
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A family of N-heterocyclic carbene-stabilized borenium ions for metal-free imine hydrogenation catalysis†
Jeffrey M.
Farrell
a,
Roy T.
Posaratnanathan
a and
Douglas W.
Stephan
*ab
aDepartment of Chemistry, University of Toronto, 80 St. George St., Toronto, ON M5H3H6, Canada. E-mail: dstephan@chem.utoronto.ca
bDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 26th January 2015
Abstract
This manuscript probes the steric and electronic attributes that lead to “frustrated Lewis pair” (FLP)-type catalysis of imine hydrogenation by borenium ions. Hydride abstraction from (ItBu)HB(C6F5)22 prompts intramolecular C–H bond activation to give (CHN)2(tBu) (CMe2CH2)CB(C6F5)23, defining an upper limit of Lewis acidity for FLP hydrogenation catalysis. A series of seven N-heterocyclic carbene–borane (NHC–borane) adducts ((R′CNR)2C)(HBC8H14) (R′ = H, R = dipp 4a, Mes 5a, Me 8a; R = Me R′ = Me 9a, Cl, 10a) and ((HC)2(NMe)(NR)C)(HBC8H14) (R = tBu, 6a, Ph 7a) are prepared and converted to corresponding borenium salts. These species are evaluated as catalysts for metal-free imine hydrogenation at room temperature. Systematic tuning of the carbene donor for the hydrogenation of archetypal substrate N-benzylidene-tert-butylamine achieves the highest reported turn-over frequencies for FLP-catalyzed hydrogenation at amongst the lowest reported catalyst loadings. The most active NHC–borenium catalyst of this series, derived from 10a, is readily isolable, crystallographically characterized and shown to be effective in the hydrogenation catalysis of functional group-containing imines and N-heterocycles.
Introduction
As the global consciousness awakens to the environmental and fiscal costs associated with energy and material-intensive chemical processes, the development of new and effective catalytic strategies grows ever more significant. The hydrogenation of unsaturated bonds is one such chemical transformation that is employed on a terrific industrial scale.1–4 Currently these processes employ highly effective transition metal catalysts despite oft associated high cost, toxicity and significant carbon footprint. These drawbacks have led to the intense pursuit of alternative or complimentary technologies. For example, hydrogenation catalysis by cheap and non-toxic transition metals such as iron5–8 and cobalt,9,10 as well as early metals such as titanium11,12 and calcium13 has drawn considerable attention. Our group14–23 and others24–40 have focused on main-group alternatives motivated by our report of metal-free hydrogen activation by a linked phosphino-borane.41 Indeed, soon after this initial report, we described the use of “frustrated Lewis pairs” (FLPs) in the catalytic hydrogenation of imines and protected nitriles.23 This prompted a flurry of developments in FLP hydrogenation catalysis. While substrate scope has since been dramatically broadened, the catalytic activities of FLP systems and the catalyst loadings required are not yet competitive with transition metal catalysts. Perhaps more importantly, the synthetic challenge of preparing electrophilic boranes limits the range of potential catalysts that are readily accessible for a systematic evaluation of structure–activity relationships. Indeed, although studies involving families of closely related FLP hydrogenation catalysts are rare,29,38,39,42 some examples in the literature suggests that subtle changes to FLP catalysts can have dramatic impact on activity and selectivity. For example, the group of Soós and co-workers has shown that substitution of one C6F5 in B(C6F5)3 with the bulkier mesityl group effects selectivity control through size exclusion.34,35 Moreover, careful choice of Lewis base in FLP hydrogenation catalysis has extended the substrate scope to include silyl enol ethers,27 olefins19,33 and most recently ketones and aldehydes.43,44Borenium ions are three-coordinate boron cations.45–47 These relatively underexplored Lewis acids have attracted recent attention for use in catalysis48–55 and selective carboborations and haloborations.56–66 In an earlier communication, our group showed that an N-heterocyclic carbene-stabilized borenium salt [(IiPr)BC8H14] [B(C6F5)4] (IiPr = 1,3-di-iso-propylimidazol-2-ylidene) can be used as a catalyst for the metal-free hydrogenation of imines and enamines.67 In this case, the borenium cation and the imine act as an FLP to cleave H2. This affords an NHC–borane that delivers hydride to a transient iminium ion (Scheme 1). Borenium-based catalyst 1b derives its Lewis acidity from a cationic charge rather than electron-withdrawing fluoroaryl groups on boron. Moreover, the precursor NHC–borane adduct is robust and easily accessible. During the review process of this paper Crudden and co-workers described triazolium derived borenium cations as catalysts.80
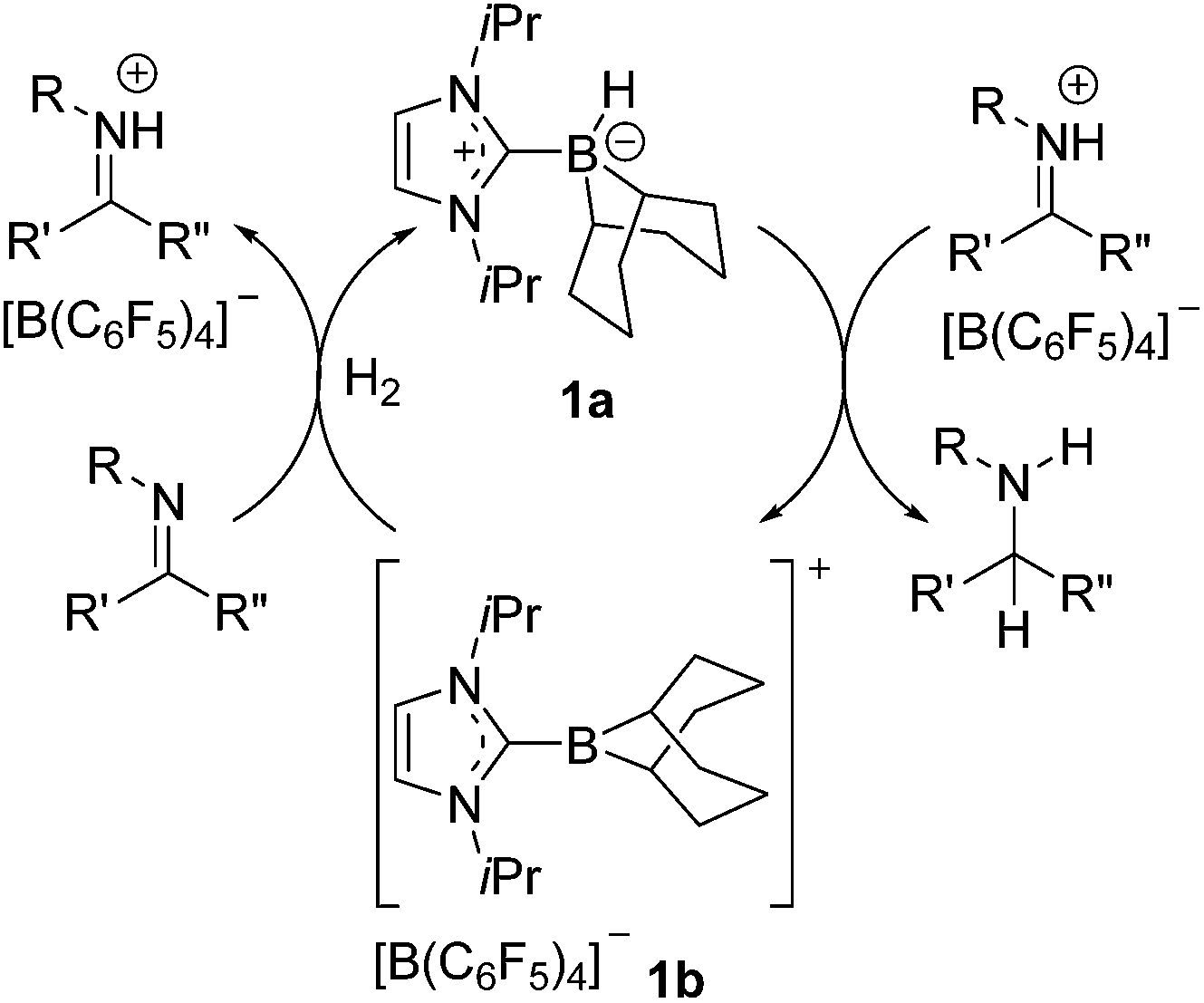 | ||
| Scheme 1 Hydrogenation catalysis by an NHC–borenium ion. | ||
Herein, we exploit our previous findings to access a family of FLP catalysts. The reactivity of these borenium cations is evaluated in the metal-free hydrogenation catalysis of imines and N-heterocycles. This systematic study of the steric and electronic attributes of NHC–borenium catalysts provides insight into the structure–activity relationship of this new class of FLP hydrogenation catalyst.
Results and discussion
Our initial efforts to enhance reactivity with respect to previously reported catalyst 1b focused on the incorporation of electron withdrawing C6F5 substituents in NHC–borenium ions. To this end, the reaction of 1,3-di-tert-butylimidazol-2-ylidene (ItBu) with HB(C6F5)2 led to the formation of NHC–borane adduct (ItBu)HB(C6F5)22 (Scheme 2), which was isolated in 76% yield. Crystallographic characterization revealed the anticipated pseudo-tetrahedral geometry about boron, an average CNHC–B bond length of 1.645(4) Å and an average B–CC6F5 bond length of 1.639(4) Å (Fig. 1(a)). The expected upfield doublet for 2 is observed by 11B NMR spectroscopy at −22.9 ppm with a 1JB–H coupling of 88 Hz. The 1H NMR and 19F NMR spectra indicate hindered rotation about the CNHC–B bond in 2 on the NMR time scale. Broad and inequivalent resonances were observed for the tert-butyl protons and fluorine atoms at room temperature; however, these were resolved upon cooling to −40 °C. | ||
| Scheme 2 Synthesis and reactivity of NHC–borane 2. | ||
 | ||
| Fig. 1 POV-ray depiction of (a) 2 and (b) 3. C: black, B: yellow-green, N: blue, F: pink, H: grey. H-atoms except for borohydride omitted for clarity. | ||
Attempts to generate an NHC–borenium ion derived from 2via treatment with the hydride abstraction reagents [Ph3C][B(C6F5)4], Me3SiOTf or HOTf showed no reaction. This stands in contrast to the facile hydride donation typically demonstrated by NHC–boranes.68,69 However, upon heating 2 with HNTf2 in toluene to >100 °C for four days the clean conversion to a new product was evident from the appearance of the 11B resonance at −14.8 ppm. 1H NMR spectroscopy showed sharp singlet resonances at 0.86 ppm and 1.04 ppm and a broad singlet resonance at 1.80 ppm integrating in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:2 ratio. These combined NMR data suggest the new species (CHN)2(tBu) (CMe2CH2)CB(C6F5)23 is derived from C–H activation of a tert-butyl substituent (Scheme 2). A crystallographic study of 3 confirmed its bicyclic nature (Fig. 1(b)). This species is similar to compounds (CHN)2(tBu)(CMe2CH2)CBBr270 and (CHN)2(tBu)(CMe2CH2) CB(tBu)Cl71 recently reported by Braunschweig and co-workers. The formation of 3 is thought to proceed via transient generation of a cation and subsequent C–H activation (Scheme 2). Similar borylations of aliphatic groups by donor stabilized borenium ions have been reported by Prokofjevs and Vedejs.72
:6:2 ratio. These combined NMR data suggest the new species (CHN)2(tBu) (CMe2CH2)CB(C6F5)23 is derived from C–H activation of a tert-butyl substituent (Scheme 2). A crystallographic study of 3 confirmed its bicyclic nature (Fig. 1(b)). This species is similar to compounds (CHN)2(tBu)(CMe2CH2)CBBr270 and (CHN)2(tBu)(CMe2CH2) CB(tBu)Cl71 recently reported by Braunschweig and co-workers. The formation of 3 is thought to proceed via transient generation of a cation and subsequent C–H activation (Scheme 2). Similar borylations of aliphatic groups by donor stabilized borenium ions have been reported by Prokofjevs and Vedejs.72
The C–H activation that yields 3 suggests that the proposed C6F5-substituted borenium ion derived from 2 is too Lewis acidic for application in catalysis. This prompted us to further examine 9-BBN based borenium cations. To this end 9-BBN was reacted with 1,3-bis(2,6-di-iso-propylphenyl)imidazol-2-ylidene (Idipp) at 60 °C for one hour to afford Idipp–borane adduct 4a in 79% yield (Scheme 3). Compound 4a exhibits a broad 11B NMR signal at −15.3 ppm. Reaction of 4a with [Ph3C][B(C6F5)4] at 45 °C overnight results in the generation of Ph3CH and the quantitative conversion of the NHC–borane to a new species as evidenced by 11B NMR signals at 82.6 ppm and −16.6 ppm. These are consistent with the formation the borenium–borate salt [(Idipp)BC8H14][B(C6F5)4] 4b. Alternatively, treatment of 4a with tBuN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh and the addition of a stoichiometric equivalent of [tBu3PH]][B(C6F5)4] results in generation of 4b with concurrent reduction of the imine as evidenced by 1H NMR spectroscopy. This observation prompted efforts to employ 4b in an FLP hydrogenation of tBuNCHPh. However combination of excess tBuNCHPh and 4b under 102 atm H2(g) showed no evidence of reduction of the imine (Table 1, entry 2). Thus while the steric bulk of 4a does not deter hydride delivery, it does preclude H2 activation by the corresponding borenium/imine FLP.
CHPh and the addition of a stoichiometric equivalent of [tBu3PH]][B(C6F5)4] results in generation of 4b with concurrent reduction of the imine as evidenced by 1H NMR spectroscopy. This observation prompted efforts to employ 4b in an FLP hydrogenation of tBuNCHPh. However combination of excess tBuNCHPh and 4b under 102 atm H2(g) showed no evidence of reduction of the imine (Table 1, entry 2). Thus while the steric bulk of 4a does not deter hydride delivery, it does preclude H2 activation by the corresponding borenium/imine FLP.
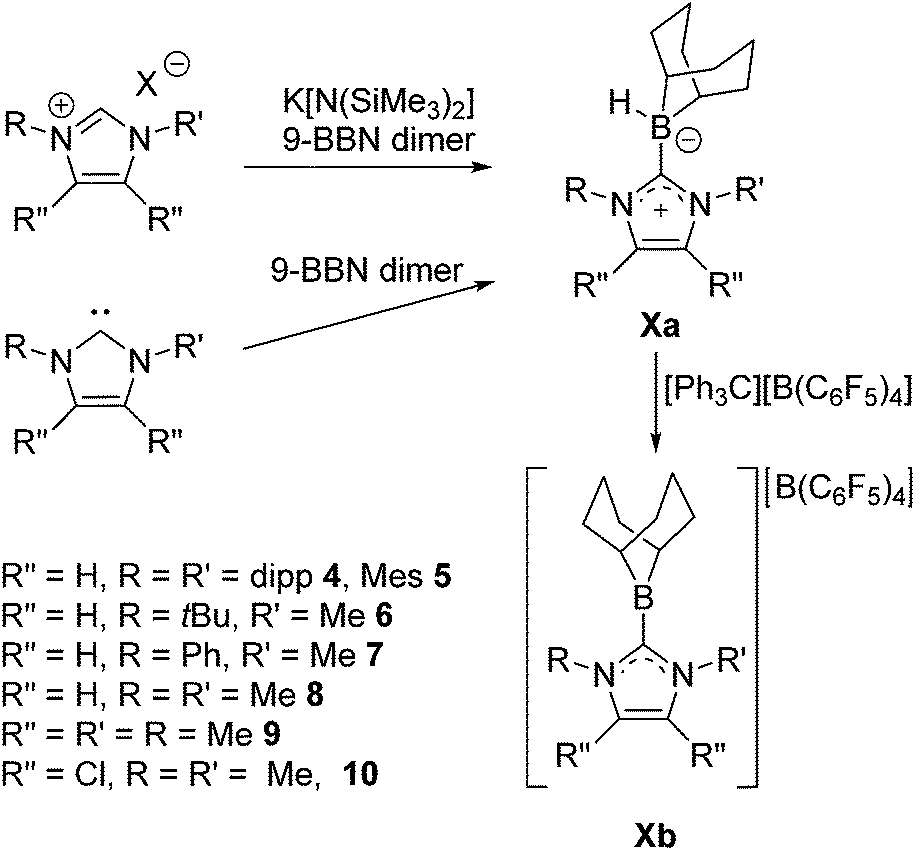 | ||
| Scheme 3 General syntheses of NHC–boranes 4a–10a and generation of borenium salts 4b–10b. | ||
|
|
||
|---|---|---|
| Entry | Cat. (mol%) | Yieldb (%) |
| a Borenium salts were generated in situ by addition of [Ph3C][B(C6F5)4] to the corresponding borohydride precursor. Isolated 10b was used in entries 11–13. b Determined by 1H NMR spectroscopy, isolated yields in parentheses. All reaction times were 30 min, except: c 2 h reaction time. | ||
| 1 | 1b (1) | 35 |
| 2 | 4b (5) | 0 |
| 3 | 5b (1) | 0 |
| 4 | 6b (1) | Trace |
| 5 | 7b (1) | 100 |
| 6 | 7b (0.5) | 35 |
| 7 | 8b (1) | 100 |
| 8 | 8b (0.5) | 67 |
| 9 | 9b (0.5) | 21 |
| 10 | 10b (0.5) | 100 |
| 11 | 10b (0.25) | 100 |
| 12 | 10b (0.1) | 47 |
| 13 | 10b (0.15) | 100 (83)c |
To further probe the steric and electronic factors impacting on the reactivity of NHC–borenium cations, a series of NHC-9-BBN adducts were prepared exercising judicious variation of the NHC. This was achieved by either directly reacting 9-BBN dimer with the isolated carbene or by reacting 9-BBN dimer with a carbene generated in situ through the combination of an imidazolium salt with K[N(SiMe3)2]. This latter one-pot approach is similar to that described by Brahmi et al. to prepare a series of NHC–BH3 compounds.73 A series of seven adducts including ((R′CNR)2C)HBC8H14 (R′ = H, R = dipp 4a, Mes 5a,74 Me 8a;50 R = Me R′ = Me 9a, Cl, 10a) and ((HC)2(NMe)(NR)C)HBC8H14 (R = tBu, 6a, Ph 7a) were prepared (Scheme 3). The NHC–borane adducts 4a–10a were isolated and purified via recrystallization from pentane or toluene in yields ranging from 72–95%. The spectral data reported for these compounds were as expected and crystallographic data for 5a, 7a, (see ESI†) and 8a–10a (Fig. 2) further corroborated these formulations.
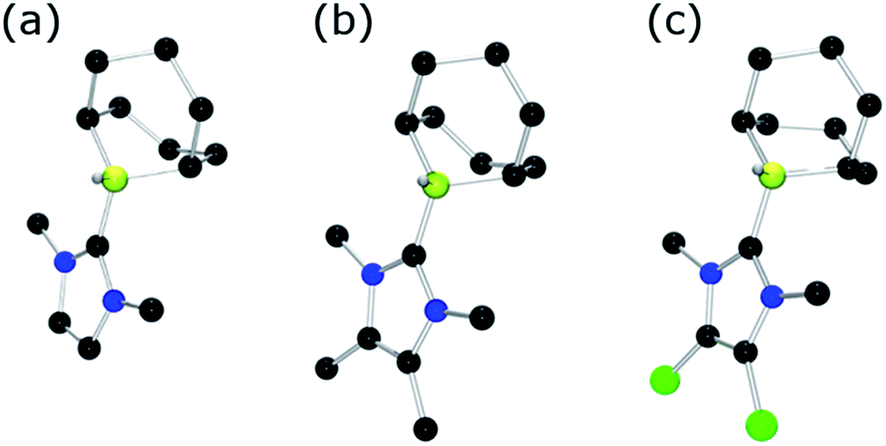 | ||
| Fig. 2 POV-ray depiction of (a) 8a (b) 9a (c) 10a; C: black, B: yellow-green, N: blue, H: grey, Cl: green. H-atoms except BH omitted for clarity. | ||
Each of these adducts reacts with [Ph3C][B(C6F5)4] to give the corresponding borenium salts [((R′CNR)2C)BC8H14] [B(C6F5)4] (R′ = H, R = dipp 4b, Mes 5b,75,76 Me 8b; R = Me R′ = Me 9b, Cl, 10b) and [((HC)2(NMe)(NR)C)BC8H14] [B(C6F5)4] (R = tBu, 6b, Ph 7b) concomitant with the generation of a stoichiometric amount of Ph3CH (Scheme 3). The most diagnostic spectroscopic change in each case is the appearance of a broad 11B resonance in the range of 81–88 ppm attributable to a three-coordinate B center. The expected resonances for the [B(C6F5)4]− anion were seen at −16.7 ppm. The species 10b was isolated as colorless crystals in 72% yield via recrystallization from CH2Cl2/pentane at −35 °C. Crystallographic data (Fig. 3) revealed trigonal planar geometry about the B center in the cation with a B–CNHC bond length of 1.5768(3) Å similar to that observed for 1b (1.580(3) Å).67
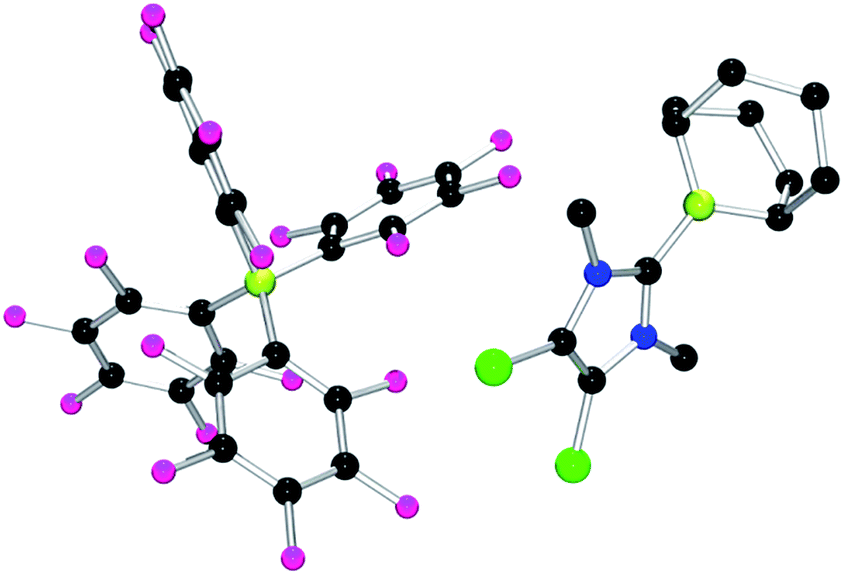 | ||
| Fig. 3 POV-ray depiction of 10b. C: black, B: yellow-green, N: blue, F: pink, Cl: green. H-atoms omitted for clarity. | ||
For comparative purposes the phosphine–borane adduct (Me3P)(HBC8H14) (11) was also synthesized and isolated as colorless crystals in 82% yield. The 11B NMR signal was observed at −14.9 ppm and exhibited both B–H coupling of 88 Hz and B–P coupling of 48 Hz. The 31P{1H} resonance for 11 is at −13.0 ppm and possesses similar B–P coupling. Single crystal X-ray diffraction confirmed the formulation (see ESI†). In contrast to the carbene complexes described above, treatment of 11 with stoichiometric [Ph3C][B(C6F5)4] gave a complex mixture of products as evidenced by 31P{1H} and 11B NMR-spectroscopy.
Hydrogenation catalysis
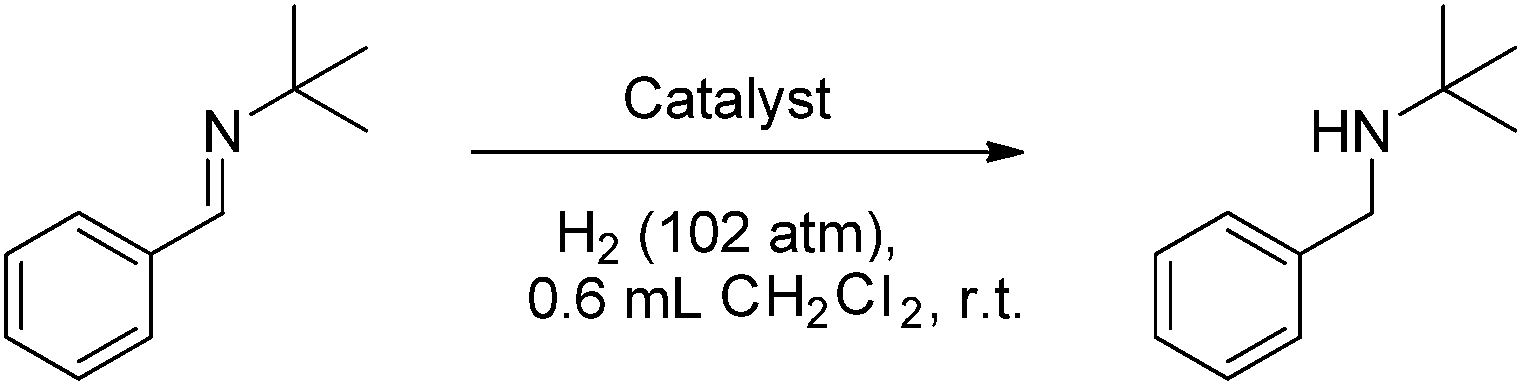
Compounds 4b–10b were tested for catalytic activity using the hydrogenation of tBuNCHPh as a comparative screen. A solution of each was generated in situ, added to the imine substrate and pressurized with 102 atm H2(g) for 30 minutes. After the reaction, the extent of conversion to amine was assessed by 1H NMR spectroscopy. These data reveal an inverse correlation between the steric demands of the NHC and the hydrogenation activity of the borenium catalyst (Table 1). The bulkiest catalyst 5b shows no catalytic activity, while the slightly less bulky catalyst 6b shows only trace conversion of imine to amine at 1 mol% catalyst loading after 30 minutes of reaction time. A sharp increase of catalytic activity is observed as the steric demands of the catalyst are further reduced. The previously reported catalyst 1b allows for 35% conversion to amine at 1 mol% catalyst loading after 30 minutes while catalysts 7b and 8b show quantitative conversion.
Reducing the loadings of these catalysts to 0.5 mol% under otherwise identical conditions reduced the conversions and demonstrated that the least bulky catalyst 8b effects 68% conversion while 7b and 9b reach only 35% and 21% conversion, respectively. In contrast, 10b gave complete conversion. Even when the loading was dropped to 0.25 mol% under otherwise identical conditions 10b gave complete conversion of imine to amine. Further reduction to 0.1 mol% gave 47% conversion representing a turn-over frequency (TOF) of 940 h−1. A slight increase of catalyst loading to 0.15 mol% and an extension of the reaction time to 2 h at room temperature under 102 atm H2(g) led to complete conversion to tBuNHCH2Ph and the product could be isolated in 83% yield (Table 1, entry 13).
These observations reveal that the least sterically encumbered NHCs stabilize the most active borenium catalysts despite the fact that FLP reactivity hinges upon the steric protection of an acidic center. This suggests that the bulkier catalysts impede either H2 activation or hydride delivery in the catalytic cycle. Since bulky NHC–borane 4a readily delivers hydride to an iminium ion it seems most likely that the bulkiest borenium ions are sterically prevented from generating the “encounter complex” with the imine that is required for H2 activation. Similarly diminished reactivity has been observed for FLPs incorporating excessively bulky boranes.34 It is noteworthy that computations suggest that a donor–boron distance of 4.2 Å is necessary to effect heterolytic cleavage of H2.77 Thus, it is reasonable to suggest that bulky peripheral substituents inhibit such a close approach.
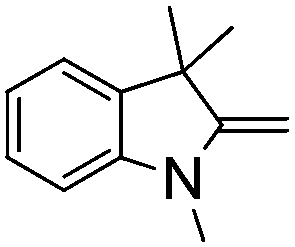
Comparison of the isosteric catalysts 8b–10b reveals that reduced donation from the NHC78 to the B center has a positive impact on the catalytic activity. This is thought to result from an increase in the Lewis acidity at B. That being said, further reduction of the donor ability of the stabilizing ligand79 jeopardizes the stability of the borenium cation as evidenced by the efforts to abstract hydride from 11. Apparently the donor ability and steric demands of the NHC are suitably balanced in 10b as it provides, to our knowledge, the highest TOF reported to date for the metal-free hydrogenation of imines. With the optimized catalyst 10b in hand, a variety of N-containing unsaturated substrates were reduced affording products in high isolated yields (Table 2). In these cases a catalyst loading of 5 mol% was employed to ensure high conversions in 30 minutes and to overcome the impact of adventitious water. The imine o-ClC6H4CHNtBu, is readily reduced (Table 2, entry 1) as is p-(MeO2C)C6H4CHNtBu (Table 2, entry 2). The latter stands in contrast to previous FLP hydrogenations where sterically unencumbered esters preclude or inhibit reductions using the borane B(C6F5)3.20 While the steric demands of C6H5CHNCHPh2 slow imine reduction (Table 2, entry 3), the aniline-derived ketimines Ph(Me)CNPh and p-EtOC6H4(Me)CNPh are readily hydrogenated to corresponding amines (Table 2, entries 4 and 5). In stark contrast, no hydrogenation of Ph(Me)CNCH2Ph was observed (Table 2, entry 6). This was attributed to the greater basicity and lesser steric demands about the N-donor. 1,3,3-Trimethyl-2-methylideneindoline is hydrogenated to afford 1,2,3,3-tetramethylindoline (Table 2, entry 7), however 2,3,3-trimethylindolenine is not reduced (Table 2, entry 8). Nonetheless, in contrast to 1b,6710b smoothly catalyzes the hydrogenation of 8-methylquinoline to 1,2,3,4-tetrahydro-8-methylquinoline (Table 2, entry 9), illustrating the subtlety of steric and electronic effects on substrate scope.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yield |
| a Yields determined by 1H NMR spectroscopy, isolated yields in parentheses. All reactions were carried out using 0.500 mmol substrate in CH2Cl2. Reaction times were 30 minutes. Catalyst loadings: 5 mol% except: b 2.5 mol%. | |||
| 1 |
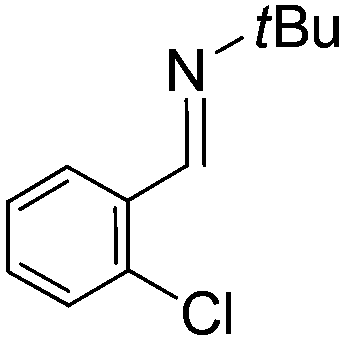
|

|
100b (98) |
| 2 |

|
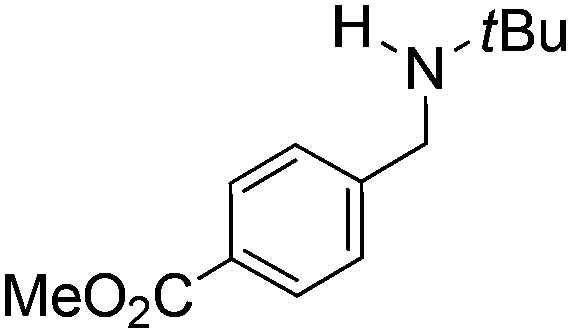
|
100 (82) |
| 3 |

|

|
39 |
| 4 |
|
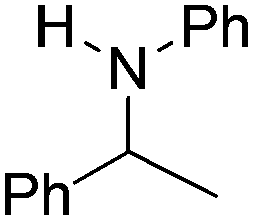
|
100 (71) |
| 5 |

|

|
100 (95) |
| 6 |

|

|
0 |
| 7 |
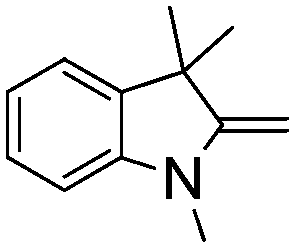
|
|
100 (91) |
| 8 |

|

|
0 |
| 9 |

|
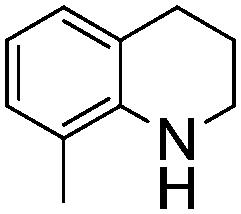
|
100 (87) |

Conclusions
In this manuscript we have probed the electronic and steric parameters that impact on the ability of ligand stabilized borenium cations to act as metal-free hydrogenation catalysts. Although this catalysis proceeds via an FLP mechanism, perturbations that enhance the Lewis acidity at B or the steric demands of the NHC ligand can serve to deactivate the catalyst. At the same time, sterically unencumbered NHCs bearing electron withdrawing substituents enhance catalyst activity. Crudden and co-workers80 have very recently described related triazolium derived borenium cations and their use as catalysts for hydrogenation. Nonetheless, the present systematic examination of NHC stabilized borenium ion has led to catalysts that are highly efficient. Indeed the isolable catalyst 10b is an effective catalyst for imine and N-heterocycle reduction at low catalyst loadings and it affords the highest TOF yet reported for metal-free hydrogenation catalysis. Efforts are continuing to systematically develop borenium-based metal-free hydrogenation catalysts and to further broaden their applications.Acknowledgements
The authors acknowledge the financial support from NSERC of Canada and DWS is grateful for the award of a Canada Research Chair.Notes and references
- H.-U. Blaser, F. Spindler and M. Thommen, in The Handbook of Homogeneous Hydrogenation, Wiley-VCH Verlag GmbH, 2008, pp. 1279–1324, DOI:10.1002/9783527619382.ch37.
- W. A. Herrmann and B. Cornils, Angew. Chem., Int. Ed. Engl., 1997, 36, 1048–1067 CrossRef.
- H. U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385–1393 CrossRef CAS PubMed.
- W. W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS PubMed.
- R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC.
- R. V. Jagadeesh, A. E. Surkus, H. Junge, M. M. Pohl, J. Radnik, J. Rabeah, H. M. Huan, V. Schunemann, A. Bruckner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076–1080 CrossRef CAS PubMed.
- S. Monfette, Z. R. Turner, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 4561–4564 CrossRef CAS PubMed.
- C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1992, 114, 7562–7564 CrossRef CAS.
- C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 8952–8965 CrossRef CAS.
- J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434–9438 CrossRef CAS PubMed.
- J. A. Hatnean, J. W. Thomson, P. A. Chase and D. W. Stephan, Chem. Commun., 2014, 50, 301–303 RSC.
- T. Mahdi, J. N. del Castillo and D. W. Stephan, Organometallics, 2013, 32, 1971–1978 CrossRef CAS.
- T. Mahdi, Z. M. Heiden, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 4088–4091 CrossRef CAS PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed.
- L. J. Hounjet, C. Bannwarth, C. N. Garon, C. B. Caputo, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 7492–7495 CrossRef CAS PubMed.
- L. Greb, P. Oña-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 51, 10164–10168 CrossRef CAS PubMed.
- D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geier, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338–12348 CrossRef CAS PubMed.
- S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884–4886 RSC.
- P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701–1703 RSC.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- S. Schwendemann, T. A. Tumay, K. V. Axenov, I. Peuser, G. Kehr, R. Fröhlich and G. Erker, Organometallics, 2010, 29, 1067–1069 CrossRef CAS.
- K. V. Axenov, G. Kehr, R. Fröhlich and G. Erker, J. Am. Chem. Soc., 2009, 131, 3454–3455 CrossRef CAS PubMed.
- H. D. Wang, R. Fröhlich, G. Kehr and G. Erker, Chem. Commun., 2008, 5966–5968, 10.1039/b813286k.
- P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543–7546 CrossRef CAS PubMed.
- V. Sumerin, K. Chernichenko, M. Nieger, M. Leskelä, B. Rieger and T. Repo, Adv. Synth. Catal., 2011, 353, 2093–2110 CrossRef CAS.
- V. Sumerin, F. Schulz, M. Atsumi, C. Wang, M. Nieger, M. Leskelä, T. Repo, P. Pyykkö and B. Rieger, J. Am. Chem. Soc., 2008, 130, 14117–14118 CrossRef CAS PubMed.
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001–6003 CrossRef CAS PubMed.
- K. Chernichenko, Á. Madarász, I. Pápai, M. Nieger, M. Leskelä and T. Repo, Nat. Chem., 2013, 5, 718–723 CrossRef CAS PubMed.
- L. Greb, C. G. Daniliuc, K. Bergander and J. Paradies, Angew. Chem., Int. Ed., 2013, 52, 5876–5879 CrossRef CAS PubMed.
- G. Erõs, H. Mehdi, I. Pápai, T. A. Rokob, P. Király, G. Tárkányi and T. Soós, Angew. Chem., Int. Ed., 2010, 49, 6559–6563 CrossRef PubMed.
- G. Erõs, K. Nagy, H. Mehdi, I. Pápai, P. Nagy, P. Király, G. Tárkányi and T. Soós, Chem.–Eur. J., 2012, 18, 574–585 CrossRef PubMed.
- D. Chen, Y. Wang and J. Klankermayer, Angew. Chem., Int. Ed., 2010, 49, 9475–9478 CrossRef CAS PubMed.
- D. Chen and J. Klankermayer, Chem. Commun., 2008, 2130–2131, 10.1039/801806e.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 6810–6813 CrossRef CAS PubMed.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 6810–6813 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 332, 85–110 CrossRef CAS.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- J. A. Nicasio, S. Steinberg, B. Ines and M. Alcarazo, Chemistry, 2013, 19, 11016–11020 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- P. Koelle and H. Nöth, Chem. Rev., 1985, 85, 399–418 CrossRef CAS.
- W. E. Piers, S. C. Bourke and K. D. Conroy, Angew. Chem., Int. Ed., 2005, 44, 5016–5036 CrossRef CAS PubMed.
- T. S. De Vries, A. Prokofjevs and E. Vedejs, Chem. Rev., 2012, 112, 4246–4282 CrossRef CAS PubMed.
- E. J. Corey, Angew. Chem., Int. Ed., 2009, 48, 2100–2117 CrossRef CAS PubMed.
- A. Boussonnière, X. Pan, S. J. Geib and D. P. Curran, Organometallics, 2013, 32, 7445–7450 CrossRef.
- X. Pan, A. Boussonniere and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 14433–14437 CrossRef CAS PubMed.
- X. Pan, X. Chen, T. Li, Y. Li and X. Wang, J. Am. Chem. Soc., 2013, 135, 3414–3417 CrossRef CAS PubMed.
- A. Prokofjevs, A. Boussonniere, L. Li, H. Bonin, E. Lacôte, D. P. Curran and E. Vedejs, J. Am. Chem. Soc., 2012, 51, 5958–5961 Search PubMed.
- P. Eisenberger, A. M. Bailey and C. M. Crudden, J. Am. Chem. Soc., 2012, 134, 17384–17387 CrossRef CAS PubMed.
- J. Chen, R. A. Lalancette and F. Jäkle, Chem. Commun., 2013, 49, 4893–4895 RSC.
- E. J. Lawrence, V. S. Oganesyan, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, J. Am. Chem. Soc., 2014, 136, 6031–6036 CrossRef CAS PubMed.
- V. Bagutski, A. Del Grosso, J. A. Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474–487 CrossRef CAS PubMed.
- I. A. Cade and M. J. Ingleson, Chem.–Eur. J., 2014, 20, 12874–12880 CrossRef CAS PubMed.
- E. R. Clark, A. Del Grosso and M. J. Ingleson, Chem.–Eur. J., 2013, 19, 2462–2466 CrossRef CAS PubMed.
- E. R. Clark and M. J. Ingleson, Organometallics, 2013, 32, 6712–6717 CrossRef CAS.
- A. Del Grosso, M. D. Helm, S. A. Solomon, D. Caras-Quintero and M. J. Ingleson, Chem. Commun., 2011, 47, 12459–12461 RSC.
- A. Del Grosso, P. J. Singleton, C. A. Muryn and M. J. Ingleson, Angew. Chem., Int. Ed., 2011, 50, 2102–2106 CrossRef CAS PubMed.
- M. J. Ingleson, Synlett, 2012, 1411–1415, DOI:10.1055/s-0031–1291147.
- J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon and M. J. Ingleson, Angew. Chem., Int. Ed., 2013, 52, 7518–7522 CrossRef CAS PubMed.
- S. A. Solomon, A. Del Grosso, E. R. Clark, V. Bagutski, J. J. W. McDouall and M. J. Ingleson, Organometallics, 2012, 31, 1908–1916 CrossRef CAS.
- A. Prokofjevs, J. W. Kampf and E. Vedejs, Angew. Chem., Int. Ed., 2011, 50, 2098–2101 CrossRef CAS PubMed.
- A. Del Grosso, R. G. Pritchard, C. A. Muryn and M. J. Ingleson, Organometallics, 2009, 29, 241–249 CrossRef.
- J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS PubMed.
- D. P. Curran, A. Solovyev, M. M. Brahmi, L. Fensterbank, M. Malacria and E. Lacote, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed.
- M. Horn, H. Mayr, E. Lacote, E. Merling, J. Deaner, S. Wells, T. McFadden and D. P. Curran, Org. Lett., 2012, 14, 82–85 CrossRef CAS PubMed.
- P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044–19047 CrossRef CAS PubMed.
- H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kramer, T. Kupfer, K. Radacki, E. Siedler, A. Trumpp, K. Wagner and C. Werner, J. Am. Chem. Soc., 2013, 135, 8702–8707 CrossRef CAS PubMed.
- (a) A. Prokofjevs and E. Vedejs, J. Am. Chem. Soc., 2011, 133, 20056–20059 CrossRef CAS PubMed; (b) A. Prokofjevs, J. Jermaks, A. Borovika, J. W. Kampf and E. Vedejs, Organometallics, 2013, 32, 6701–6711 CrossRef CAS PubMed.
- M. M. Brahmi, J. Monot, M. D.-E. Murr, D. P. Curran, L. Fensterbank, E. Lacôte and M. Malacria, J. Org. Chem., 2010, 75, 6983–6985 CrossRef CAS PubMed.
- D. M. Lindsay and D. McArthur, Chem. Commun., 2010, 46, 2474–2475 RSC.
- The corresponding triflate salt has been reported in the following reference.
- D. McArthur, C. P. Butts and D. M. Lindsay, Chem. Commun., 2011, 47, 6650–6652 RSC.
- T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435–2438 CrossRef CAS PubMed.
- D. G. Gusev, Organometallics, 2009, 28, 6458–6461 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313 CrossRef CAS.
- P. Eisenberger, B. P. Bestvater, E. C. Keske and C. M. Crudden, Angew. Chem., Int. Ed., 2015, 54, 1–6 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and spectral details are deposited. CCDC 1035566–1035574. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03675a |
| This journal is © The Royal Society of Chemistry 2015 |