
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rh(III)-catalyzed C–H olefination of N-pentafluoroaryl benzamides using air as the sole oxidant†
Yi
Lu
*a,
Huai-Wei
Wang
a,
Jillian E.
Spangler
b,
Kai
Chen
a,
Pei-Pei
Cui
a,
Yue
Zhao
a,
Wei-Yin
Sun
*a and
Jin-Quan
Yu
*b
aCoordination Chemistry Institute, State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing National Laboratory of Microstructures, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, China. E-mail: luyi@nju.edu.cn; sunwy@nju.edu.cn
bDepartment of Chemistry, The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, California 92037, USA. E-mail: yu200@scripps.edu
First published on 8th January 2015
Abstract
The oxidative olefination of a broad array of arenes and heteroarenes with a variety of activated and unactivated olefins has be achieved via a rhodium(III)-catalyzed C–H activation reaction. The use of an N-pentafluorophenyl benzamide directing group is crucial for achieving catalytic turnovers in the presence of air as the sole oxidant without using a co-oxidant.
Introduction
The direct coupling of unactivated aryl C–H bonds with olefins provides a step- and atom-economical method for the functionalization of arenes.1 While a number of transition metals have been explored in this capacity, Rh(III) has emerged as an effective catalyst for the olefination of aryl C–H bonds under mild reaction conditions.2,3 Many research groups have sought to improve the utility of Rh(III)-catalyzed aryl C–H olefinations by increasing reactivity of the catalytic systems and improving site selectivity of the transformation. This has been achieved via the use of proximal directing groups to promote ortho-C–H bond cleavage. However, in the previously reported Rh(III)-catalyzed aryl C–H olefination reactions, stoichiometric oxidants such as peroxides, hypervalent iodonium salts, fluorinating agents, NXS (X = F, Cl, Br, or I) or inorganic salts are generally required to sustain the catalytic cycle.4 The use of any of these oxidants results in the undesired generation of stoichiometric reaction byproducts, reducing the utility and practicality of these transformations. An alternative approach has been the use of oxidizing directing groups CONHOR, which generally feature a cleavable N–O bond that can serve to reoxidize the Rh(I) catalyst.5 A greener and more environmentally benign approach is the use of molecular oxygen (O2) or air as the terminal oxidant, which generates water as the sole reaction byproduct.6 The goal of using O2 as the sole oxidant for C–H olefination reactions has been achieved in several cases using palladium.7 More recently Huang and coworkers reported the Rh(III)-catalyzed C–H activation reactions using O2 as the sole oxidant in alkynylation reactions for the synthesis of isoquinolinium salts8 and indoles9 and the direct C-2 olefination of electron-rich indoles.10 However, to date, we are unaware of any other instances of a Rh(III)-catalyzed C–H functionalization reaction of inert arenes using air as the sole oxidant without using a metal co-oxidant.3uWhile the use of molecular O2 as oxidant is fundamentally important, the use of air as the sole oxidant for Rh(III)/Rh(I) catalysis is practically desirable in terms of operational safety. Herein we report a Rh(III)-catalyzed C–H activation/olefination reaction of aryl and heteroaryl benzamides under air at atmospheric pressure without the addition of an external oxidant. This reaction utilizes an N-pentafluorophenyl benzamide directing group, which has not previously been explored as a directing group for Rh(III)-catalyzed reactions. The transformation is compatible with a range of olefin coupling partners, including simple styrenes (Scheme 1).
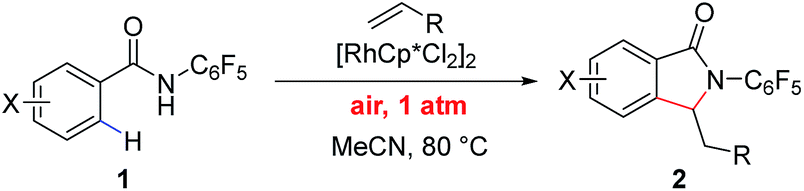 | ||
| Scheme 1 Aerobic Rh(III)-catalyzed C–H olefination. | ||
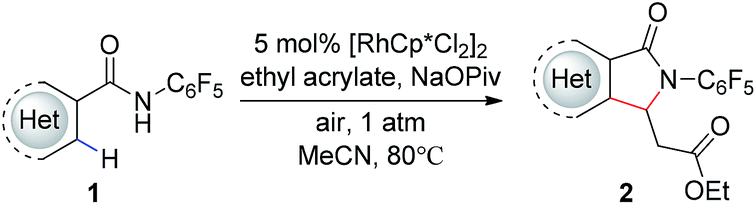
Our group has recently described the effectiveness of N-polyfluoroaryl benzamide directing groups in Pd(II)-catalyzed C(sp3)–H activation/olefination reactions.11 We hoped to extrapolate the use of this directing group to Rh(III)-catalyzed C–H olefination reactions. As shown in Table 1, treatment of pentafluorobenzamide 1a and ethyl acrylate with 5 mol% [RhCp*Cl2]2 in the presence of AgOAc or Cu(OAc)2 as a stoichiometric oxidant provided product 2a in good yield (Table 1, entries 1 and 2), verifying the efficacy of this directing group in rhodium-catalyzed transformations. Cyclization of the pentafluorophenyl benzamide onto the pendant enoate to form a γ-lactam product was previously described in a ruthenium-catalyzed oxidative C–H olefination.12 Acetonitrile was identified as the ideal solvent for achieving high levels of mono-selective olefination (see ESI†). However, when we conducted this reaction under an atmosphere of oxygen in the absence of an additional oxidant, only a trace amount of the desired product was observed (Table 1, entry 3).
|
|
|||
|---|---|---|---|
| Entrya | Oxidant | Base | Yieldb (%) |
| a Reaction conditions: benzamide (0.2 mmol, 1.0 eq.), ethyl acrylate (0.5 mmol, 2.5 eq.), [RhCp*Cl2]2 (0.01 mmol, 0.05 eq.), oxidant (0.4 mmol, 2.0 eq.), base (0.2 mmol, 1.0 eq.), MeCN (2 mL), 80 °C, 24 h. b Determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard. c [RhCp*Cl2]2 (0.004 mmol, 0.02 eq.), 48 h. d Isolated yield. e Amino acid (0.02 mmol, 0.1 eq.). | |||
| 1 | AgOAc | — | 67 |
| 2 | Cu(OAc)2 | — | 60 |
| 3 | O2 | — | Trace |
| 4 | O2 | Na2CO3 | 27 |
| 5 | O2 | K2CO3 | 26 |
| 6 | O2 | NaOAc | 64 |
| 7 | O2 | NaOPiv | 99 |
| 8 | Air | NaOPiv | 92 |
| 9 | Air | NaOPiv | 99(92) |
| 10e | Air | Na2CO3 + Boc–Leu–OH | 93 |
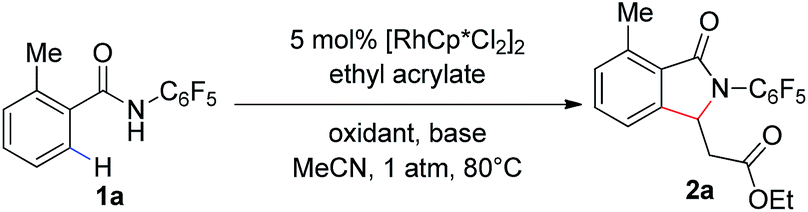
The addition of a base such as Na2CO3 or K2CO3 led to improved yields of the 2a (Table 1, entries 4 and 5), indicating that the acidity of the polyfluorinated benzamide may be critical for the success of transformation in the absence of a stoichiometric oxidant. The use of a carboxylate base, such as NaOAc or NaOPiv, provided a dramatic improvement in reaction efficiency (Table 1, entries 6 and 7). As such, the reaction of benzamide 1a with 5 mol% [RhCp*Cl2]2 and 1.0 eq. of NaOPiv under an atmosphere of O2 provided the desired product 2a in 99% yield (Table 1, entry 7). Conducting the reaction under air instead of oxygen led to a slight decrease in product yield (Table 1, entry 8).‡ However, an increased reaction time improved the yield of the transformation under air, under these reaction conditions product 2a was formed in 99% yield with only 2 mol% catalyst (Table 1, entry 9, 92% isolated yield). Interestingly, we found that a similar level of reactivity could be obtained with Na2CO3 as the base with the addition of 10 mol% of a mono-protected amino acid ligand (MPAA) (Table 1, entry 10).13
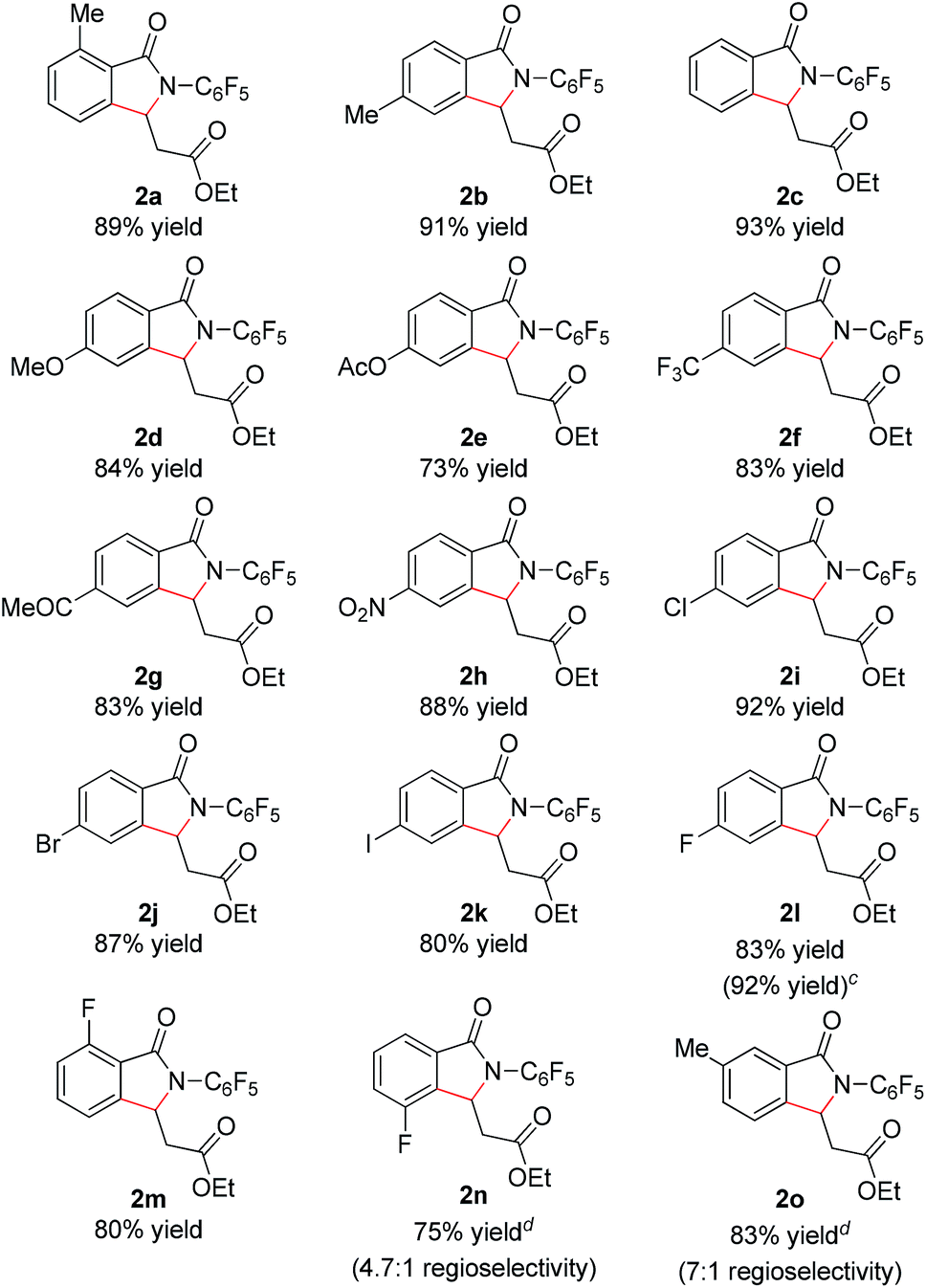
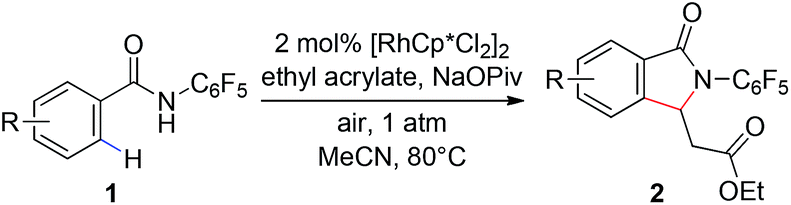
With these optimized reaction conditions in hand we set out to analyze the scope of this transformation with respect to the benzamide substrate. N-Pentafluorophenyl benzamides with electron-donating (Table 2, entries 2a–b and 2d) or electron-withdrawing groups (Table 2, entries 2e–h) react to provide corresponding olefinated products in excellent yields. Halide-substituted substrates, which bear a handle for further chemical manipulation, are also tolerated in this transformation (Table 2, entries 2i–n). It is worth noting that the meta-substituted benzamides (Table 2, entries 2n and 2o) provide a regioisomeric mixture of olefination products. For meta-fluorinated substrate, the major product in most cases with Pd(OAc)2-catalyzed C–H activation occurs para to F with one exceptional example.14 Interestingly, meta-fluorinated substrate affords the ortho position olefination product 2n, while meta-Me substrate affords the para position olefination product 2o, illustrating that the acidity of the C–H bond plays a role in this C–H olefination reaction. The reaction can be conducted on gram scale with no decrease in isolated yield (Table 2, entry 2l). The catalyst loading can also be lowered to 2 mol% [RhCp*Cl2]2 without significantly affecting the reactivity (Table 3).
|
|
|---|
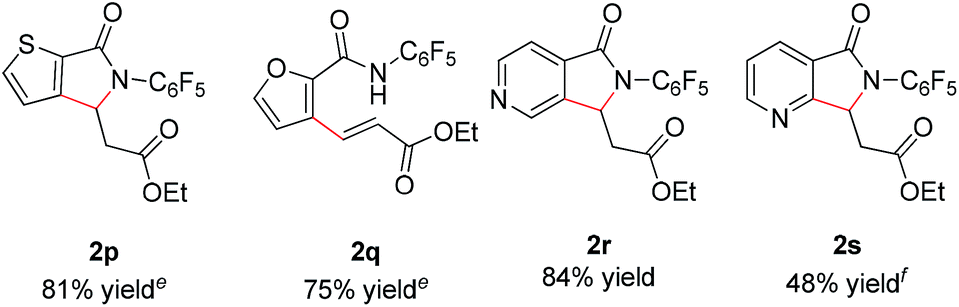
| a Reaction conditions: benzamide (0.2 mmol, 1.0 eq.), ethyl acrylate (0.5 mmol, 2.5 eq.), [RhCp*Cl2]2 (0.01 mmol, 0.05 eq.), air, 1 atm, NaOPiv (0.2 mmol, 1.0 eq.), MeCN (2 mL), 80 °C, 24 h. b Isolated yield. c Reaction conducted on 3.5 mmol scale. d isolated yields of the major isomers 2n and 2o. e Reaction temperature is increased to 100 °C. f Boc–Leu–OH (0.02 mmol, 0.1 eq.) and Na2CO3 (0.2 mmol, 1.0 eq.) instead of NaOPiv. |
|
|
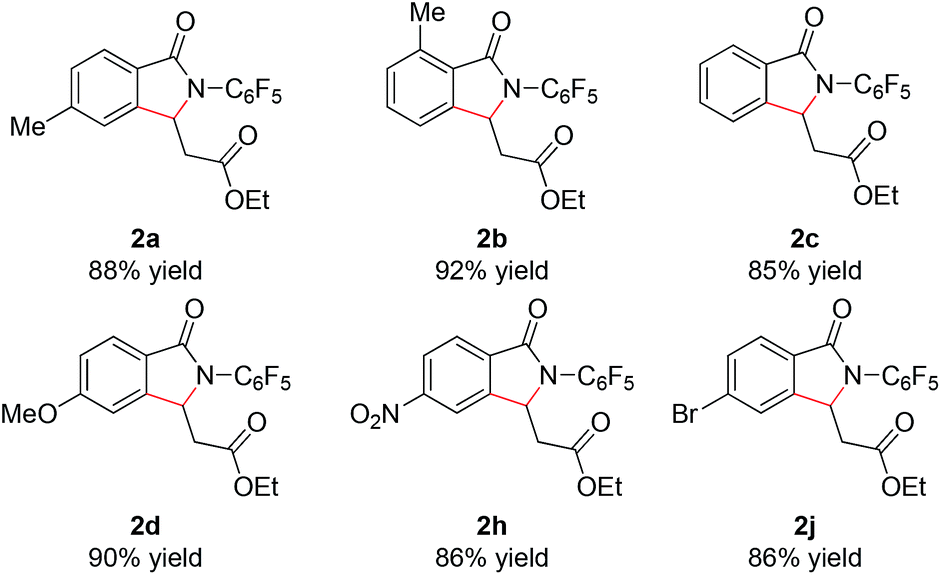
In addition, we were pleased to find that heterocyclic amides, including thiophene, furan, and pyridine are competent substrates and can be olefinated in good yield (Table 3, entry 2p–2r) with just a slight increase in reaction temperature. Interestingly, although the N-aryl thiophene-2-carboxamide reacts to provide the cyclized product 2p in 81% yield (reaction time is increased to 48 hours), the N-aryl furan-2-carboxamide provides only the uncyclized product 2q (75% yield) even with higher reaction temperatures and an extended reaction time. While olefination of pyridine-4-carboxamide gave the desired product 2r in excellent yield, pyridine-3-carboxamide is less reactive, affording lower yield (Table 2, entry 2s, 48%). 4-(Pyridine-2-yl)benzamide was also subjected to the standard conditions and the olefination occurred ortho to the amide rather than the pyridyl, albeit affording low yield (28%, see ESI†). Not surprisingly, pyridine-2-carboxamide is not reactive under these conditions due to the bis-dentate coordination of the substrate with Rh(III).
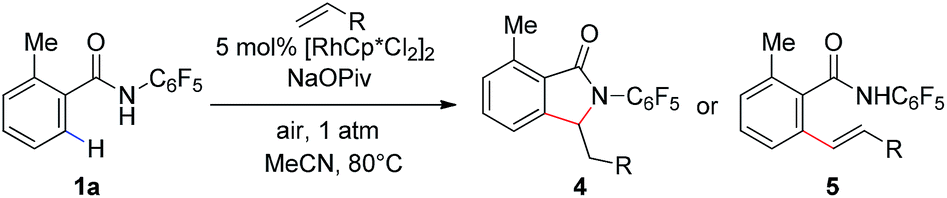
We subsequently analyzed the scope of the olefin coupling partner in this transformation. As shown in Table 4, electron-deficient olefins, including ethyl vinyl ketone and acrylonitrile, can be coupled in good yield to provide lactam products 4a–b. A variety of styrenes substituents are also coupled in good yields to provide the corresponding uncyclized products 5a–5c. In addition, the unactivated olefin pentene can also be coupled, albeit in modest yield, to provide amide 5d. However, di-substituted olefins are not reactive under these conditions.
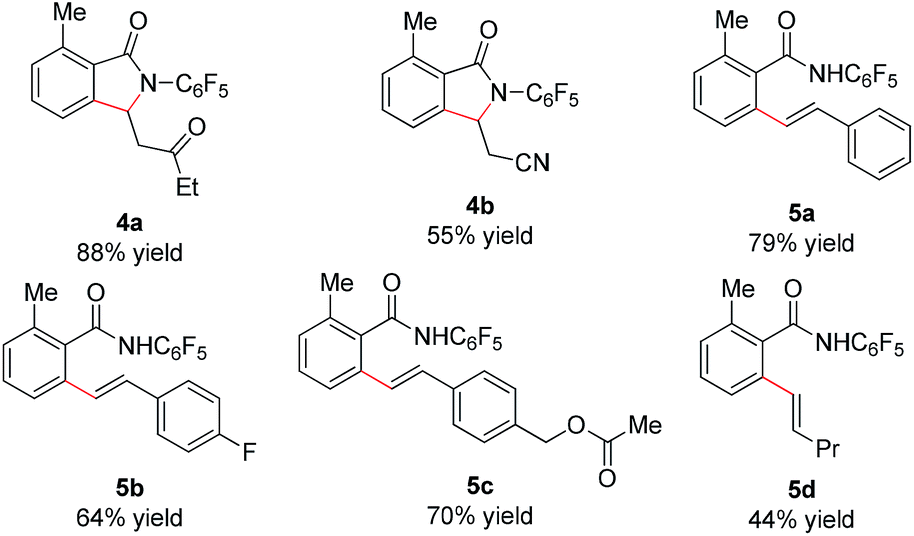
As shown in Scheme 2, the γ-lactam products formed in this transformation are readily converted to the olefinated products. Treatment of lactam 2d with LiHMDS, Boc2O, and EtONa step by step in one-pot provides enoate 6d in good yield.
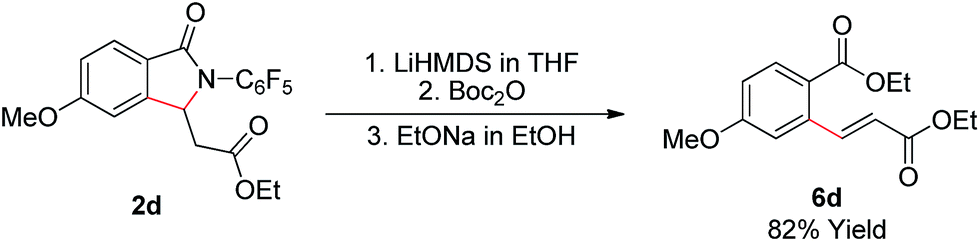 | ||
| Scheme 2 Removal of the auxiliary. | ||
Our proposed mechanism for this transformation is shown in Scheme 3. Thus coordination of the amide to the [Rh(III)] catalyst is followed by ortho-C–H bond activation to give a corresponding [Rh(III)–Ar] intermediate. Subsequent coordination to the olefin coupling partner and 1,2-migratory insertion provides an complex which can undergo β-hydride elimination to provide the uncyclized product 6. Reoxidation of the [Rh(I)] to [Rh(III)] by molecular oxygen would complete the catalytic cycle. This reoxidation step is likely facilitated by the acetate base, addition of which provides a substantial increase in reaction conversion. Subsequent base mediated 1,4-conjugate addition of the acidic N-pentafluorophenyl benzamide onto the pendant enoate provides the γ-lactam product 2. The isolation of furan 2q and alkenes 5a–5d, which have undergone olefination but not cyclization, provide some evidence for the formation of 6 during the catalytic cycle.
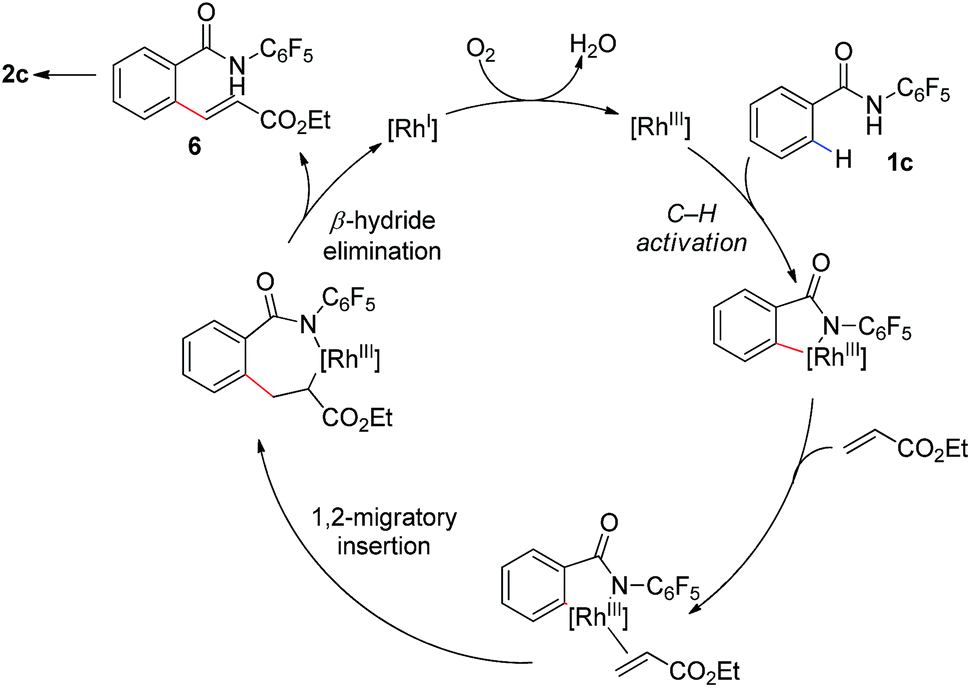 | ||
| Scheme 3 Proposed reaction mechanism. | ||
In summary, we have developed a Rh(III)-catalyzed C–H olefination of aryl C–H bonds using the N-pentaflurophenyl amide auxiliary and air as the sole oxidant. The reaction conditions can be applied to a variety of both aryl and heteroaryl benzamides.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (grant no. 21201100 and 21331002) and NSF under the CCI Center for Selective C–H Functionalization, CHE-1205646 for financial support. This work was also supported by a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- For a review, see: C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed.
- For recent reviews, see: (a) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (b) C. Zhu, R. Wang and J. R. Falck, Chem.–Asian J., 2012, 7, 1502 CrossRef CAS PubMed; (c) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS; (d) A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef PubMed; (e) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed.
- (a) N. K. Mishar, J. Park, S. Sharma, S. Han, M. Kim, Y. Shin, J. Jang, J. H. Kwak, Y. H. Jung and I. S. Kim, Chem. Commun., 2014, 50, 2350 RSC; (b) N.-J. Wang, S.-T. Mei, L. Shuai, Y. Yuan and Y. Wei, Org. Lett., 2014, 16, 3040 CrossRef CAS PubMed; (c) X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem.–Eur. J., 2013, 19, 11898 CrossRef CAS PubMed; (d) B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, Chem.–Eur. J., 2013, 19, 11863 CrossRef CAS PubMed; (e) B. Liu, Y. Fu, Y. Gao, C. Sun, C. Xu and J. Zhu, J. Am. Chem. Soc., 2013, 135, 468 CrossRef CAS PubMed; (f) C. Feng, D. Feng and T.-P. Loh, Org. Lett., 2013, 15, 3670 CrossRef CAS PubMed; (g) J. Zhou, B. Li, F. Hu and B.-F. Shi, Org. Lett., 2013, 15, 3460 CrossRef CAS PubMed; (h) M. Presset, D. Oehlrich, F. Rombouts and G. A. Molander, Org. Lett., 2013, 15, 1528 CrossRef CAS PubMed; (i) S. Cui, Y. Zhang and Q. Wu, Chem. Sci., 2013, 4, 3421 RSC; (j) Y. Shen, G. Liu, Z. Zhou and X. Lu, Org. Lett., 2013, 15, 3366 CrossRef CAS PubMed; (k) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed; (l) C. Zhu and J. R. Falck, Chem. Commun., 2012, 48, 1674 RSC; (m) G. Li, Z. Ding and B. Xu, Org. Lett., 2012, 14, 5338 CrossRef CAS PubMed; (n) K.-H. Kwon, D. W. Lee and C. S. Yi, Organometallics, 2012, 31, 495 CrossRef CAS PubMed; (o) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed; (p) A. S. Tsai, M. Brasse, R. G. Bergman and J. A. Ellman, Org. Lett., 2011, 13, 540 CrossRef CAS PubMed; (q) S. Park, J. Y. Kim and S. Chang, Org. Lett., 2011, 13, 2372 CrossRef CAS PubMed; (r) X. Li and M. Zhao, J. Org. Chem., 2011, 76, 8530 CrossRef CAS PubMed; (s) F. Wang, G. Song and X. Li, Org. Lett., 2010, 12, 5430 CrossRef CAS PubMed; (t) F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9982 CrossRef CAS PubMed; (u) K. Ueura, T. Satoh and M. Miura, Org. Lett., 2007, 9, 1407 CrossRef CAS PubMed.
- For some examples of using peroxides as stoichiometric oxidants, see: (a) H. X. Dai and J. Q. Yu, J. Am. Chem. Soc., 2012, 134, 134 CrossRef CAS PubMed; (b) A. J. Canty, H. Jin, A. S. Roberts, B. W. Skelton and A. H. White, Organometallics, 1996, 15, 5713 CrossRef CAS; for some examples of using hypervalent iodonium salts as stoichiometric oxidants, see: (c) M. H. Emmert, A. K. Cook, Y. J. Xie and M. Sanford, Angew. Chem., Int. Ed., 2011, 50, 9409 CrossRef CAS PubMed; (d) A. R. Dick, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 12790 CrossRef CAS PubMed; (e) M. C. Lagunas, R. A. Gossage, A. L. Spek and G. van Koten, Organometallics, 1998, 17, 731 CrossRef CAS; for some examples using fluorinating agents as stoichiometric oxidants, see: (f) N. D. Ball, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 2878 CrossRef CAS PubMed; (g) T. S. Mei, X. S. Wang and J. Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806 CrossRef CAS PubMed; for some examples of using NXS (X = F, Cl, Br, or I) as stoichiometric oxidants, see: (h) A. John and K. M. Nicholas, J. Org. Chem., 2012, 77, 5600 CrossRef CAS PubMed; (i) T. Lv, X. L. Zhang, J. S. Han and P. Zhong, J. Fluorine Chem., 2012, 137, 44 CrossRef CAS PubMed; (j) S. R. Whitfield and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 15142 CrossRef CAS PubMed; for some examples of using inorganic salts as stoichiometric oxidants, see: (k) M. Miura, C.-G. Feng, S. Ma and J.-Q. Yu, Org. Lett., 2013, 15, 5258 CrossRef CAS PubMed; (l) S. Sharma, E. Park, J. Park and I. S. Kim, Org. Lett., 2012, 14, 906 CrossRef CAS PubMed; (m) F. Wang, G. Y. Song and X. W. Li, Org. Lett., 2010, 12, 5430 CrossRef CAS PubMed.
- (a) For a review, see: F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977 CrossRef CAS PubMed; (b) For initial development of CONHOMe directing group for Pd-catalyzed C–H activation reactions, see: D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS PubMed.
- (a) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed; (b) W. Wu and H. Jiang, Acc. Chem. Res., 2012, 45, 1736 CrossRef CAS PubMed; (c) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (d) K. M. Gligorich and M. S. Sigman, Chem. Commun., 2009, 3854 RSC; (e) J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506 CrossRef CAS PubMed; (f) T. Punniyamurthy, S. Veusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed; (g) S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400 CrossRef CAS PubMed.
- (a) M. Ito, R. Namie, J. Krishnamurthi, H. Miyamae, K. Takeda, H. Nambu and S. Hashimoto, Synlett, 2014, 25, 288 CAS; (b) B. Liu, H.-Z. Jiang and B.-F. Shi, J. Org. Chem., 2014, 79, 1521 CrossRef CAS PubMed; (c) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567 CrossRef CAS PubMed; (d) A. Vasseur, D. Harakat, J. Muzart and J. L. Bras, Adv. Synth. Catal., 2013, 355, 59 CrossRef CAS; (e) R. Sallio, S. Lebrun, N. Schifano-Faux, J.-F. Goossens, F. Agbossou-Niedercorn, E. Deniau and C. Michon, Synlett, 2013, 24, 1785 CrossRef CAS PubMed; (f) X. Cong, H. Tang, C. Wu and X. Zeng, Organometallics, 2013, 32, 6565 CrossRef CAS; (g) G. Yang and W. Zhang, Org. Lett., 2012, 14, 268 CrossRef CAS PubMed; (h) A. K. Ghosh, X. Cheng and B. Zhou, Org. Lett., 2012, 14, 5046 CrossRef CAS PubMed; (i) D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 33, 5767 CrossRef PubMed; (j) C. Zhu and J. R. Falck, Org. Lett., 2011, 13, 1214 CrossRef CAS PubMed; (k) J. W. Wigglesworth, B. Cox, G. C. Lloyd-Jones and K. I. Booker-Milburn, Org. Lett., 2011, 13, 5326 CrossRef PubMed; (l) K. M. Engle, D. H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137 CrossRef CAS PubMed; (m) K. M. Engle, D. H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 6169 CrossRef CAS PubMed; (n) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 5072 CrossRef CAS PubMed.
- G. Zhang, L. Yang, Y. Wang, Y. Xie and H. Huang, J. Am. Chem. Soc., 2013, 135, 8850 CrossRef CAS PubMed.
- G. Zhang, H. Yu, G. Qin and H. Huang, Chem. Commun., 2014, 50, 4331 RSC.
- L. Yang, G. Zhang and H. Huang, Adv. Synth. Catal., 2014, 356, 1509 CrossRef CAS.
- (a) M. Wasa, K. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 3680 CrossRef CAS PubMed; (b) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2013, 343, 1216 CrossRef PubMed.
- L. Ackermann, L. Wang, R. Wolfram and A. V. Lygin, Org. Lett., 2012, 14, 728 CrossRef CAS PubMed.
- For optimization of the amino acid additive, see ESI.†.
- J.-J. Li, R. Giri and J.-Q. Yu, Tetrahedron, 2008, 64, 6979 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. CCDC 1042327. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03350g |
| ‡ General procedure: to a 350 mL Schlenk-type sealed tube equipped with a magnetic stirring bar, were added the substrate (0.2 mmol, 1.0 eq.), [RhCp*Cl2]2 (0.01 mmol, 0.05 eq.), NaOPiv (0.2 mmol, 1.0 eq.), MeCN (2.0 mL) and olefin coupling partner (0.5 mmol, 2.5 eq.). The tube was capped and heated to 80 °C for 24 h. After cooling to room temperature, the reaction mixture was filtered through a pad of Celite. The filtrate was concentrated in vacuo to afford the crude product, which was purified by flash column chromatography (SiO2) gel to provide the desired product. |
| This journal is © The Royal Society of Chemistry 2015 |