, N. Jux and D. M. Guldi, Acc. Chem. Res., 2013, 46, 53–64 CrossRef CAS PubMed ;
(c) G. Bottari, G. De la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed .
;
(b) J. Malig, N. Jux and D. M. Guldi, Acc. Chem. Res., 2013, 46, 53–64 CrossRef CAS PubMed ;
(c) G. Bottari, G. De la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed .
-
(a) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed ;
(b) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed ;
(c) J. Frey, G. Kodis, S. D. Straight, T. A. Moore, A. L. Moore and D. Gust, J. Phys. Chem. A, 2013, 117, 607–615 CrossRef CAS PubMed .
-
(a) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed ;
(b) S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283–2297 RSC ;
(c) K. Ohkubo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2009, 82, 303–315 CrossRef CAS ;
(d) S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 177–195 CrossRef CAS ;
(e) S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609–620 RSC .
-
(a) Q. Yan, Z. Luo, K. Cai, Y. Ma and D. Zhao, Chem. Soc. Rev., 2014, 43, 4199–4221 RSC ;
(b) P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC ;
(c) Y. K. Kang, P. M. Iovine and M. J. Therien, Coord. Chem. Rev., 2011, 255, 804–824 CrossRef CAS ;
(d) B. Albinsson and J. Mårtensson, J. Photochem. Photobiol., C, 2008, 9, 138–155 CrossRef CAS .
-
(a) M. E. El-Khouly, S. Fukuzumi and F. D'Souza, ChemPhysChem, 2014, 15, 30–47 CrossRef CAS PubMed ;
(b) F. D'Souza and O. Ito, Chem. Soc. Rev., 2012, 41, 86–96 RSC ;
(c) S. Fukuzumi, K. Ohkubo, F. D'Souza and J. L. Sessler, Chem. Commun., 2012, 48, 9801–9815 RSC ;
(d) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913–4928 RSC ;
(e) R. Chitta and F. D'Souza, J. Mater. Chem., 2008, 18, 1440–1471 RSC .
-
(a) M. Natali, S. Campagna and F. Scandola, Chem. Soc. Rev., 2014, 43, 4005–4018 RSC ;
(b) A. Arrigo, A. Santoro, M. T. Indelli, M. Natali, F. Scandola and S. Campagna, Phys. Chem. Chem. Phys., 2014, 16, 818–826 RSC ;
(c) D. Hanss, M. E. Walther and O. S. Wenger, Coord. Chem. Rev., 2010, 254, 2584–2592 CrossRef CAS .
-
(a) D. M. Guldi and R. D. Costa, J. Phys. Chem. Lett., 2013, 4, 1489–1501 CrossRef CAS PubMed ;
(b) K. Tambara and G. D. Pantos, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2012, 108, 186–201 RSC .
- G. Bottari, O. Trukhina, M. Ince and T. Torres, Coord. Chem. Rev., 2012, 256, 2453–2477 CrossRef CAS .
-
(a) S. Fukuzumi and K. Ohkubo, Dalton Trans., 2013, 42, 15846–15858 RSC ;
(b) S. Fukuzumi and K. Ohkubo, J. Mater. Chem., 2012, 22, 4575–4587 RSC ;
(c) S. Fukuzumi, T. Honda and T. Kojima, Coord. Chem. Rev., 2012, 256, 2488–2502 CrossRef CAS .
-
(a) J. L. Sessler, B. Wang and A. Harriman, J. Am. Chem. Soc., 1993, 115, 10418–10419 CrossRef CAS ;
(b) F. Wessendorf, J.-F. Gnichwitz, G. H. Sarova, K. Hager, U. Hartnagel, D. M. Guldi and A. Hirsch, J. Am. Chem. Soc., 2007, 129, 16057–16071 CrossRef CAS PubMed ;
(c) H. Kar and S. Ghosh, Chem. Commun., 2014, 50, 1064–1066 RSC ;
(d) S. Verma, A. Ghosh, A. Das and H. N. Ghosh, Chem.–Eur. J., 2011, 17, 3458–3464 CrossRef CAS PubMed ;
(e) S. Murphy, L. Huang and P. V. Kamat, J. Phys. Chem. C, 2011, 115, 22761–22769 CrossRef CAS .
-
(a) M. Gallego, J. Calbo, J. Aragó, R. M. Krick Calderon, F. H. Liquido, T. Iwamoto, A. K. Greene, E. A. Jackson, E. M. Pérez, E. Ortí, D. M. Guldi, L. T. Scott and N. Martín, Angew. Chem., Int. Ed., 2014, 53, 2170–2175 CrossRef CAS PubMed ;
(b) K. S. Suslick and R. A. Watson, New J. Chem., 1992, 16, 633–642 CAS ;
(c) I. Duchemin and X. Blase, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 245412 CrossRef .
-
(a) N. L. Bill, M. Ishida, Y. Kawashima, K. Ohkubo, Y. M. Sung, V. M. Lynch, J. M. Lim, D. Kim, J. L. Sessler and S. Fukuzumi, Chem. Sci., 2014, 5, 3888–3896 RSC ;
(b) N. L. Bill, M. Ishida, S. Bähring, J. M. Lim, S. Lee, C. M. Davis, V. M. Lynch, K. A. Nielsen, J. O. Jeppesen, K. Ohkubo, S. Fukuzumi, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2013, 135, 10852–10862 CrossRef CAS PubMed ;
(c) C. M. Davis, Y. Kawashima, K. Ohkubo, J. M. Lim, D. Kim, S. Fukuzumi and J. L. Sessler, J. Phys. Chem. C, 2014, 118, 13503–13513 CrossRef CAS .
-
(a) P. Li, S. Amirjalayer, F. Hartl, M. Lutz, B. de Bruin, R. Becker, S. Woutersen and J. N. H. Reek, Inorg. Chem., 2014, 53, 5373–5383 CrossRef CAS PubMed ;
(b) P. K. Poddutoori, N. Zarrabi, A. G. Moiseev, R. Gumbau-Brisa, S. Vassiliev and A. van der Est, Chem.–Eur. J., 2013, 19, 3148–3161 CrossRef CAS PubMed ;
(c) E. Iengo, G. Dan Pantosx, J. K. M. Sanders, M. Orlandi, C. Chiorboli, S. Fracasso and F. Scandola, Chem. Sci., 2011, 2, 676–685 RSC .
-
(a) T. Kamimura, K. Ohkubo, Y. Kawashima, H. Nobukuni, Y. Naruta, F. Tani and S. Fukuzumi, Chem. Sci., 2013, 4, 1451–1461 RSC ;
(b) H. Nobukuni, Y. Shimazaki, H. Uno, Y. Naruta, K. Ohkubo, T. Kojima, S. Fukuzumi, S. Seki, H. Sakai, T. Hasobe and F. Tani, Chem.–Eur. J., 2010, 16, 11611–11623 CrossRef CAS PubMed .
-
(a) Y. Kawashima, K. Ohkubo, K. Mase and S. Fukuzumi, J. Phys. Chem. C, 2013, 117, 21166–21177 CrossRef CAS ;
(b) K. Ohkubo, Y. Kawashima and S. Fukuzumi, Chem. Commun., 2012, 48, 4314–4316 RSC ;
(c) S. Fukuzumi, K. Ohkubo, Y. Kawashima, D. S. Kim, J. S. Park, A. Jana, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 15938–15941 CrossRef CAS PubMed .
-
(a) S. Fukuzumi, T. Honda and T. Kojima, Coord. Chem. Rev., 2012, 256, 2488–2502 CrossRef CAS ;
(b) M. Kanematsu, P. Naumov, T. Kojima and S. Fukuzumi, Chem.–Eur. J., 2011, 17, 12372–12384 CrossRef CAS PubMed ;
(c) T. Honda, T. Nakanishi, K. Ohkubo, T. Kojima and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 10155–10163 CrossRef CAS PubMed ;
(d) T. Kojima, T. Honda, K. Ohkubo, M. Shiro, T. Kusukawa, T. Fukuda, N. Kobayashi and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 6712–6716 CrossRef CAS PubMed .
-
(a) F. D'Souza, A. N. Amin, M. E. El-Khouly, N. K. Subbaiyan, M. E. Zandler and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 654–664 CrossRef PubMed ;
(b) S. Fukuzumi, K. Saito, K. Ohkubo, T. Khoury, Y. Kashiwagi, M. A. Absalom, S. Gadde, F. D'Souza, Y. Araki, O. Ito and M. J. Crossley, Chem. Commun., 2011, 47, 7980–7982 RSC ;
(c) M. E. El-Khouly, D. K. Ju, K.-Y. Kay, F. D'Souza and S. Fukuzumi, Chem.–Eur. J., 2010, 16, 6193–6202 CrossRef CAS PubMed ;
(d) A. Takai, M. Chkounda, A. Eggenspiller, C. P. Gros, M. Lachkar, J.-M. Barbe and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 4477–4489 CrossRef CAS PubMed ;
(e) F. D'Souza, E. Maligaspe, K. Ohkubo, M. E. Zandler, N. K. Subbaiyan and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 8787–8797 CrossRef PubMed ;
(f) F. D'Souza, N. K. Subbaiyan, Y. Xie, J. P. Hill, K. Ariga, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 16138–16146 CrossRef PubMed ;
(g) V. Bandi, M. E. El-Khouly, K. Ohkubo, V. N. Nesterov, M. E. Zandler, S. Fukuzumi and F. D'Souza, J. Phys. Chem. C, 2014, 118, 2321–2332 CrossRef CAS ;
(h) C. B. K. C., S. K. Das, K. Ohkubo, S. Fukuzumi and F. D'Souza, Chem. Commun., 2012, 48, 11859–11861 RSC .
-
(a) S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427–1439 RSC ;
(b) T. Kojima, T. Nakanishi, T. Honda and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2009, 13, 14–21 CrossRef CAS ;
(c) S. Fukuzumi, T. Honda, K. Ohkubo and T. Kojima, Dalton Trans., 2009, 3880–3889 RSC ;
(d) T. Kojima, K. Hanabusa, K. Ohkubo, M. Shiro and S. Fukuzumi, Chem.–Eur. J., 2010, 16, 3646–3655 CrossRef CAS PubMed ;
(e) T. Honda, T. Nakanishi, K. Ohkubo, T. Kojima and S. Fukuzumi, J. Phys. Chem. C, 2010, 114, 14920 CrossRef .
-
(a) P. Mondal, A. Chaudhary and S. P. Rath, Dalton Trans., 2013, 42, 12381–12394 RSC ;
(b) A. Chaudhary and S. P. Rath, Chem.–Eur. J., 2012, 18, 7404–7417 CrossRef CAS PubMed ;
(c) S. Fukuzumi, I. Amasaki, K. Ohkubo, C. P. Gros, R. Guilard and J.-M. Barbe, RSC Adv., 2012, 2, 3741–3747 RSC ;
(d) S. S. Gayathri, M. Wielopolski, E. M. Pérez, G. Fernández, L. Sánchez, R. Viruela, E. Ortí, D. M. Guldi and N. Martín, Angew. Chem., Int. Ed., 2009, 48, 815–819 CrossRef CAS PubMed ;
(e) M. Tanaka, K. Ohkubo, C. P. Gros, R. Guilard and S. Fukuzumi, J. Am. Chem. Soc., 2006, 128, 14625–14633 CrossRef CAS PubMed .
-
(a)
P. D. Harvey, C. Stern and R. Guilard, in Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing, Singapore, 2011, vol. 11, p. 1–177 Search PubMed ;
(b) M. K. Panda, K. Ladomenou and A. G. Coutsolelos, Coord. Chem. Rev., 2012, 256, 2601–2627 CrossRef CAS ;
(c) J. Yang, M.-C. Yoon, H. Yoo, P. Kim and D. Kim, Chem. Soc. Rev., 2012, 41, 4808–4826 RSC ;
(d) V. K. Praveen, C. Ranjith, E. Bandini, A. Ajayaghosh and N. Armaroli, Chem. Soc. Rev., 2014, 43, 4222–4242 RSC .
-
(a) N. Aratani, D. Kim and A. Osuka, Acc. Chem. Res., 2009, 42, 1922–1934 CrossRef CAS PubMed ;
(b) A. Uetomo, M. Kozaki, S. Suzuki, K.-i. Yamanaka, O. Ito and K. Okada, J. Am. Chem. Soc., 2011, 133, 13276–13279 CrossRef CAS PubMed ;
(c) J.-M. Camus, S. M. Aly, D. Fortin, R. Guilard and P. D. Harvey, Inorg. Chem., 2013, 52, 8360–8368 CrossRef CAS PubMed .
-
(a) M. Beyler, L. Flamigni, V. Heitz, J.-P. Sauvage and B. Ventura, Photochem. Photobiol., 2014, 90, 275–286 CrossRef CAS PubMed ;
(b) A. Satake and Y. Kobuke, Tetrahedron, 2005, 61, 13–41 CrossRef CAS ;
(c) F. Hajjaj, Z. S. Yoon, M.-C. Yoon, J. Park, A. Satake, D. Kim and Y. Kobuke, J. Am. Chem. Soc., 2006, 128, 4612–4623 CrossRef CAS PubMed .
- D. Adam, P. Schuhmacher, J. Simmerer, L. Haeussling, K. Siemensmeyer, K. H. Etzbach, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141–143 CrossRef CAS .
- Y. Wang, C. Zhang, H. Wu and J. Pu, J. Mater. Chem. C, 2014, 2, 1667–1674 RSC .
-
(a) K. S. Mali, M. G. Schwab, X. Feng, K. Müllen and S. De Feyter, Phys. Chem. Chem. Phys., 2013, 15, 12495–12503 RSC ;
(b) F. Hu, Y. Gong, X. Zhang, J. Xue, B. Liu, T. Lu, K. Deng, W. Duan, Q. Zeng and C. Wang, Nanoscale, 2014, 6, 4243–4249 RSC .
- R. Nasielski-Hinkens, M. Benedek-Vamos, D. Maetens and J. Nasielski, J. Organomet. Chem., 1981, 217, 179–182 CrossRef CAS .
- R. Juárez, M. M. Oliva, M. Ramos, J. L. Segura, C. Alemán, F. Rodríguez-Ropero, D. Curcó, F. Montilla, V. Coropceanu, J. L. Brédas, Y. Qi, A. Kahn, M. C. Ruiz Delgado, J. Casado and J. T. López Navarre, Chem.–Eur. J., 2011, 17, 10312–10322 CrossRef PubMed .
- G. Aragay, A. Frontera, V. Lloveras, J. Vidal-Gancedo and P. Ballester, J. Am. Chem. Soc., 2013, 135, 2620–2627 CrossRef CAS PubMed .
- T. Chiba, Y.-J. Pu, R. Miyazaki, K.-i. Nakayama, H. Sasabe and J. Kido, Org. Electron., 2011, 12, 710–715 CrossRef CAS .
- K. Pieterse, P. A. van Hal, R. Kleppinger, J. A. J. M. Vekemans, R. A. J. Janssen and E. W. Meijer, Chem. Mater., 2001, 13, 2675–2679 CrossRef CAS .
- D. Dubois, K. M. Kadish, S. Flanagan, R. E. Haufler, L. P. F. Chibante and L. J. Wilson, J. Am. Chem. Soc., 1991, 113, 4364–4366 CrossRef CAS .
- L. M. Klivansky, D. Hanifi, G. Koshkakaryan, D. R. Holycross, E. K. Gorski, Q. Wu, M. Chai and Y. Liu, Chem. Sci., 2012, 3, 2009–2014 RSC .
- K. Kanakarajan and A. W. Czarnik, J. Org. Chem., 1986, 51, 5241–5243 CrossRef CAS .
- K. Kanakarajan and A. W. Czarnik, J. Heterocycl. Chem., 1988, 25, 1869–1872 CrossRef CAS .
- M. J. Crossley, P. Thordarson, J. P. Bannerman and P. J. Maynard, J. Porphyrins Phthalocyanines, 1998, 2, 511–516 CrossRef CAS .
- Z.-G. Tao, X. Zhao, X.-K. Jiang and Z.-T. Li, Tetrahedron Lett., 2012, 53, 1840–1842 CrossRef CAS .
- S. Hirayama, H. Sakai, Y. Araki, M. Tanaka, M. Imakawa, T. Wada, T. Takenobu and T. Hasobe, Chem.–Eur. J., 2014, 20, 9081–9093 CrossRef CAS PubMed .
- K. Ida, H. Sakai, K. Ohkubo, Y. Araki, T. Wada, T. Sakanoue, T. Takenobu, S. Fukuzumi and T. Hasobe, J. Phys. Chem. C, 2014, 118, 7710–7720 CAS .
-
(a)
I. B. Berlman, in Handbook of Fluorescence Spectra of Aromatic Molecules, Academic Press, New York, Second Edn, 1971, p. 473 Search PubMed ;
(b)
Handbook of Photochemistry, ed. M. Motalti, A. Credi, L. Prodi and M. T. Gandolfi, CRC Press, Boca Raton, 3rd edn, 2006, pp. 83–157 Search PubMed .
- O. Reiser, B. Koenig, K. Meerholz, J. Heinze, T. Wellauer, F. Gerson, R. Frim, M. Rabinovitz and A. de Meijere, J. Am. Chem. Soc., 1993, 115, 3511–3518 CrossRef CAS .
-
S. L. Mattes and S. Farid, in Organic Photochemistry, ed. A. Padwa, Marcel Dekker, New York, 1983, vol. 6, p. 233 Search PubMed .
- J. Yin, H. Qu, K. Zhang, J. Luo, X. Zhang, C. Chi and J. Wu, Org. Lett., 2009, 11, 3028–3031 CrossRef CAS PubMed .
- L. Tan-Sien-Hee and A. Kirsch-De Mesmaeker, J. Chem. Soc., Dalton Trans., 1994, 3651–3658 RSC .
- Z. Gasyna, W. R. Browett and M. Stillman, J. Inorg. Chem., 1985, 24, 2440–2447 CrossRef CAS .
- S. Fukuzumi, T. Suenobu, M. Patz, T. Hirasaka, S. Itoh, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 1998, 120, 8060–8068 CrossRef CAS .
- S. Fukuzumi, K. Ohkubo, Y. Kawashima, D. S. Kim, J. S. Park, A. Jana, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 15938–15941 CrossRef CAS PubMed .
- No additional emission band was observed even though we excited the higher energy band (i.e., Soret band at ca. 420 nm).
- P. V. Bernhardt and E. Hayes, J. Inorg. Chem., 2003, 42, 1371–1377 CrossRef CAS PubMed .
- The stabilization energy (Estab) of porphyrin and HAT-TIm was estimated from the following equation: Estab = EDA − (ED + EA), where EDA is the total energy of the D–A assembly, ED and EA are the total energies of each component. The calculated energies at the B3LYP/6-31(d) level of theory are summarized in Table S20 in the ESI.† For example, the difference of stabilization energies between ZnP–C3HAT-TIm and ZnP–C6HAT-TIm was 0.06 kcal mol−1. Similarly, the difference was also estimated to be 0.13 kcal mol−1 in the case of H2P–C3HAT-TIm and H2P–C12HAT-TIm. These results indicate that hydrophobic interactions formed between alkyl chains can be neglected. See the following reference paper: P. R. Bangal, J. Phys. Chem. A, 2007, 111, 5536–5543 CrossRef CAS PubMed .
- Dispersion-corrected calculation methods (e.g. WB97XD) were inconsistent with the absorption of the CT π-complex (Fig. 6A).
- T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, S. K. Kim, D. Kim, A. Fujimoto, T. Hirakawa and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216–1228 CrossRef CAS PubMed .
- J. L. Sessler, E. Karnas, S. K. Kim, Z. Ou, M. Zhang, K. M. Kadish, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 15256–15257 CrossRef CAS PubMed .
- K. Ohkubo, K. Mase, E. Karnas, J. L. Sessler and S. Fukuzumi, J. Phys. Chem. C, 2014, 118, 19436 Search PubMed .
- T. Honda, T. Nakanishi, K. Ohkubo, T. Kojima and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 10155–10163 CrossRef CAS PubMed .
- T. Hasobe, M. G. Rabbani, A. S. D. Sandanayaka, H. Sakai and T. Murakami, Chem. Commun., 2010, 46, 889–891 RSC .
-
Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed .
-
R. Dennington II, T. Keith, J. Millam, K. Eppinnett, W. L. Hovell and R. Gilliland, GaussView, Semichem, Shawnee Mission, KS, 2003 Search PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and MALDI-TOF mass spectra, cyclic voltammograms, fluorescence spectra, fluorescence titration spectra, 1H NMR titration, femtosecond laser-induced transient absorption spectral measurement data, and DFT data. See DOI: 10.1039/c4sc02787f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence