
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrophilic bis-fluorophosphonium dications: Lewis acid catalysts from diphosphines†
Michael H.
Holthausen
a,
Rashi R.
Hiranandani
a and
Douglas W.
Stephan
*ab
aDepartment of Chemistry, University of Toronto, 80 St. George St, Toronto, Ontario M5S3H6, Canada. E-mail: dstephan@chem.utoronto.ca
bChemistry Department-Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 27th January 2015
Abstract
Stepwise oxidation of 1,8-bis(diphenylphosphino)naphthalene and a series of (oligo)methylene-linked diphosphines with XeF2 followed by fluoride abstraction yields a family of compounds featuring phosphine, phosphonium and phosphorane moieties in close proximity. The bisphosphonium ions [(C10H6)(Ph2PF)2]2+ (5) and [CH2(Ph2PF)2]2+ (9a) exhibit remarkable Lewis acidity arising from the proximity of the phosphonium centers. The effectiveness of bisphosphonium dications (5, 9a–e) is examined in a series of Lewis acid catalysed transformations.
Introduction
Main group based Lewis acids are used as co-catalysts in polymerization catalysis or as Lewis acid catalysts in a wide range of organic transformations.1 As such reactivity is predicated only on being electron deficient, group XIII compounds such as BH3, BF3, or AlMe3 are the quintessential examples of main-group Lewis acids. Group XIV-centered Lewis acids (C,2 Si3) have also been exploited although as these compounds are generally very strong Lewis acids and coordinatively unsaturated, they tend to be employed as stoichiometric reagents. Moving further to the right to group XV, derivatives of these elements are most commonly Lewis bases and the electrophilic character of group XV species has drawn lesser attention. Nonetheless, P(III) phosphenium ions [R2P]+ have been shown to be ambiphilic effecting C–C, C–H4 and P–P5 bond activation. P(V) phosphonium cations of the form [R4P]+ (R = aryl, alkyl) are weakly Lewis acidic and have been exploited as fluoride ion sensors6 or as catalysts for (cyclo)addition reactions to polar or activated unsaturates.7 Exploring more electron deficient phosphonium cations, we found that [(C6F5)3PF]+ is a highly electrophilic phosphonium cation (EPC). This Lewis acidity is attributed to the presence of the strongly electron-withdrawing substituents and the energetically accessible σ*(P–F) orbital.8 In an alternative approach towards EPCs, the dicationic phosphonium salt [(SIMes)PPh2F][B(C6F5)4]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 was shown to be even more Lewis acidic than [(C6F5)3PF]+, demonstrating the impact of the additional positive charge. Nonetheless both [(C6F5)3PF]+ and [(SIMes)PPh2F]2+ were shown to be effective catalysts for the hydrodefluorination of fluoroalkanes and the hydrosilylation or transfer-hydrogenation of olefins and alkynes.8–10
9 was shown to be even more Lewis acidic than [(C6F5)3PF]+, demonstrating the impact of the additional positive charge. Nonetheless both [(C6F5)3PF]+ and [(SIMes)PPh2F]2+ were shown to be effective catalysts for the hydrodefluorination of fluoroalkanes and the hydrosilylation or transfer-hydrogenation of olefins and alkynes.8–10
In group XIII11 and group XIV chemistry,12 the proximity of two Lewis acidic centers has been shown to enhance the Lewis acidity, providing even more potent Lewis acids and thus impact on reactivity. Only recently, a group XV-based bidentate stiborane was introduced as strong, chelating Lewis acid for fluoride complexation.13 Exploring these notions in phosphonium chemistry prompted us to target bidentate bis-phosphonium salts. In this manuscript we prepare a series of such compounds exploiting a naphthalene backbone which locks two P moieties in the close vicinity of the peri-positions. In addition a series of more flexible (oligo)methylene-linked derivatives ((CH2)n, n = 1–5) are prepared. The impact of the proximity of two EPC centers is gauged by an examination of these species in a series of Lewis acid catalyses.
Results and discussion
The naphthalene-based diphosphine (C10H6)(PPh2)2 reacts with XeF2 at low temperatures (−30 °C) affording the selective formation of the phosphine–phosphorane species (C10H6)(Ph2PF2)(PPh2) (1) in high yields (87%, Scheme 1). The 31P{1H} NMR spectrum of 1 shows two triplet of doublet resonances at −55.5 ppm (1JPF = 742 Hz, 4JPP = 10 Hz) and −18.5 ppm (5JPF = 17 Hz, 4JPP = 10 Hz) which are assigned to the phosphorane and phosphine moieties respectively.14 The associated doublet of doublet resonance of the fluorine atoms shows a chemical shift of −42.0 ppm in the 19F{1H} NMR spectrum.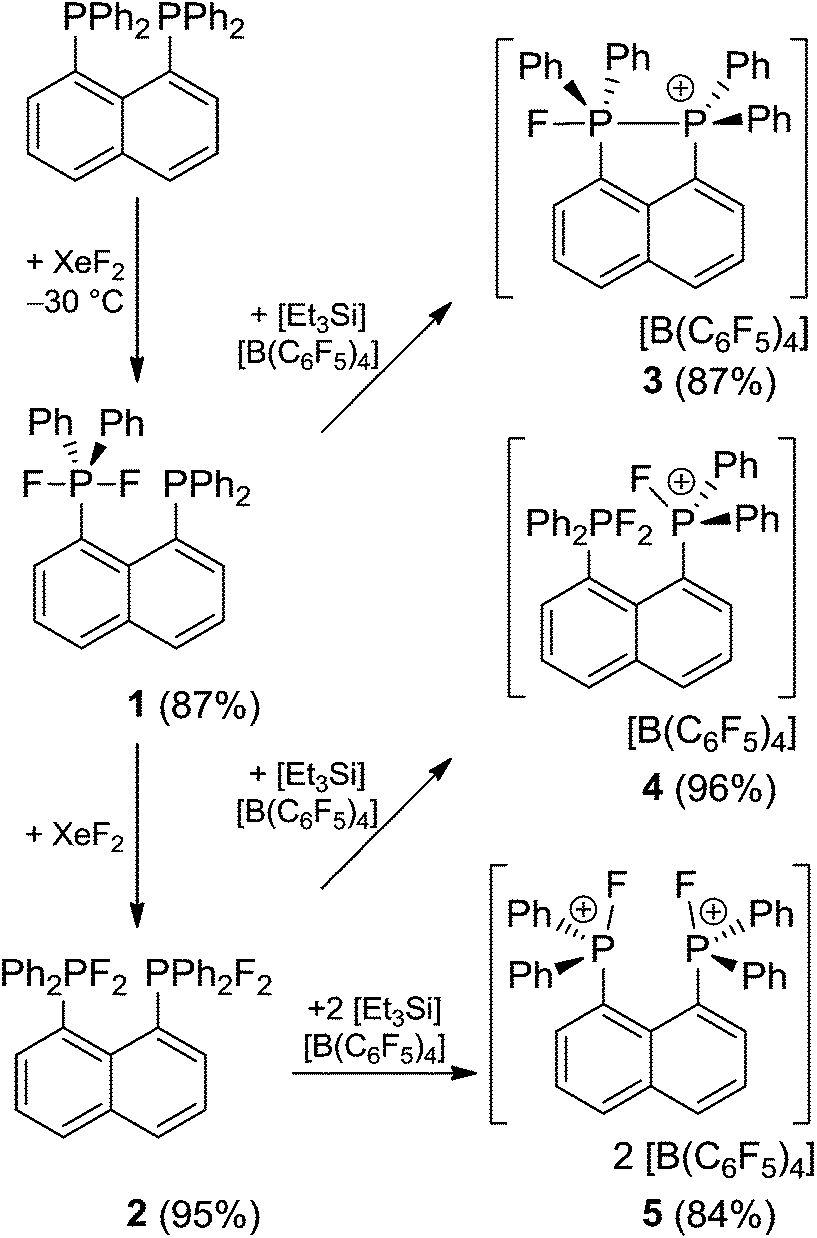 | ||
| Scheme 1 Synthetic routes to 1–5. | ||
Reacting (C10H6)(PPh2)2 with XeF2 in a 1:2 stoichiometry at ambient temperature yields quantitatively the bis-difluorophosphorane (C10H6)(Ph2PF2)2 (2, 95%). An AA′X2X′2 spin system is observed in the 31P{1H} (δA = −49.4 ppm) and 19F{1H} NMR (δX = −34.3 ppm) spectra of 2 and its parameters were evaluated by full line shape iteration.15 Both resonances of 2 are shifted to lower field compared to the phosphorane moiety in 1. A relatively large 5JPF (5JA′X = 5JAX′ = −18 Hz) and a comparatively small 4JPP coupling constant (4JAA′ = 3 Hz) might indicated a through space interaction between both phosphonium moieties.16
The molecular structure of 2 shows two trigonal bipyramidal phosphorus moieties with fluorine atoms occupying the axial positions (av. F–P–F: 177.0(1)°, Fig. 1). The P–F bond length (av. 1.67(3) Å) is typical for difluorotriarylphosphoranes.14,17 The close proximity of the phosphorane moieties in 2 leads to significant steric strain. This results in a buckling of its naphthalene-backbone as evidenced by the mean C-atom deviation from the least-squares C10 plane of 0.120(2) Å. The corresponding deviation in the parent diphosphine (C10H6)(Ph2P)2 is 0.006 Å.18 The steric strain also causes the phosphorus moieties of 2 to exhibit a strong bending out of the naphthalene plane (av. 0.73(1) Å vs. (C10H6)(Ph2P)2av. 0.42 Å). The shortest P⋯F interactions between the phosphorane moieties is 2.825(4) Å which is well within the sum of the respective van der Waal's radii (PF: 3.27 Å).19 The corresponding P⋯P distance of 3.823(3) Å is significantly longer than that in the parent diphosphine (3.052 Å).18 Earlier reports have described difficulties in effecting the quaternisation of both P atoms in (C10H6)(Ph2P)2 due to steric congestion,20 and, to the best of our knowledge, 2 represents the first compound featuring two penta-coordinated P atoms in peri-positions on a naphthalene framework.
 | ||
| Fig. 1 POV-ray depiction of 2. P: orange, F: yellow-green, C: black. Hydrogen atoms are omitted for clarity. | ||
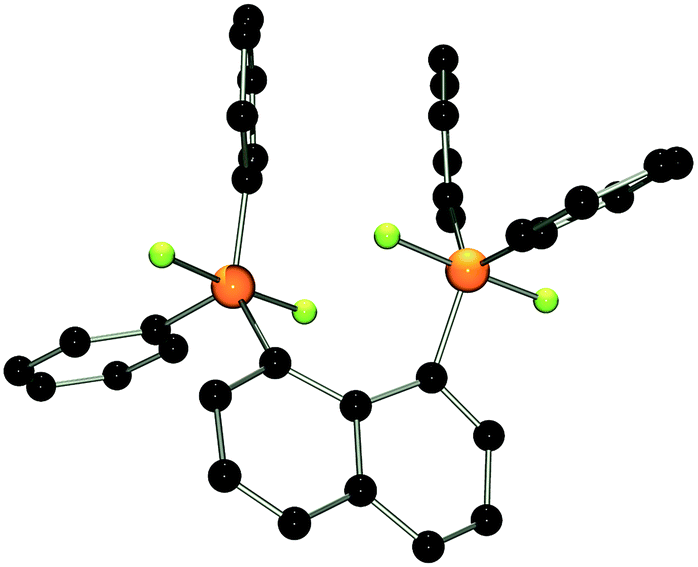
The reactions of phosphoranes 1 and 2 with [Et3Si][B(C6F5)4]·2(C7H8) in 1:1 stoichiometries yielded the respective salts [(C10H6)(Ph2PF)(PPh2)][B(C6F5)4] (3) and [(C10H6)(Ph2PF2)(Ph2PF)][B(C6F5)4] (4) in good to excellent yields (Scheme 1). The 31P{1H} NMR spectrum of 3 shows doublet of doublet resonances at −17.4 and −5.5 ppm. The former is assigned to the Ph2PF-moiety on basis of its 1JPF value of 783 Hz. A very large coupling constant between both P atoms (138 Hz) indicates coordination of the lone pair of the phosphine-moiety to the fluorophosphonium centre.21 This also results in a large coupling constant between the fluorine atom and the distal P atom (164 Hz; vs.5JPF = 17 Hz in 1). The indicated PP interaction was confirmed by single crystal X-ray crystallography (Fig. 2) as the P–P bond length was found to be 2.530(1) Å. This is longer than the P–P distance in P4 (2.2 Å)22 indicating that the interaction in 3 is weak. The fluoro-substituted P atom shows a trigonal bipyramidal bonding environment with the fluorine and the coordinating phosphine-moiety adopting axial positions. The P–F bond distance (1.637(2) Å) is significantly longer than in other fluorophosphonium species (av. 1.54(1) Å)8,9,14 which is consistent with the donation of electron density from the phosphine unit into the σ*-orbital of the P–F bond. In contrast to 2, the naphthalene backbone of 3 show no evidence of steric strain (mean C-atom deviation from the least-squares C10 plane: 0.006(1) Å).
 | ||
| Fig. 2 POV-ray depiction of the cation in 3. P: orange, F: yellow-green, C: black. Hydrogen atoms are omitted for clarity. | ||
It is noteworthy that interactions between the P atoms of adjacent phosphine and alkyl or aryl phosphonium centers in the related cations [(C10H6)(Ph2PR)(Ph2P)]+ (R = alkyl, aryl) is not observed,17,20a,23 reflecting the Lewis acidity of the fluorophosphonium cation. While we have previously reported the intramolecular coordination of an amide fragment to a fluorophosphonium moiety,24 compound 3 appears to be the first example of phosphine coordinating to a fluoro-phosphonium center. It is important to note that such an interaction is not observed in a 1:1 mixture of Ph3P and [Ph3PF][B(C6F5)4] indicating that the close proximity between Lewis acid and base imposed by the naphthalene backbone is crucial for adduct formation.
The 31P{1H} NMR spectrum of [(C10H6)(Ph2PF2)(Ph2PF)]+ (4) shows a broad triplet resonance at −51.7 ppm (1JPF = 717 Hz) assigned to the phosphorane moiety and a broad doublet at 96.6 ppm (1JPF = 1012 Hz) corresponding to the phosphonium centre. A crystallographic study revealed that the P–F bond lengths at the penta-coordinated P atom of 4 (av. 1.664(2) Å) are longer than the P–F bond distance at the adjacent fluorophosphonium centre (1.543(1) Å; Fig. 3). Similar to 2, a weak P⋯F interaction between the F on the phosphonium and P of the phosphorane fragment is observed (2.995(2) Å). Compared to 2 there is a decreased buckling of the naphthalene moiety (mean C-atom deviation from the least-squares C10 plane: 0.097(1) Å), a shorter P⋯P distance (3.668(2) Å), and the phosphonium P atom in 4 shows a reduced bending out of the naphthalene plane (0.057(1) Å) compared to its phosphorane neighbour (0.076(1) Å).
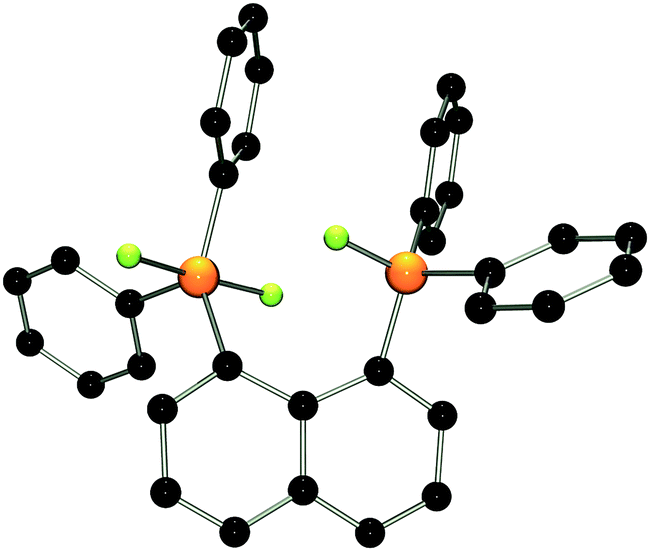 | ||
| Fig. 3 POV-ray depiction of the cation in 4·(CH2Cl2). P: orange, F: yellow-green, C: black. Hydrogen atoms are omitted for clarity. The solvate molecule was removed by the squeeze routine of the program PLATON. | ||
The bis-difluorophosphorane 2 also reacts with two equivalents of [Et3Si][B(C6F5)4]·2(C7H8) to give the salt [(C10H6)(Ph2PF)2][B(C6F5)4]2 (5) in high yields (84%, Scheme 1). The 31P{1H} and 19F{1H} NMR spectra of 5 reveal an AA′XX′ spin system. The 19F{1H} NMR spectrum reveals a very broad doublet resonance (δX = −117.1 ppm, Δν1/2 = 450 Hz). The 31P{1H} NMR resonance (δA = 96.8 ppm) shows a relatively large 1JPF coupling constant (1JAX = 1JA′X′ = 1004 Hz) which has also been observed for the related cations [(C6F5)3PF]+ (1062 Hz),8 and [(SIMes)PPh2F]2+ (1040 Hz9). Similar to 2, dication 5 also features a relative large 5JPF (5JA′X = 5JAX′ = −13 Hz) and a comparatively small 4JPP coupling constant (4JAA′ = 3 Hz). The related doubly protonated naphthalene-based bis-phosphine was previously studied by NMR spectroscopy.25 While dications incorporating a P–P bond21b,26 or a bridging substituent23,27 have been previously isolated.
In an effort to probe the Lewis acidity of 5, it was reacted with an equivalent of Et3PO employing the Gutmann–Beckett method (Scheme 2).28 The 31P{1H} NMR spectrum of the reaction mixture shows a broad resonance at 60.5 ppm corresponding to Et3PO interacting with 5. This results in an acceptor number of 21.4, which suggests that 5 is approximately half as Lewis acidic as B(C6F5)3. In comparison, combination of the salt [Ph3PF][B(C6F5)4] and Et3PO in a 2:1 stoichiometry showed only a very small shift in the resonance of Et3PO. These observations infer that the Lewis acidity of a phosphonium center in 5 is enhanced by the proximity of a second fluorophosphonium moiety. Monitoring the 1:1 mixture of 5 and Et3PO over time revealed slow transformation to [Et3PF]+ (6)9 and [(C6H10)(Ph2PO)(Ph2PF)]+ (7). The 31P{1H} NMR spectrum of (7) shows the expected doublet of doublet resonances at −34.5 ppm (1JPF = 697 Hz, JPP = 23 Hz) and 47.1 ppm (JPF = 2 Hz). The high field shift of the fluorophosphonium moiety and the low field shift of the phosphine oxide indicate some degree of donation of electron density from the PO fragment to the electrophilic fluorophosphonium cation. A similar but faster fluoride oxide exchange reaction was previously reported for the more Lewis acidic dication [(SIMes)PPh2F]2+.9
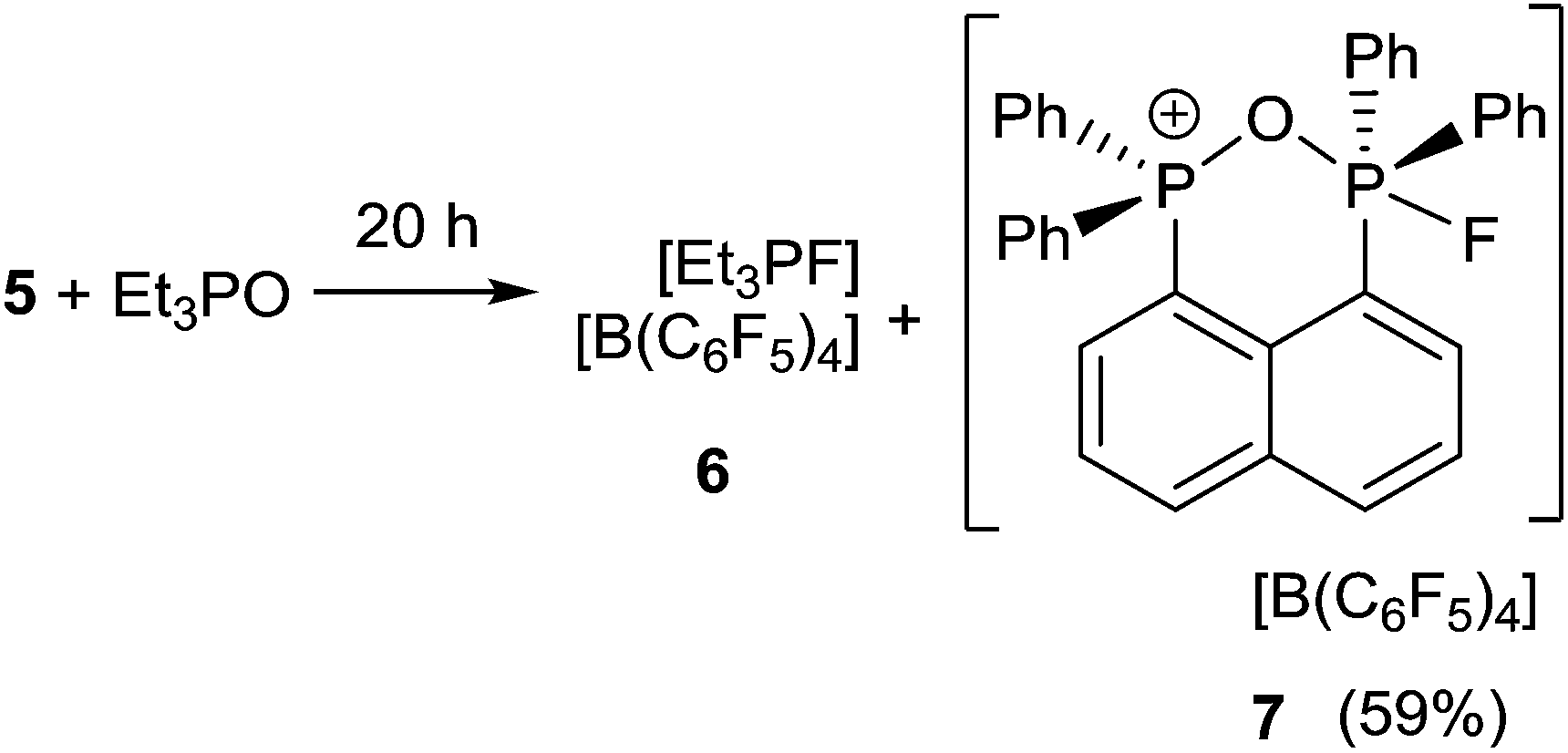 | ||
| Scheme 2 Reaction of 5 with Et3PO. | ||
A family of related bis-phosphonium dication salts was envisioned and thus reactions of (oligo)methylene-bridged diphosphines (CH2)n(Ph2P)2 (n = 1 (a), 2 (b), 3 (c), 4 (d), 5 (e)) and XeF2 were performed in CH2Cl2 to yield the respective bis-difluorophosphoranes (CH2)n(Ph2PF2)2 (8a–e) in high to almost quantitative isolated yields (Scheme 3). The species 8a–d have been previously prepared by using a range of oxidation reagents including F2,29 SF4,30 Me2NSF3,31 (CF3)2CO32 COF233 and NF3O34 or by halide exchange from the respective bromophosphoranes30 or by hydrofluorination of the respective silyl phosphinimines with HF.35 However, most of these methods suffer from low yields, laborious workup procedures and the use of hazardous reagents. The reaction of phosphoranes 8a–e with two equivalents of [Et3Si][B(C6F5)4]·2(C7H8) resulted in the formation of corresponding bis-phosphonium dication salts 9a–e (Scheme 3). All five derivatives were isolated and fully characterized. It is curious to note that the salts 9b and 9d, featuring linkers with an even number of CH2 units, are only sparingly soluble in CD2Cl2 whereas those with an odd numbers of methylene carbons in the linker (9a, 9c, 9e) show good solubility. The respective NMR spectra of 9a–e exhibit AA′XX′ spin systems (see ESI†). Compounds 9a–c show higher order effects due to coupling between the nuclei of the magnetically inequivalent phosphonium moieties and thus the NMR parameters were obtained via a full line shape iteration procedure.15 Interestingly 9a exhibits a large P–F coupling constant (1JAX = 1024 Hz) and significant coupling between both fluorine atoms (4JXX′ = 12 Hz) suggesting enhanced Lewis acidity. Remarkable large P–P coupling constant was observed for 9b (3JAA′ = 70 Hz) suggesting a weak through space interaction between the phosphorus centers (see ESI†).36
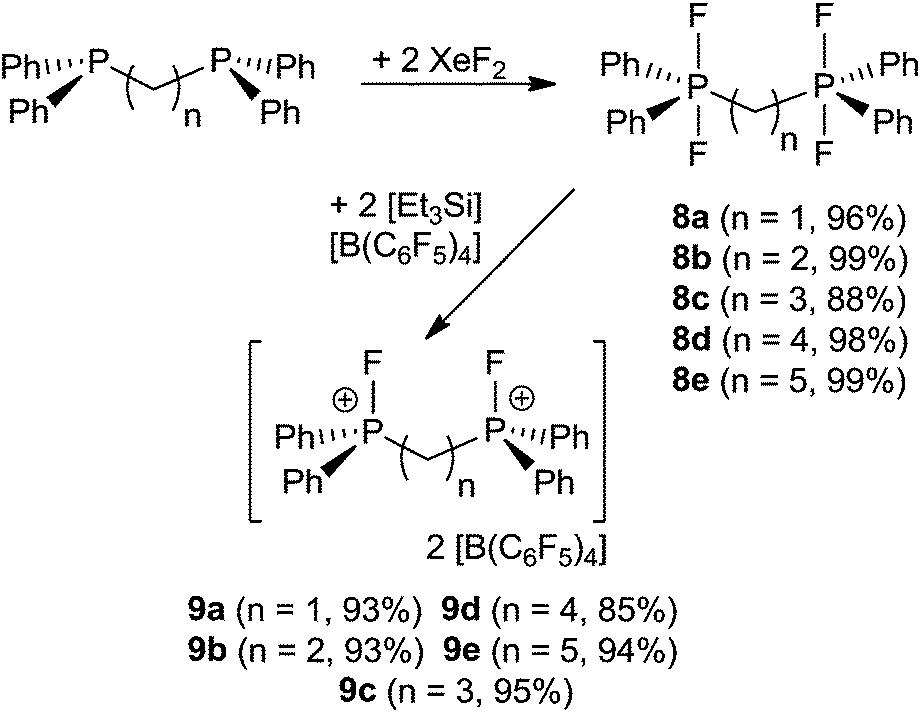 | ||
| Scheme 3 Synthetic route to 8a–e and 9a–e. | ||
Lewis acidity tests of this series of bis-fluorophosphonium ion salts 9a–e by the Gutmann–Beckett method28 were performed (Scheme 4). Determination of an acceptor number for 9a was hampered by deprotonation of the central CH2 moiety, affording [Et3POH][B(C6F5)4] (10) and [(CH)(Ph2PF)2][B(C6F5)4] (11). Compound (10) showed a broad 31P{1H} NMR resonance at 74.1 ppm, while (11) reveals an AA′XX′ spin system in the 31P{1H} (δA = 81.2 ppm) and 19F{1H} NMR spectrum (δX = −99.4 ppm). The formation of 11 was further supported by its independent synthesis from 9a and t-Bu3P.
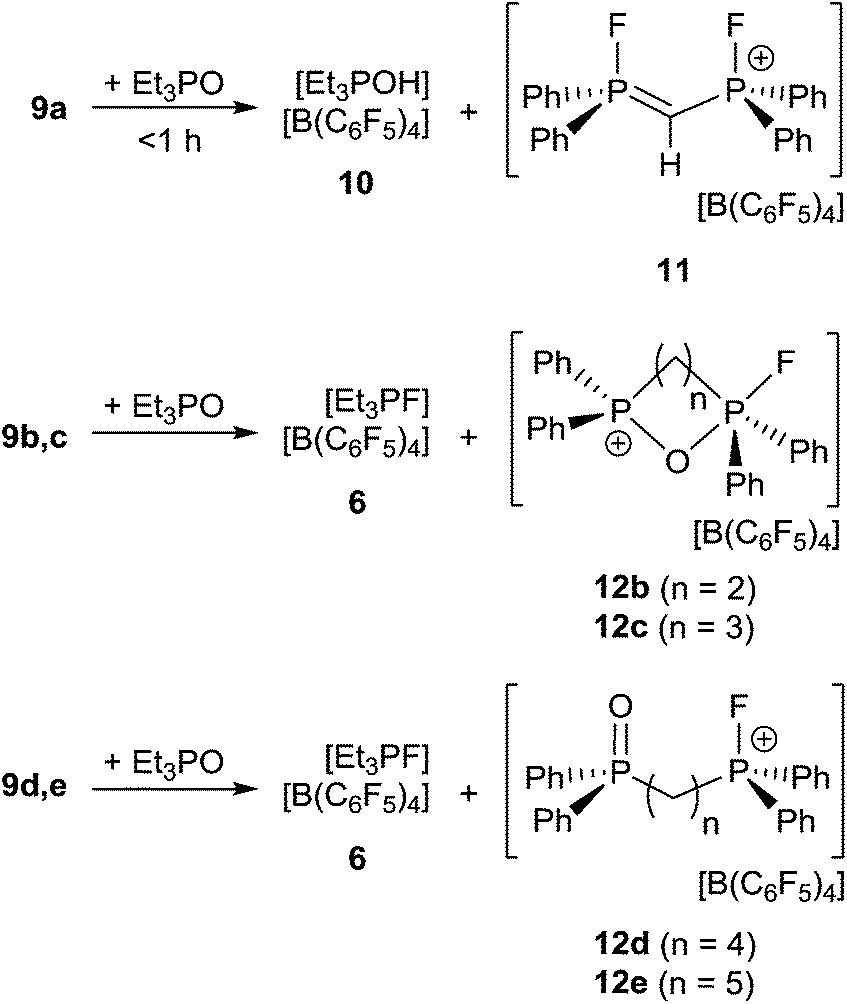 | ||
| Scheme 4 Reaction of 9a–e with Et3PO. | ||
The cations of 9b–e interact weakly with Et3PO resulting in acceptor numbers of ∼5. However, in all cases slow conversion to the monocationic species [(CH2)n(Ph2PO)(Ph2PF)][B(C6F5)4] (12b–e) and [Et3PF]+-salt 6 was observed.9 The shorter linkers in the case of 12b and 12c result in a chelate interaction of the phosphine oxide and the electrophilic fluorophosphonium center as evidenced by the high field shift of the fluoro-substituted P atom (12b: 2.2 ppm, 1JPF = 717 Hz, 12c: 45.2 ppm, 1JPF = 823 Hz). This notion is also supported by the observation of comparatively low field 31P NMR resonances arising for the PO fragments (12b: 53.1 ppm, 12d: 44.1 ppm).28 In contrast 12d–e show 31P{1H} NMR resonances in the typical range for discrete phosphine oxide37 and fluorophosphonium moieties.
The utility of the dications 5 and 9a–e in a variety of Lewis acid catalysed reactions was probed (Scheme 5). 1,1-Diphenylethylene in CD2Cl2 was treated with 2 mol% of 5 or 9a–e. In the case of 5 almost quantitative Friedel–Crafts dimerization was observed within 24 h at ambient temperature and the resulting product was isolated in 94% yield.38 Complete conversion was also achieved using 9a–c, however, 9a required only one hour reaction time, while 9b,c were complete in 24 h. In the case of 9d–e under the same conditions after 24 h, the Friedel–Crafts product was formed in lesser yields (9d: 50%, 9e: 25%). The corresponding reaction of 1,1-diphenylethylene with Et3SiH in the presence of these Lewis acid catalysts was assessed (Scheme 5).9,10a Using 9a as the catalyst gave 89% of the hydrosilylated product in one hour at ambient temperature whereas 9b and 5 achieve comparable yields after 24 h at 50 °C. No conversion is observed in the case of less Lewis acidic catalysts 9c–e.
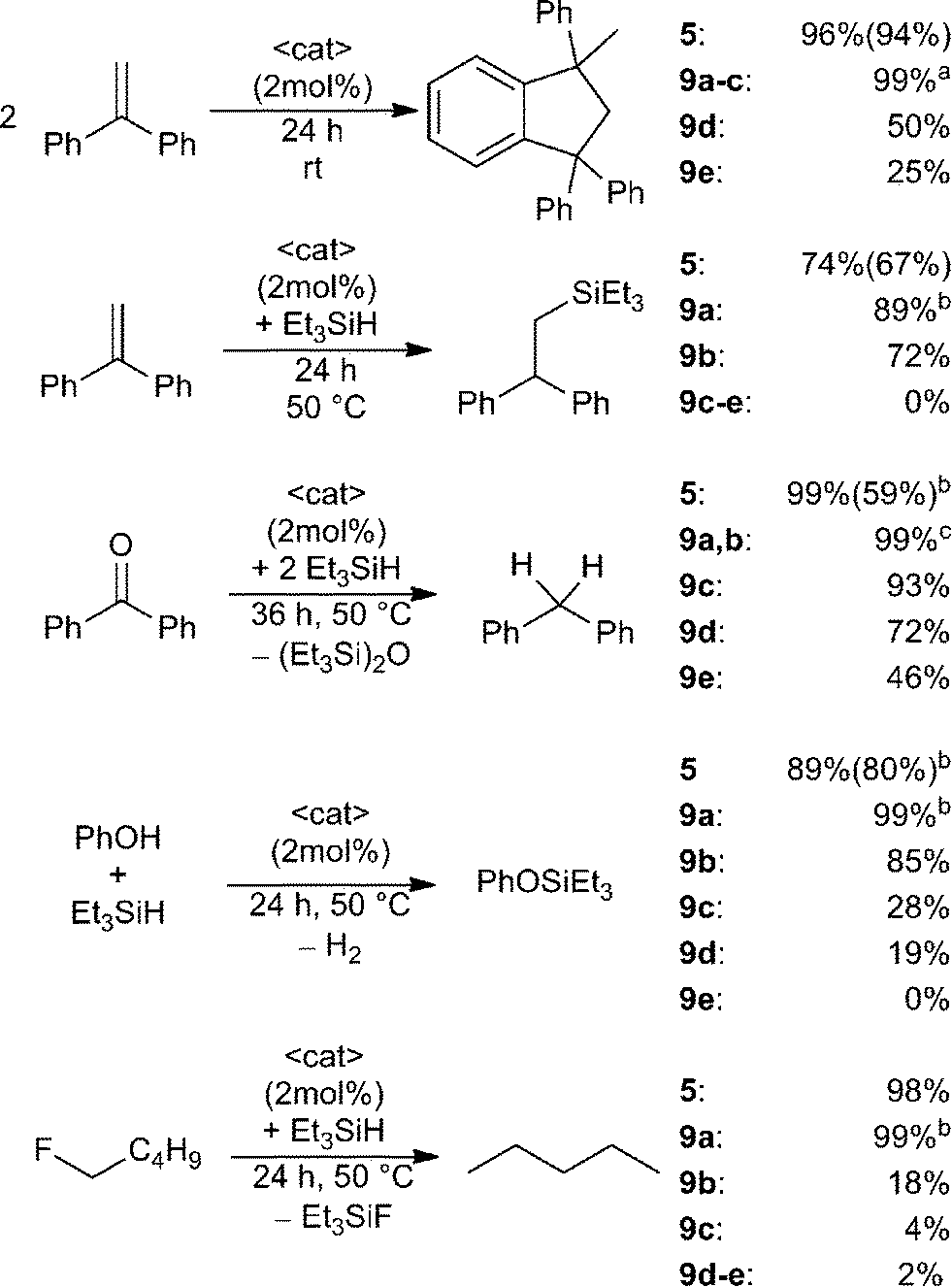 | ||
| Scheme 5 Lewis acid catalysis by salts 5 and 9a–e as catalysts for selected Lewis acid mediated transformations. Conversions were determined by 1H NMR or 19F NMR spectroscopy and isolated yields are given in parenthesis. (a) Ambient temperature and one hour reaction time (only for 9a). (b) Ambient temperature and one hour reaction time. (c) Ambient temperature and one hour (9a) or three hours (9b) reaction time. | ||
In a similar fashion treatment of benzophenone with two equivalents of Et3SiH in the presence of Lewis acid catalysts 5 and 9a,b led to hydrodeoxygenation and complete conversion to diphenylmethane and Et3SiOSiEt3 after one hour (5, 9a) or three hours (9b) respectively (Scheme 5). Compounds 9c–e required elevated temperatures and 36 h reaction time and afforded product in decreased yields (9c: 93%, 9d: 72%, 9e: 46%).
A fourth test involved the utility of these catalysts in the dehydrocoupling of Si–H bonds in silanes with phenol (Scheme 5).10b Compounds 5 and 9a result in rapid dehydrogenative coupling which gives PhOSiEt3 in high yields within one hour at ambient temperature. In contrast, elevated temperature (50 °C) and 24 h reaction time were required to observe similar reactivity for catalyst 9b. With an increase in methylene-linker length a decrease in conversion was observed (9c: 30%, 9d: 19%) and only traces of the coupling product are detected using 9e.
Finally, these EPCs were evaluated as catalysts for the hydrodefluorination of fluoropentane using Et3SiH as the hydride source (Scheme 5).8,9 Compounds 9a and 5 catalyse the hydrodefluorination affording almost complete conversion after one hour at ambient temperature for (9a) or 24 h at 50 °C for (5). The less Lewis acidic species 9b–e effect incomplete to trace conversions (9b: 18%, 9c: 4%, 9d: 2%, 9e: 2%) after 24 h at 50 °C.
The above results reveal that these dications are generally effective catalysts for a variety or Lewis acid mediated reactions. Moreover the proximity of two fluorophosphonium centers as in 5 and 9a–b acts to enhance the Lewis acidity and the resulting reactivity by a distal P–F interaction. Indeed, the longer chain species (9c–e) show diminished reactivity behaving more like independent [Ph2PF(alkyl)]+ cations. In this regard it is also noteworthy that in each of the above Lewis acid-catalysed reactions the species [Ph3PF][B(C6F5)4] was completely ineffective, which further supports the view that Lewis acidity of the present species is enhanced by spatial proximity.
Conclusions
In summary, stepwise oxidations of 1,8-bis-(diphenylphosphino)naphthalene and a series of bidentate (oligo)methylene-linked phosphines with XeF2 provide mono- and bisphosphorane species. Subsequent stepwise fluoride abstraction yielded phosphine/phosphonium, phosphonium/phosphorane and bisphosphonium species. The close spatial proximity in the naphthalene derived compound and the mono-methylene-linked species gives rise to Lewis acidity that is enhanced by the neighbouring fluorophosphonium moieties as evidenced by the reactivity of these species in the Lewis acid catalysed transformations including Friedel Crafts-type dimerization, hydrosilylation, dehydrocoupling, hydrodeoxygenation and hydrodefluorination. We are continuing to explore the facile and high yielding synthetic protocol that converts readily available bidentate donors providing Lewis acids of tuneable strength. New applications in Lewis acid catalysis and applications of these P(V) Lewis acids in frustrated Lewis pair (FLP) chemistry are the subject of on-going efforts.Acknowledgements
D.W.S. gratefully acknowledges the financial support of the NSERC of Canada and is grateful for the award of a Canada Research Chair. M.H.H. thanks the Alexander von Humboldt Foundation for a Feodor Lynen Research Fellowship. R.R.H. is grateful for the support of an NSERC CGS-M scholarship.Notes and references
- (a) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345 RSC; (b) G. Erker, Dalton Trans., 2005, 1883 RSC; (c) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578 CrossRef CAS; (d) F. J. Fernández-Alvarez, A. M. Aitani and L. A. Oro, Catal. Sci. Technol., 2014, 4, 611 RSC; (e) E. Dimitrijević and M. S. Taylor, ACS Catal., 2013, 3, 945 CrossRef; (f) K. Ishihara and H. Yamamoto, Eur. J. Org. Chem., 1999, 527 CrossRef CAS; (g) F. Blank and C. Janiak, Coord. Chem. Rev., 2009, 253, 827 CrossRef CAS PubMed; (h) S. V. Kostjuk and F. Ganachaud, Acc. Chem. Res., 2010, 43, 357 CrossRef CAS PubMed; (i) W. Kaminsky, Macromolecules, 2012, 45, 3289 CrossRef CAS.
- (a) B. Inés, S. Holle, R. Goddard and M. Alcarazo, Angew. Chem., Int. Ed., 2010, 49, 8389 CrossRef PubMed; (b) M. H. Holthausen, T. Mahdi, C. Schlepphorst, J. J. Weigand and D. W. Stephan, Chem. Commun., 2014, 50, 10038 RSC; (c) E. R. Clark and M. J. Ingleson, Organometallics, 2013, 32, 6712 CrossRef CAS; (d) E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2014, 53, 11306 CrossRef CAS PubMed; (e) M. P. Boone and D. W. Stephan, Chem.–Eur. J., 2014, 20, 3333 CrossRef CAS PubMed.
- (a) A. Schäfer, M. Reißmann, A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2011, 50, 12636 CrossRef PubMed; (b) A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2012, 51, 2981 CrossRef PubMed; (c) T. J. Herrington, B. J. Ward, L. R. Doyle, J. McDermott, A. J. P. White, P. A. Hunt and A. F. Ashley, Chem. Commun., 2014, 50, 12753 RSC.
- A. H. Cowley and R. A. Kemp, Chem. Rev., 1985, 85, 367 CrossRef CAS.
- (a) C. A. Dyker and N. Burford, Chem.–Asian J., 2008, 3, 28 CrossRef CAS PubMed; (b) M. H. Holthausen and J. J. Weigand, Chem. Rev., 2014, 43, 15201 CAS.
- (a) T. W. Hudnall, Y. M. Kim, M. W. P. Bebbington, D. Bourissou and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 10890 CrossRef CAS PubMed; (b) C. W. Chiu, Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 60 CrossRef CAS PubMed.
- (a) A. Werner, Adv. Synth. Catal., 2009, 351, 1469 CrossRef; (b) M. Terada and M. Kouchi, Tetrahedron, 2006, 62, 401 CrossRef CAS PubMed.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS PubMed.
- M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 65387 CrossRef PubMed.
- (a) M. Pérez, L. J. Hounjet, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 18308 CrossRef PubMed; (b) M. Pérez, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10917 CrossRef PubMed.
- (a) C. Jiang, O. Blacque and H. Berke, Chem. Commun., 2009, 5518 RSC; (b) X. Zhao and D. W. Stephan, Chem. Commun., 2011, 47, 1833 RSC; (c) M. J. Sgro, J. Dömer and D. W. Stephan, Chem. Commun., 2012, 48, 7253 RSC; (d) W. E. Piers, G. J. Irvine and V. C. Williams, Eur. J. Inorg. Chem., 2000, 2131 CrossRef CAS.
- (a) T. J. Karol, J. P. Hutchinson, J. R. Hyde, H. G. Kuivilla and J. A. Zubieta, Organometallics, 1983, 2, 106 CrossRef CAS; (b) H. G. Kuivilla, T. J. Karol and K. Swami, Organometallics, 1983, 2, 909 CrossRef; (c) M. Gielen and K. Jurkschat, J. Organomet. Chem., 1984, 273, 303 CrossRef CAS; (d) M. Austin, K. Gebreyes, H. G. Kivilla, K. Swami and J. A. Zubieta, Organometallics, 1987, 6, 834 CrossRef CAS; (e) K. Jurkschat, F. Hesselbarth, M. Dagatz, J. Lehmann, E. Kleinpeter and A. Tzschach, J. Organomet. Chem., 1990, 388, 259 CrossRef CAS; (f) D. Dakternieks, K. Jurkschat, H. Zhu and E. R. T. Tiekink, Organometallics, 1995, 14, 251 CrossRef; (g) N. Asao, P. Liu and K. Maruoka, Angew. Chem., Int. Ed., 1997, 36, 2507 CrossRef CAS.
- M. Hirai and F. P. Gabaï, Angew. Chem., Int. Ed., 2015, 54, 1205 CrossRef CAS PubMed.
- L. J. Hounjet, C. B. Caputo and D. W. Stephan, Dalton Trans., 2013, 42, 2629 RSC.
- P. H. M. Budzelaar, gNMR for Windows (5.0.6.0) NMR Simulation Program, Ivory Soft, 2006 Search PubMed.
- (a) G. R. Miller, A. W. Yankowsky and S. O. Grim, J. Chem. Phys., 1969, 51, 3185 CrossRef CAS PubMed; (b) E. Fluck and D. Wachtler, Liebigs Ann. Chem., 1979, 1125 CrossRef CAS.
- (a) R. R. Holmes, J. M. Holmes, R. O. Day, K. C. Kumara Swamy and V. Chandrasekhar, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 103, 153 CrossRef CAS; (b) G. M. Sheldrick, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 1776 CrossRef.
- R. D. Jackson, S. James, A. G. Orpen and P. G. Pringle, J. Organomet. Chem., 1993, 458, C3 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- (a) T. Costa and H. Schmidbaur, Chem. Ber., 1982, 115, 1374 CrossRef CAS; (b) D. M. Upulani, K. Somisara, M. Bühl, T. Lebl, N. V. Richardson, A. M. Z. Slawin, J. D. Woollins and P. Kilian, Chem.–Eur. J., 2011, 17, 2666 CrossRef PubMed; (c) A. Karacar, M. Freytag, H. Thönnessen, J. Omelańczuk, P. G. Jones, R. Bartsch and R. Schmutzler, Heteroat. Chem., 2001, 12, 102 CrossRef CAS PubMed.
- (a) M. Sanchez, R. Réau, F. Dahan, M. Regitz and G. Bertrand, Angew. Chem., Int. Ed., 1996, 35, 2228 CrossRef CAS; (b) M. Sanchez, R. Réau, H. Gornitzka, F. Dahan, M. Regitz and G. Bertrand, J. Am. Chem. Soc., 1997, 119, 9720 CrossRef CAS; (c) P. Kilian, D. Philp, A. M. Z. Slawin and J. D. Woollins, Eur. J. Inorg. Chem., 2003, 249 CrossRef CAS.
- (a) I. Krossing, J. Am. Chem. Soc., 2001, 123, 4603 CrossRef CAS; (b) H. Tai, I. Krossing, M. Seth and D. V. Deubel, Organometallics, 2004, 23, 2343 CrossRef CAS.
- K. Owsianik, L. Vendier, J. Błaszczyk and L. Sieroń, Tetrahedron, 2013, 69, 1628 CrossRef CAS PubMed.
- L. J. Hounjet, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4714 CrossRef CAS PubMed.
- A. Karaçar, V. Klaukien, M. Freytag, H. Thönnessen, J. Omelańzuk, P. G. Jones, R. Bartsch and R. Schmutzler, Z. Anorg. Allg. Chem., 2001, 627, 2589 CrossRef.
- (a) B. A. Surgenor, M. Bühl, A. M. Z. Slawin, J. D. Woollins and P. Kilian, Angew. Chem., Int. Ed., 2012, 51, 10150 CrossRef CAS PubMed; (b) M. J. Ray, A. M. Z. Slawin, M. Bühl and P. Kilian, Organometallics, 2013, 32, 3481 CrossRef CAS.
- (a) P. Kilian, A. M. Z. Slawin and J. D. Woollins, Dalton Trans., 2006, 2175 RSC; (b) K. Owsianik, R. Chauvin, A. Baliń, M. Wieczorek, M. Cypryk and M. Mikołajczyk, Organometallics, 2009, 28, 4929 CrossRef CAS.
- V. Gutmann, Coord. Chem. Rev., 1976, 18, 225 CrossRef CAS.
- I. Ruppert and V. Bastian, Angew. Chem., Int. Ed. Engl., 1977, 16, 718 CrossRef.
- E. Fluck and R. Braun, Synth. React. Inorg. Met.-Org. Chem., 1988, 18, 727 CrossRef CAS.
- R. R. Holmes, J. M. Holmes, R. O. Day, K. C. Kumara Swamy and V. Chandrasehkar, Phosphorus, Sulfur Silicon Relat. Elem., 1995, 103, 153 CrossRef CAS.
- I. V. Shevchenko, R. N. Mikolenko, E. Lork and G.-V. Röschenthaler, Eur. J. Inorg. Chem., 2001, 9, 2377 CrossRef.
- (a) O. D. Gupta and J. M. Shreeve, J. Chem. Soc., Chem. Commun., 1984, 416 RSC; (b) O. D. Gupta, R. L. Kirchmeier and J. M. Shreeve, J. Fluorine Chem., 1991, 52, 1 CrossRef CAS.
- O. D. Gupta, R. L. Kirchmeier and J. M. Shreeve, Inorg. Chem., 1990, 29, 573 CrossRef CAS.
- R. Appelt and I. Ruppert, Chem. Ber., 1975, 108, 919 CrossRef.
- (a) K.-O. Feldmann and J. J. Weigand, J. Am. Chem. Soc., 2012, 134, 15443 CrossRef CAS PubMed; (b) M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078 CrossRef CAS PubMed.
- (a) W. Peng and J. M. Shreeve, J. Fluorine Chem., 2005, 126, 1054 CrossRef CAS PubMed; (b) G. E. Maciel and R. V. James, Inorg. Chem., 1964, 3, 1650 CrossRef CAS.
- (a) A. G. Evans and E. D. Owen, J. Chem. Soc., 1959, 4123 CAS; (b) L. J. Hounjet, C. Bannwarth, C. N. Garon, C. B. Caputo, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 7492 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Preparative, spectroscopic and analytical details for the experiments described herein. CCDC 1041558–1041565. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00051c |
| This journal is © The Royal Society of Chemistry 2015 |