
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and reactivity of cyclo-tetra(stibinophosphonium) tetracations: redox and coordination chemistry of phosphine–antimony complexes†
Saurabh S.
Chitnis
a,
Alasdair P. M.
Robertson
a,
Neil
Burford
*a,
Jan J.
Weigand
*b and
Roland
Fischer
c
aDepartment of Chemistry, University of Victoria, Victoria, BC V8W 3V6, Canada. E-mail: nburford@uvic.ca; Fax: +1 250 721 7147; Tel: +1 250 721 7150
bDepartment of Chemistry and Food Chemistry, TU Dresden, 01062, Dresden, Germany. E-mail: jan.weigand@tu-dresden.de; Tel: +49 351 46842800
cInstitute for Inorganic Chemistry, TU Graz, 98010, Graz, Austria. E-mail: roland.fischer@tugraz.at; Tel: +43 316 87332109
First published on 3rd February 2015
Abstract
Reductive elimination of [R3PPR3]2+, [11(R)]2+, from the highly electrophilic SbIII centres in [(R3P)3Sb]3+, [8(R)]3+, gives SbI containing cations [(R3P)Sb]1+, [9(R)]1+, which assemble into frameworks identified as cyclo-tetra(stibinophosphonium) tetracations, [(R3P)4Sb4]4+, [10(R)]4+. A phosphine catalyzed mechanism is proposed for conversion of fluoroantimony complexes [(R3P)2SbF]2+, [7(R)]2+, to [10(R)]4+, and the characterization of key intermediates is presented. The results constitute evidence of a novel ligand activation pathway for phosphines in the coordination sphere of hard, electron deficient acceptors. Characterization of the associated reactants and products supports earlier, albeit less definitive, detection of analogous phosphine ligand activation in CuIII and TlIII complexes, demonstrating that these prototypical ligands can behave simultaneously as reducing agents and σ donors towards a variety of hard acceptors. The reactivity of the parent cyclo-tetra(stibinophosphonium) tetracation, [10(Me)]4+, is directed by high charge concentration and strong polarization of the P–Sb bonds. The former explains the observed facility for reductive elimination to yield elemental antimony and the latter enabled activation of P–Cl and P–H bonds to give phosphinophosphonium cations, [Me3PPR′2]1+, including the first example of an H-phosphinophosphonium, [(Me3P)P(H)R′]1+, and 2-phosphino-1,3-diphosphonium cations, [(Me3P)2PR′]2+. Exchange of a phosphine ligand in [10(Me)]4+ with [nacnac]1− gives [(Me3P)3Sb4(nacnac)]3+, [15(Me)]3+, and with dmap gives [(Me3P)3Sb4(dmap)]4+, [16]4+. The lability of P–Sb or Sb–Sb interactions in [10(Me)]4+ has also been illustrated by characterization of heteroleptically substituted derivatives featuring PMe3 and PEt3 ligands.
Introduction
Phosphines are prototypical ligands in the coordination chemistry of d-block metals. While the chemistry of p-block elements is primarily defined by covalent bonding as typified by organic frameworks, an array of phosphine adducts has also been characterized for main group element acceptors.1–5 Beyond their versatile ligand properties as neutral, two-electron donors (L-type),6 phosphines also exhibit redox reactivity within the coordination sphere of an acceptor. For example, reductive elimination of tetraorgano- or halotriorganophosphonium cations (Scheme 1a),7 and oxidative addition of PR–X bonds (Scheme 1b),8 or P–R bonds (Scheme 1c)9 are all known pathways of tertiary phosphine activation in transition metal chemistry. One report10 hints at the reductive elimination of a diphosphonium dication from a phosphine–metal complex (Scheme 1d). In this instance, spectroscopic studies indicate that the reaction of excess PMe3 with [Cu(MeCN)x][PF6]2 or [Tl(MeCN)x][UF6]3 yields [Me3PPMe3]2+, and the reduced metal complexes [Cu(PMe3)4][PF6] and [Tl(PMe3)2][UF6], respectively.10 However, neither the high oxidation state reactants nor the reduced products have been structurally verified and three different 31P NMR chemical shifts were ascribed to [Me3PPMe3]2+ (depending upon the counterion: +65.0 ppm, +46.3 ppm, or +27.8 ppm). As reductive elimination is observed for both a transition metal (CuII) and a main group metal (TlIII) acceptor, phosphine activation may be broadly applicable to complexes exhibiting a mismatch between hard (high oxidation state/charge) acceptors and soft phosphine donors. Indeed phosphines are considered poor donors for hard acceptors and coordination to such centres generally requires enforcement by chelate or pincer ligands.11–13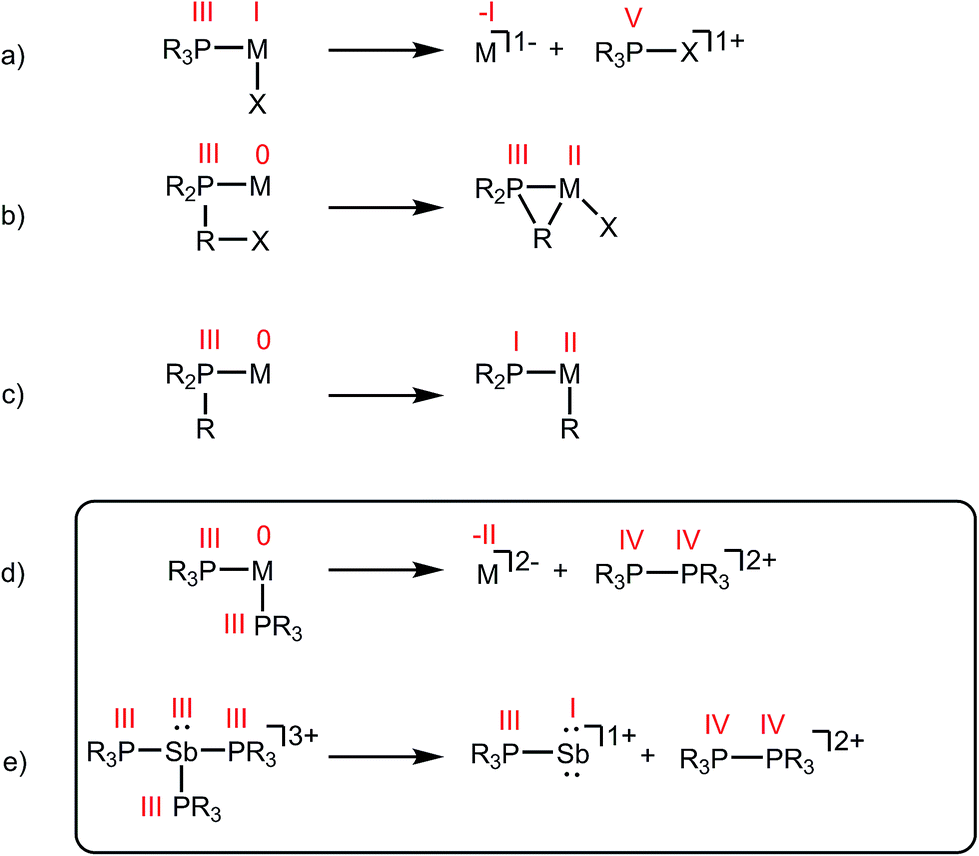 | ||
| Scheme 1 Activation of phosphine ligands in the coordination sphere of a Lewis acceptor. Numerals in red denote formal oxidation states for the element. | ||
As part of a systematic evolution of p-block element phosphine complexes, we have sought derivatives featuring multiply-charged, hard acceptors and now report evidence of a new phosphine ligand activation pathway in the coordination sphere of polycationic SbIII centres. Specifically, reductive elimination of diphosphonium dications (Scheme 1e) from trialkylphosphine complexes of SbIII has been demonstrated, comprehensively defining a fundamental P–P bond forming redox process. The reduction products are the unusual cyclo-tetra(stibinophosphonium) tetracations [10(R)]4+, representing a new catena-homocyclic framework.14 Examples of cationic homocycles for p-block metalloids are limited to unsupported selenium and tellurium dications15 and heavily substituted silicon16 or germanium17 monocations. For antimony, a number of acyclic catenated monocations ([1]1+ and [2]1+)18–20 and dications ([3]2+, [4]2+, [5]2+ and [6]2+)19,21–23 have recently been isolated (Chart 1), but generally on small scales, precluding further reactivity studies of these interesting species. Enabled by a rational and large scale synthetic protocol for cations [10(R)]4+, we now report the reaction chemistry of the prototypical derivative, [10(Me)]4+, debuting the coordination chemistry of a new catena-element framework.
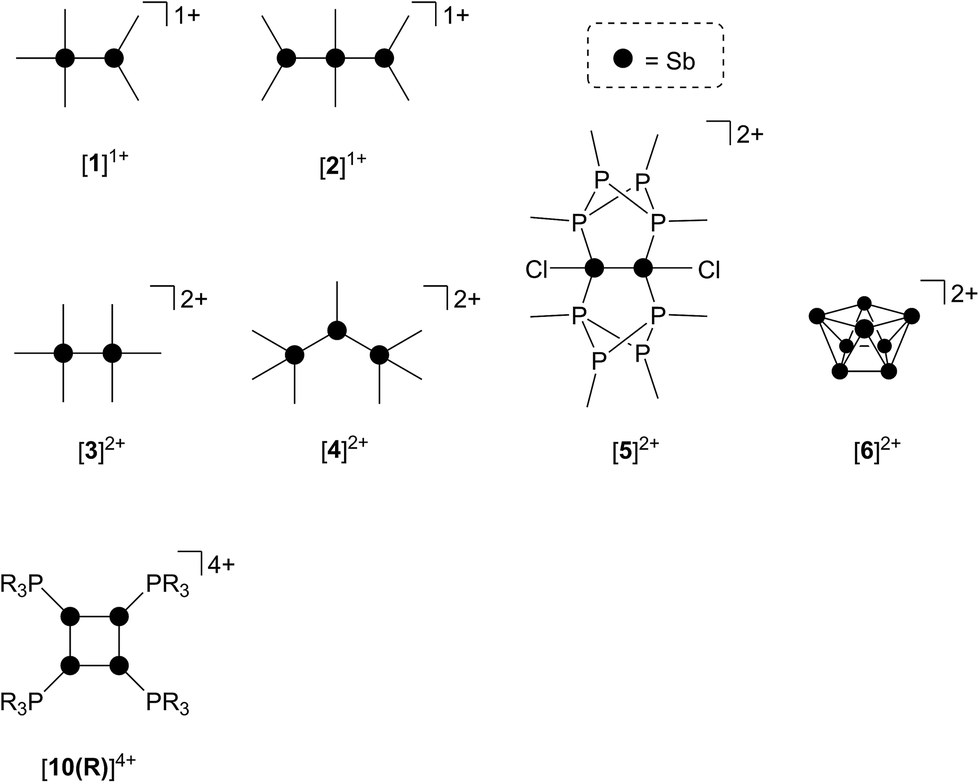 | ||
| Chart 1 Structurally confirmed cations featuring Sb–Sb bonds. See text for references. | ||
Results and discussion
Reactions of PR3 with FSb(OTf)2 and Sb(OTf)3
Combinations of FSb(OTf)2 or Sb(OTf)3 with PR3 (R = Me, Et, Pr, or Bu) in MeCN solvent at the optimized stoichiometries given in Scheme 2 have been investigated. The 31P, 13C, 19F and 1H NMR spectra of reaction mixtures indicate quantitative formation of cyclo-tetra(stibinophosphonium) triflate salts [10(R)][OTf]4 (R = Me, Et, Pr, Bu) together with derivatives of [11(R)][OTf]2 (Scheme 2a) or [12(R)][OTf] (Scheme 2b). Large lattice enthalpy differences permit separation of the monocationic salts [12(R)][OTf] from the tetracationic salts [10(R)][OTf]4 by fractional crystallization, whereas pure salts cannot be isolated from mixtures of dicationic [11(R)][OTf]2 and [10(R)][OTf]4.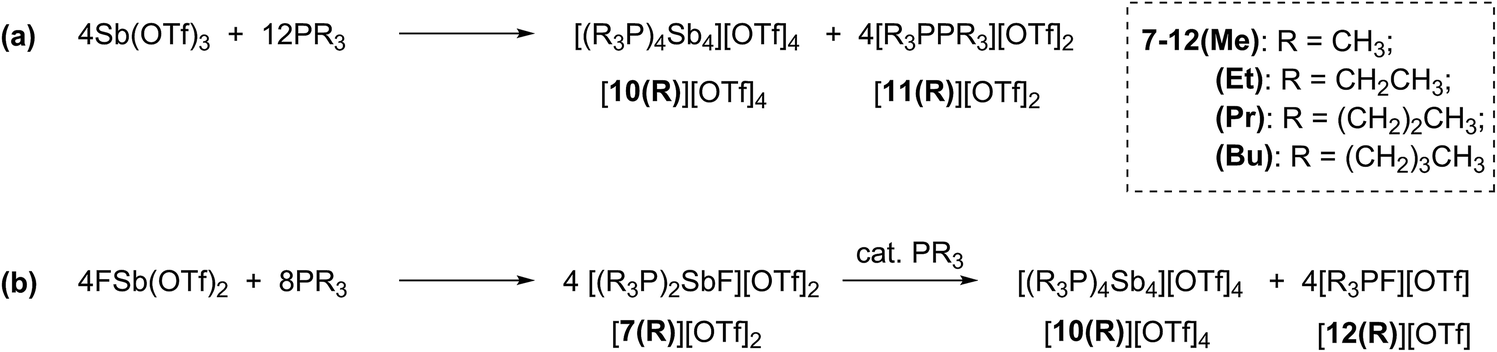 | ||
| Scheme 2 Formations of cations [10(R)]4+, [11(R)]2+, and [12(R)]1+ as triflate salts in reaction mixtures containing trialkylphosphines and (a) Sb(OTf)3 or (b) FSb(OTf)2. | ||
Four derivatives of [10(R)][OTf]4 (R = Me, Et, Pr, Bu) have been characterized spectroscopically by solution NMR spectroscopy, and two derivatives, [10(Me)][OTf]4 and [10(Et)][OTf]4, comprehensively characterized. The solid-state structures of these two salts have been determined by X-ray crystallography to confirm formulae involving a tetracation with a folded Sb4-cyclic core with four exocyclic PR3 units and four triflate anions (Fig. 1 and Table 1). The Sb–Sb bond lengths are very similar for [10(Me)]4+ [2.8354(6)–2.8797(5) Å] and [10(Et)]4+ [2.838(2)–2.884(2) Å] and their values are marginally longer than those observed in rare examples of catena-antimony cations [cf. [1]1+ = 2.8205(12) Å,18 2.8278(3) Å,19 [2]1+ = 2.8203(4) Å,20 [3]2+ = 2.7624(11) Å and 2.7867(12) Å,21 [4]2+ = 2.811(1) Å and 2.830(1) Å,19 and [5]2+ = 2.8484(12) Å and 2.8353(12) Å![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22]. Consistent with the high Lewis acidity of the Sb4 core, the P–Sb distances [2.552(2)–2.578(2) Å] are similar to those observed in other triflate salts such as [(Me3P)2SbCl][OTf]2 [2.5950(4) Å and 2.5834(4) Å] and [(Me3P)SbPh2][OTf] [2.5584(4) Å].32
22]. Consistent with the high Lewis acidity of the Sb4 core, the P–Sb distances [2.552(2)–2.578(2) Å] are similar to those observed in other triflate salts such as [(Me3P)2SbCl][OTf]2 [2.5950(4) Å and 2.5834(4) Å] and [(Me3P)SbPh2][OTf] [2.5584(4) Å].32
 | ||
| Fig. 1 Solid-state molecular structure of the cation in [10(Et)][OTf]4·(MeCN). Hydrogen atoms, anions and solvent molecules have been omitted for clarity. Thermal ellipsoids are drawn at 30% probability level. Metric parameters are given in Table 1. | ||
| [10(Me)][OTf]4·(MeCN)3 | [10(Et)][OTf]4·(MeCN) | |
|---|---|---|
| d(Sb–Sb) | 2.8354(6)–2.8797(5) | 2.838(2)–2.884(2) |
| d(Sb⋯Sb) | 3.7471(5)–3.7792(5) | 3.646(2)–3.804(2) |
| d(P–Sb) | 2.552(2)–2.564(2) | 2.553(3)–2.578(2) |
| d min(Sb⋯OOTf) | 3.210(4) | 2.871(8) |
| <(Sb–Sb–Sb) | 82.09(2)–83.36(2) | 78.62(3)–82.88(4) |
| <(P–Sb–Sb) | 93.74(4)–99.60(4) | 91.91(7)–98.68(7) |
| <(Sb–Sb–Sb–Sb) | 38.85(2)–39.27(2) | 43.30(2)–44.56(3) |
A number of Sb–OOTf contacts are also observed, the shortest of which measure 3.210(4) Å for [10(Me)]4+ and 2.871(8) Å for [10(Et)]4+. Given the high molecular charge, these values are expectedly smaller than the sum of the van der Waals radii (∑r,vdW = 3.61 Å)24 but nevertheless significantly longer than the sum of the single bond covalent radii (∑r,cov = 2.05 Å)25 for the two elements. For [10(Me)]4+, a gas-phase optimization26 of the cation at the MP2 level in the absence of the triflate anions produced a geometry that is essentially identically to that observed experimentally, and we therefore infer that the anion contacts do not distort the structural features to a measurable extent.
The reactions in Scheme 2a represent a two electron reduction of each antimony(III) center and collectively, an eight electron reductive coupling of four antimony centers to form derivatives of [10(R)]4+. In Scheme 2a, eight of the twelve equivalents of phosphine are involved in the redox process, being oxidatively coupled to give four diphosphonium cations, [11(R)]2+,27,28,31 and the remaining four equivalents represent ligands on the reduced antimony(I) centers of [10(R)]4+. Scheme 2b describes a similar redox process that involves formation of [11(R)]2+ as transients, which are converted to the corresponding fluorophosphonium cations, [12(R)]1+, in the presence of the fluoride ion, as envisaged in the mechanism outlined in Scheme 3 (left). The key feature in both processes is reductive elimination of a diphosphonium unit from a hard, tricationic SbIII centre to give a soft, monocationic SbI centre, representing a novel mode of phosphine ligand activation in the coordination sphere of metals (Scheme 1e).
 | ||
| Scheme 3 (Left) Proposed catalytic mechanism for the formation of derivatives of cations [7(R)]2+, [8(R)]3+, [9(R)]1+, [10(R)]4+, [11(R)]2+, and [12(R)]1+. See text for descriptions of a–f. (Right) Non-catalytic formation of [10(Me)]4+ from the reaction of [(Me3P)2SbCl]2+ with PMe3. | ||
31P NMR spectra (Fig. 2) of reaction mixtures containing PR3 and FSb(OTf)2 in a 2:1 stoichiometry show a broad doublet in the +20 to +40 ppm range and the signal due to the free phosphine (−60 to −20 ppm) is not observed. The 19F NMR spectra of these mixtures show a broad triplet in the range −170 to −175 ppm and no evidence of FSb(OTf)2. The broadness of peaks in the 31P and 19F NMR spectra is consistent with the connectivity of these nuclides to a quadrupolar antimony center [I = 5/2 for 121Sb (57%), 7/2 for 123Sb (43%)],40 and we assign these signals to the dicationic bis-phosphine cations [7(R)]2+, which are stable as MeCN solutions (Scheme 3a). Upon addition of ca. 5 mol% of phosphine to these solutions, the 31P NMR signals due to cations [7(R)]2+ are replaced over 16 hours by doublets corresponding to [12(R)]1+ (δ31P: +140 to +150 ppm, 1JPF = 950–1000 Hz) and a singlet in the −25 to 0 ppm range, corresponding to [10(R)]4+. Addition of ca. 15 mol% of phosphine increases the rate of the reaction and effects complete conversion of [7(R)]2+ to [12(R)]1+ and [10(R)]4+ within an hour.
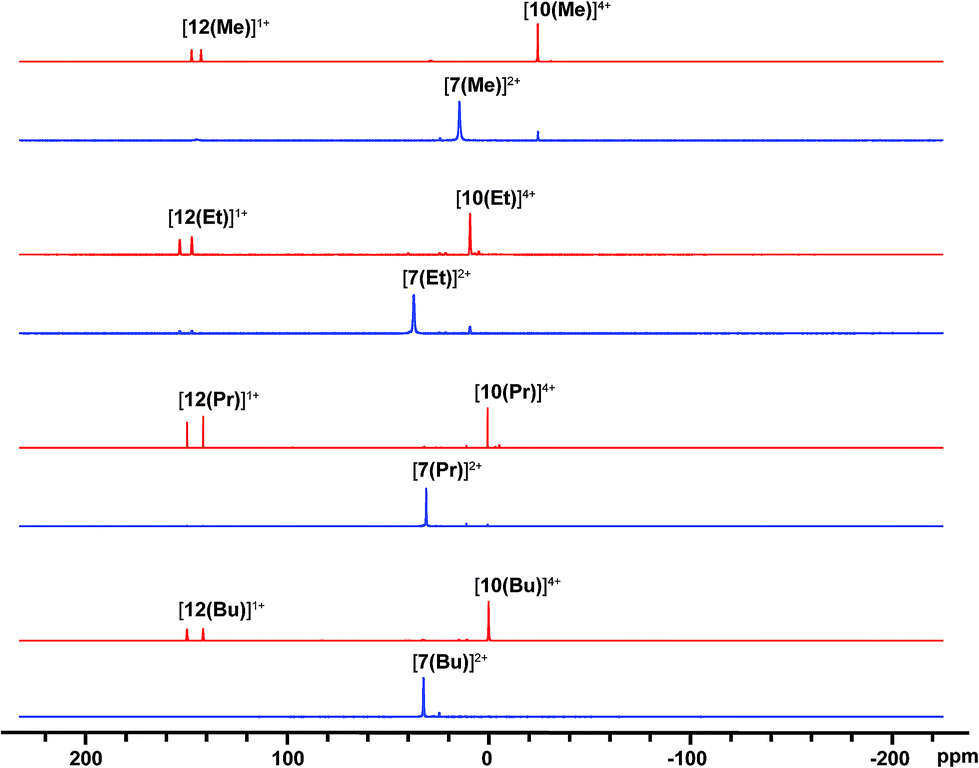 | ||
| Fig. 2 31P{1H} NMR spectra (202.5 MHz, 298 K, CD3CN) of reaction mixtures containing FSb(OTf)2 and PR3 leading to the formation of [7(R)]2+ (blue) and, upon addition of 15 mol% PR3, to [12(R)]1+, and [10(R)]4+ (red). See Table 2 for chemical shift data. | ||
We propose that displacement of fluoride from [7(R)]2+ by added phosphine yields the highly-charged trications [8(R)]3+ (Scheme 3b), which undergo reductive elimination of [11(R)]2+ and [9]1+ (Scheme 3c). Subsequent tetramerization of the six-valence electron cations [9(R)]1+ to [10(R)]4+ (Scheme 3d), and displacement of PR3 from [11(R)]2+ by fluoride gives [12(R)]1+, regenerating the phosphine catalyst (Scheme 3e). Cyclization of transients [9(R)]1+ is analogous to the formation of tetrameric (Mes-E)4 (Mes = 2,4,6-trimethylphenyl, E = As or Sb) via catalytic extrusion of Mes-AsI from a zirconium complex29 or Mes-SbI from a hafnium complex.30 Nucleophilic displacement of PMe3 has been reported31 in reaction mixtures of [11(Me)][ClO4]2 and [NEt4][F], and we have further confirmed that the equimolar reaction of [11(Me)][OTf]2 with CsF (Fig. S1, ESI†) yields a 1:1 mixture of PMe3 and [12(Me)]1+. Trications [8(R)]3+ are also implicated in the formation of [10(R)]4+ from Sb(OTf)3 (Scheme 3f) and we have previously reported26 the structure of the ternary salt [8(Me)][11(Me)][OTf]5 from a 1:3 mixture of Sb(OTf)3 and PMe3 at −30 °C (vide infra). Consistent with the role of [8(Me)]3+ as an intermediate, the same reaction stoichiometry yields only [10(Me)][OTf]4 and [11(Me)][OTf]2 at ambient temperature.
The 31P NMR spectra of reaction mixtures containing [(Me3P)2SbCl]2+ and 20 mol% PMe3 show only partial conversion to [10(Me)][OTf]4 and [11(Me)][OTf]2 after 48 hours. Additionally, a broad signal at +10.4 ppm is also observed (Fig. S2, ESI†), which is close to the average for the values in [(Me3P)2SbCl]2+ (+15.8 ppm) and [(Me3P)2SbCl2]1+ (+6.2 ppm),32 suggesting that the free chloride ion is sequestered in an equilibrium between the starting material and [(Me3P)2SbCl2]1+. Consequently, nucleophilic attack by chloride to liberate free phosphine from [11(Me)]2+ is precluded in these reaction mixtures and neither [Me3PCl]1+ nor free phosphine are detected by 31P NMR spectroscopy.
Signifying the role of free phosphine as a catalyst, formation of [10(Me)]4+ does not occur catalytically in the chloride system because the reaction is arrested upon formation of [11(Me)]2+, which is the spectroscopically detected oxidation product. Generation of free phosphine from diphosphonium, the turnover limiting step, does not take place (Scheme 3, right). In contrast, no diphosphonium is detected in reactions involving the fluoroantimony complexes [7(R)]2+ (Scheme 3, left), where, due to nucleophilic attack by fluoride anions on [11(R)]2+, only the fluorophosphoniums [12(R)]1+ are detected as the oxidation product and the formation of [10(R)]4+ occurs catalytically in the presence of free PR3. Differences in the reactivity of homologous Sb–X (X = Cl, F) complexes towards Lewis acids have been noted previously.33
Solution NMR data for derivatives of [7(R)]2+, [8(R)]3+, [10(R)]4+, [11(R)]2+, and [12(R)]1+ are summarized in Table 2, with evidence for the assignments discussed below. It has not been possible to detect or isolate derivatives of [9(R)]1+. Attempts to trap these cations, or radical intermediates arising from one-electron processes, in the presence of a twenty-fold excess of 2,3-dimethyl-1,3-butadiene were unsuccessful.
| δ 31P (ppm) | δ 19F (ppm) | n J PF (Hz) | |
|---|---|---|---|
| [7(Me)]2+ | +15.9 | −178.2 | 44 |
| [7(Et)]2+ | +38.0 [23] | −174.2 [73] | n.o. |
| [7(Pr)]2+ | +29.3 | −173.4 | 41 |
| [7(Bu)]2+ | +32.4 [62] | −173.9 [52] | n.o. |
| [13]2+ | +41.3 | −187.1 | 39 |
| [8(Me)]3+ | +21.3 | — | — |
| [8(Et)]3+ | +27.8 | — | — |
| [8(Pr)]3+ | +22.1 | — | — |
| [8(Bu)]3+ | +22.4 | — | — |
| [10(Me)]4+ | −24.4 | — | — |
| [10(Et)]4+ | +9.3 | — | — |
| [10(Pr)]4+ | +0.6 | — | — |
| [10(Bu)]4+ | +0.5 | — | — |
| [11(Me)]2+ | +28.5 (28.4)27 | — | — |
| [11(Et)]2+ | +38.5 (21)31 | — | — |
| [11(Pr)]2+ | +31.4 (32)31 | — | — |
| [11(Bu)]2+ | +31.0 (32)31 | — | — |
| [12(Me)]1+ | +148.0 | −138.0 | 948 |
| [12(Et)]1+ | +150.2 | −159.3 | 973 |
| [12(Pr)]1+ | +145.6 | −154.7 | 971 |
| [12(Bu)]1+ | +145.6 | −155.4 | 971 |
Derivatives of [7(R)]2+ represent the first examples of phosphine complexes of fluoroantimony acceptors although numerous fluoroantimony complexes with hard, oxidatively-resistant donors such as pyridines,33,34 ethers,35–38 and pnictogen oxides34,39 have been reported. The 2JPF couplings for [7(Me)]2+ and [7(Pr)]2+ are resolved as a doublet in the 31P NMR spectra and as a triplet in the 19F NMR spectra, consistent with an AX2 spin system. Fine structure could not be resolved for [7(Et)]2+ and [7(Bu)]2+ even under the dilute conditions and low temperature (−30 °C) employed to mitigate broadening due to exchange.
Although [7(Me)][OTf]2 and [7(Et)][OTf]2 have both been isolated as analytically pure substances and spectroscopically characterized, we were unable to obtain X-ray quality crystals. Moreover, to the best of our knowledge, there are no known examples of 2JPF coupling constants through an antimony centre for direct comparison with our assigned NMR data. For this reason, we prepared and isolated the analogous [(dmpe)SbF][OTf]2, [13][OTf]2, from an equimolar mixture of 1,2-bis(dimethylphosphino)ethane (dmpe) and FSb(OTf)2 in MeCN. The solid state structure of [13][OTf]2, as determined by X-ray crystallography, shows a dimeric arrangement with the cations bridged by O–S–O contacts from the triflate anions, and additional interactions with two non-bridging triflate anions, as shown in Fig. S3 (ESI).† The pyramidal geometry at Sb in the cation is retained in solution, as demonstrated by the two non-equivalent methyl group resonances in the 13C (6.1 and 7.2 ppm) and 1H NMR (1.86 and 2.10 ppm) spectra. Crucially, the expected 2JPF coupling was unambiguously observed (Fig. S4, ESI†) in signals due to [13]2+, and the chemical shift and coupling constants are comparable to those assigned to derivatives of [7(R)]2+ (Table 2).
It was not possible to isolate salts of [8(R)]3+ due to their high reactivity, consistent with their disproportionation to [11(R)]2+ and [10(R)]4+ in solution as proposed above. The 31P NMR signals assigned to derivatives of [8(R)]3+ are singlets and broadened (Δν1/2 = 90–500 Hz), presumably due to a combination of the quadrupolar antimony nuclides40 and dynamic ligand exchange. Nevertheless, [8(Me)][OTf]3 has been detected as a co-crystallate with [11(Me)][OTf]2 in a 3:1 reaction mixture of PMe3 and Sb(OTf)3 at −30 °C.26 The molecular structure of [8(Me)]3+ (Fig. S5, ESI†) in the salt [8(Me)][11(Me)][OTf]5 shows a pyramidal arrangement around the Sb atom with three P–Sb lengths in the range 2.5974(8)–2.6115(7) Å and P–Sb–P angles in the range 101.33(3)–102.40(2)°. In addition, three interion Sb–O contacts are observed in the 2.791(2)–2.960(2) Å range (cf. ∑r,vdW = 3.61 Å),24 with each contact appearing trans to a P–Sb bond, illustrating a triple displacement of triflate anions from Sb(OTf)3 by three PMe3 ligands.
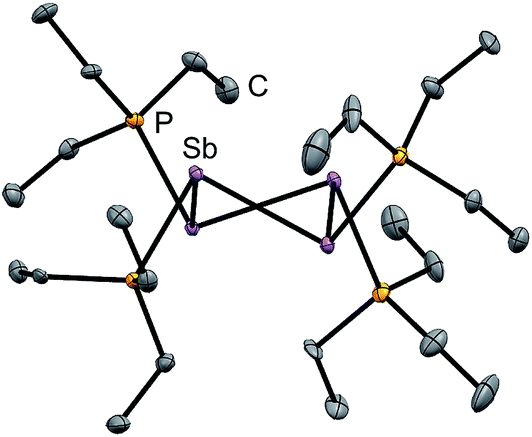
Signals attributed to derivatives of [11(R)]2+ are assigned by comparison with previously reported 31P chemical shifts for their triflate or perchlorate salts in MeCN for [11(Me)]2+, [11(Pr)]2+, and [11(Bu)]2+.27,31 Isolation of [11(Pr)][OTf]2 enabled comprehensive characterization, including X-ray structural determination and we have reported this data elsewhere.41 The salt [11(Et)][OTf]2 has been prepared independently from a 2:1 reaction of PEt3 with in situ generated Ph3Sb(OTf)2, according to Scheme 4,42 and the structure of the cation is shown in Fig. 3. The P–P bond length [2.2209(8) Å] is comparable to that in rare examples of acyclic diphosphonium dications such as [11(Me)]2+ [2.198(2) Å]27 or [Me3PPEt3]2+ [2.216(1) Å],27 and a partially eclipsed conformation is observed between the six ethyl groups. In contrast to a previously assigned 31P NMR chemical shift (+21 ppm)31 for [11(Et)][ClO4]2, which was not structurally authenticated, [11(Et)][OTf]2 exhibits a 31P NMR chemical shift of +38.5 ppm.
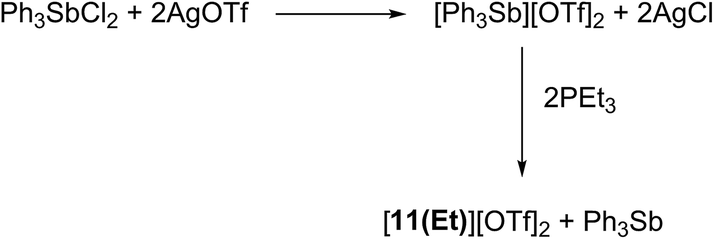 | ||
| Scheme 4 Synthesis of [11(Et)][OTf]2via oxidative coupling of PEt3 with [Ph3Sb][OTf]2. | ||
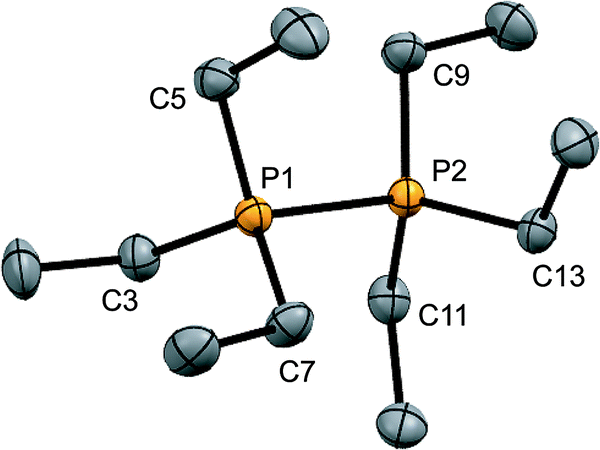 | ||
| Fig. 3 Molecular structure of the cation in [11(Et)][OTf]2 in the solid state. Hydrogen atoms and triflate anions have been omitted for clarity. Thermal ellipsoids are drawn at 30% probability level. Bond lengths (Å) and angles (°) are as follows: P1–P2 = 2.2209(8), P1–C3 = 1.800(2), P1–C5 = 1.802(2), P1–C7 = 1.806(2), P2–C9 = 1.796(2), P2–C11 = 1.809(2), P2–C13 = 1.798(2), C3–P1–P2–C9 = 28.7 (1). | ||
The 31P and 19F NMR resonances attributed to fluorophosphonium cations [12(R)]1+ were confirmed by comparison to literature values or independent synthesis of triflate salts from small-scale equimolar mixtures of the appropriate phosphine with XeF2 followed by treatment with one equivalent of TMSOTf. To the best of our knowledge, the solid-state structure of [12(Me)][OTf] (Fig. 4) represents the first structural characterization of a trialkylfluorophosphonium salt, and involves three hydrogen bonds with the triflate anion in addition to one weak contact [3.301(2) Å] between a triflate oxygen atom and the phosphorus atom, which is marginally shorter than Σr,vdw for the two elements (3.320 Å).24 The O–P–F angle generated by this contact is 177.34(8)°, representing adjustment of the D3h structure of Me3PF2.43
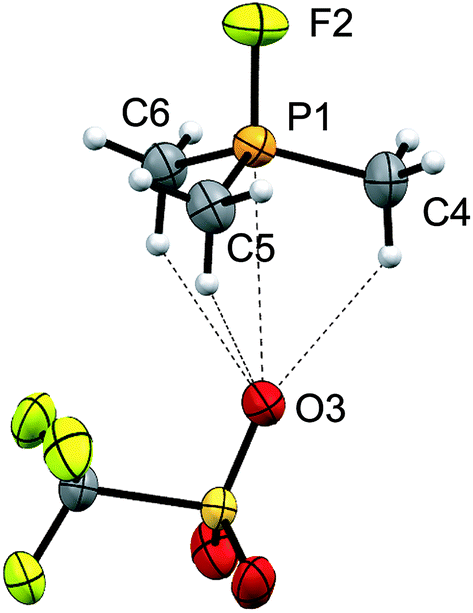 | ||
| Fig. 4 Molecular structure of [12(Me)][OTf] in the solid state. Thermal ellipsoids are drawn at 30% probability level. Bond lengths (Å) and angles (°) are as follows: P1–F2 = 1.551 (1), P1–C = 1.757(2), 1.755 (2), 1.755 (2), P1–O3 = 3.301(2), F2–P1–O3 = 177.34(8), F2–P1–C = 106.38(6), 105.2(1), 106.38(6). | ||
The 31P NMR chemical shifts for species in Table 2 over a 150 ppm range but within each class of cations, generally decrease in the order δ(R = Et) > δ(R = Pr) ≈ δ(R = Bu) > δ(R = Me). Notably, the chemical shifts of the free phosphines (range of 30 ppm) also show the same order, providing additional support for the proposed assignments (Fig. S6, ESI†).
Attempts to isolate cations [7(R)]2+ or [10(R)]4+ with bulky phosphines such as PiPr3 were unsuccessful. A 31P NMR assay of the reaction mixture containing PiPr3 and FSb(OTf)2 in a 2:1 ratio displayed numerous fluorine containing products as indicated by the observation of spin system with P–F couplings but no pure compounds could be isolated. A 3:1 mixture of PiPr3 with Sb(OTf)3 also gave a complex mixture of products at room temperature which could not be separated. Deprotonation of MeCN solvent was observed upon refluxing the reaction mixture for short periods or stirring at room temperature for 16 hours. We conclude that steric bulk at the α-carbon of the phosphine hinders the coordination required for clean transformation of bis-phosphine cations [7(R)]2+ to tris-phosphine cations [8(R)]3+.
While the initial isolation of [10(Me)][OTf]4 as a pure substance was achieved on a 150 mg scale (ca. 0.1 mmol), making it unamenable to reactivity studies, reaction conditions have now been optimized for a one-pot, three-step reaction (ESI) to give reproducible yields of analytically pure [10(Me)][OTf]4 and [10(Et)][OTf]4 on a scale up to 10 g. Consistent with the exquisite sensitivity of these compounds towards hydrolysis and oxidation, particularly in solution, the key determinant of purity and reactions yields is the rigorous drying and deoxygenation of the solvent and careful application of dynamic vacuum (ca. 10−1 mbar) in the latter stages of the reaction to avoid free phosphine-catalyzed decomposition (vide infra).
Thermolysis and photolysis of [10(Me)][OTf]4
The four-membered ring of [10(Me)]4+ contains four of the six Sb–Sb bonds required to make neutral, tetrahedral Sb4, which is directly analogous to P4 and As4. Moreover, [10(Me)]4+ also contains four phosphine ligands which may be susceptible to further reductive elimination of two diphosphonium dications, [11(Me)]2+, to yield neutral Sb4. While P4 and As4 are well characterized, Sb4 has not been isolated as a bulk solid, and only one solid-state structural determination has been made using a scanning tunnelling microscope to characterize a thin film of Sb4 under ultra-high-vacuum conditions.44 In this context, we envisioned the thermal or photochemical decomposition of [10(Me)][OTf]4 as a route to bulk solid Sb4.A sample of solid [10(Me)][OTf]4 (yellow-colored) heated under argon at 120 °C for 16 hours turned black, consistent with the formation of elemental antimony (Scheme 5). A CD3CN extract of the black product showed 31P, 1H and 13C NMR signals corresponding exclusively to [11(Me)]2+ as the sole oxidation product. A Raman spectrum of the black solid (Fig. S7, ESI†) matched that of the amorphous α-phase (110 cm−1, 150 cm−1)45 of antimony rather than the reported Raman spectrum of tetrahedral Sb4 in argon matrix (138 cm−1, 179 cm−1, 242 cm−1).46 Identical results were obtained when heating was carried out in the dark, under vacuum, or in solution (toluene). Irradiating solid [10(Me)][OTf]4 or as a solution in MeCN at 256 nm for 3 hours at room temperature had no measurable effect. It should be noted that in the gas phase tetrahedral Sb4 is the preferred allotrope of the element up to 1050 K.47 It is possible that despite its gaseous stability, tetrahedral Sb4 is thermodynamically unstable with respect to its amorphous phases in the condensed state, preventing its isolation as a solid and is, in this context, analogous to tetrahedral As4 (yellow arsenic) which spontaneously decomposes to a hexagonal allotrope (grey arsenic, α-As) at room temperature.48
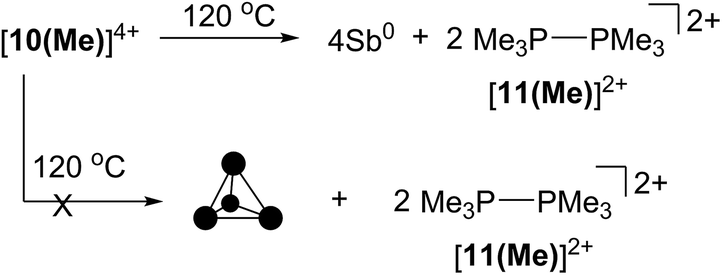 | ||
| Scheme 5 Thermolysis of [10(Me)][OTf]4 to yield [11(Me)][OTf]2 and elemental antimony. | ||
The thermolysis described above must be carried out in rigorously dried glassware, the surface of which has been treated with Me3SiCl to silanize terminal –OH groups. Samples heated without prior passivation of glassware produced elemental antimony and [11(Me)][OTf]2, but also showed resonances due to [Me3PH]1+ and a singlet at +115.6 ppm in the 31P{1H} NMR spectrum (CD3CN) of the reaction mixture, consistent with formation of [Me3POPMe3]2+. This assignment is supported by an independent synthesis from a 2:1 mixture of Me3PO and triflic anhydride, using a well-established protocol for these reagents.49 We interpret the formation of these by-products as being due to the reaction of the extremely moisture sensitive [10(Me)][OTf]4 with surface hydroxyl groups in nonsilanized glassware.
Reactions of [10(Me)][OTf]4 with RnPX(3−n); X = H, Cl; n = 1, 2
Addition of a solution of R2PH (R = C6H11 (Cy), Ph) to a clear yellow-colored solution of [10(Me)][OTf]4 in MeCN results in immediate deposition of a fine black precipitate and loss of the yellow coloration. The 31P{1H} NMR spectra of reaction supernatants show a singlet due to [Me3PH]1+ and two doublets characteristic of phosphinophosphonium cations [Me3PPR2]1+ (R = Cy, Ph) with typical 1JPP values in the 300–350 Hz range (Scheme 6).50 Cation [Me3PPPh2]1+ is known51 and the assignment of [Me3PPCy2]1+ was confirmed by comparison of chemical shifts and coupling constants with literature values50 for phosphinophosphonium salts and by elemental analysis. | ||
| Scheme 6 Formation of [Me3PH]1+ and [Me3PPCy2]1+ from the reaction of [10(Me)][OTf]4 with Cy2PH. | ||
Analogously, addition of a solution of RPH2 (R = Cy, tbutyl) to a solution of [10(Me)][OTf]4 results in immediate precipitation of elemental antimony. The 31P NMR spectra of these reaction mixtures show complete consumption of RPH2 and [10(Me)][OTf]4, and formation of a singlet due to [Me3PH]1+ and a pair of doublets assigned to [Me3PP(H)R]1+ (Scheme 7). Consistent with this formulation, the 31P–1H coupled NMR spectrum of the reaction involving CyPH2 shows (Fig. 5a) both 1JPP and 1JHP couplings for the phosphinic signal centered at −83.6 ppm. The Pα–Pβ(Hβ)Cy connectivity is also confirmed in the 1H NMR spectrum of the reaction mixture (Fig. S8, ESI†), where Hβ resonates at +3.65 ppm exhibiting 1JHβPβ, 2JHβPα, and 3JHβHγ couplings, the last of these arising from coupling to the ipso proton (Hγ) of the cyclohexyl ring. The methyl protons (Hα) around Pα also show the expected 2JHαPα and 3JHαPβ couplings, indicating a P–P bond. Finally, a two-dimensional 31P/1H HSQC (Fig. 5b) spectrum, which was optimized to show one-bond couplings, shows coupling between Hβ and Pβ but no coupling involving Hβ and Pα. The corollary two-dimensional HMBC experiment (Fig. 5c), optimized to exclude one-bond couplings, shows coupling between Hβ and Pα, but no coupling involving Hβ and Pβ. Despite numerous attempts, it was not possible to separate [Me3PP(H)Cy][OTf] from [Me3PH][OTf], precluding elemental analysis or structural determination by X-ray diffraction. Nevertheless, to the best of our knowledge this is the first spectroscopic detection of an H-phosphinophosphonium cation.
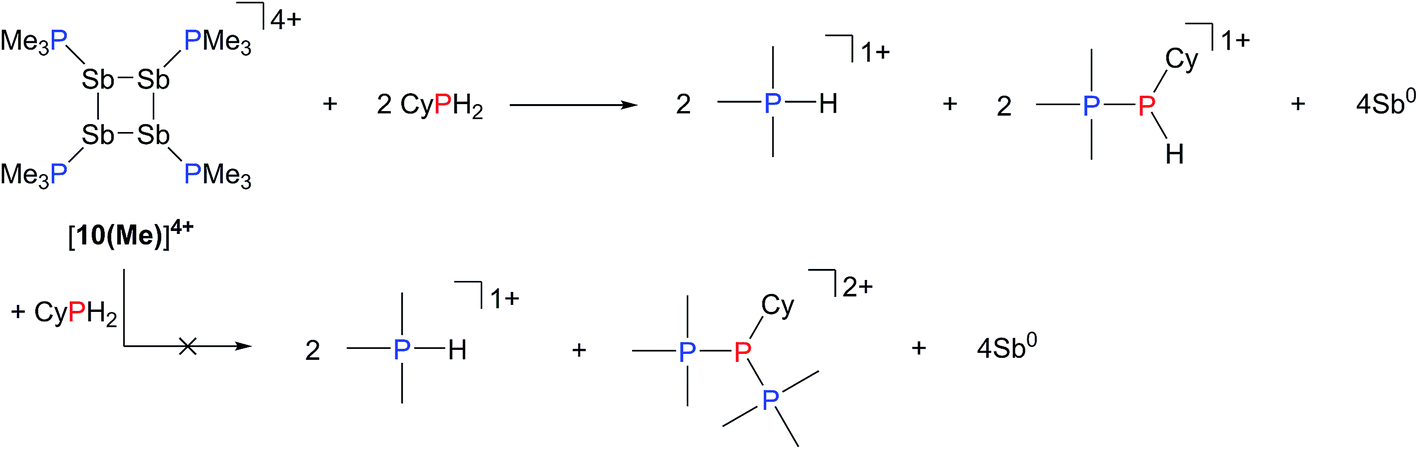 | ||
| Scheme 7 Reaction of CyPH2 with [10(Me)][OTf]4. | ||
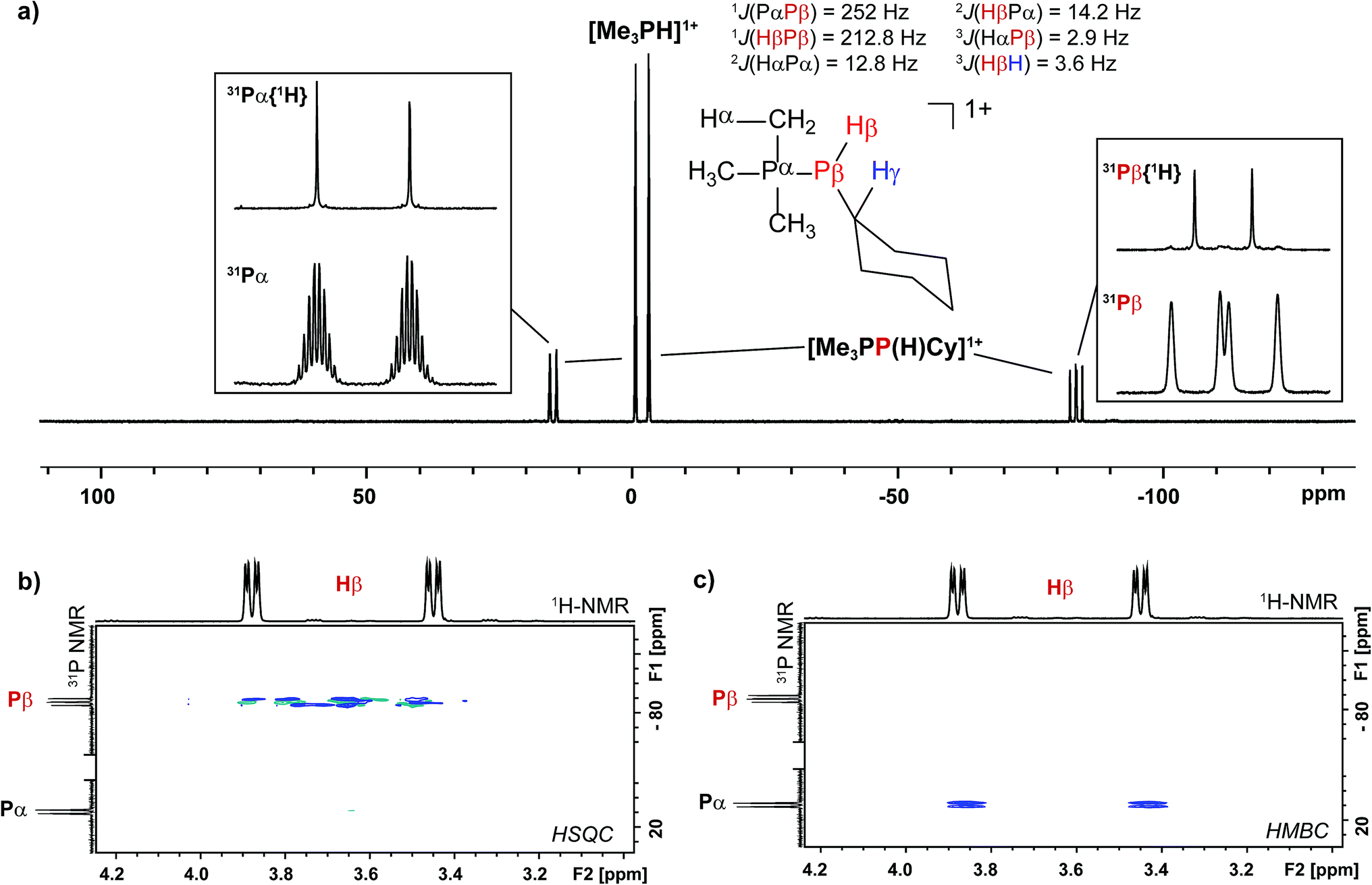 | ||
| Fig. 5 (a) 31P NMR spectrum of the crude reaction mixture containing CyPH2 and [10(Me)][OTf]4 in a 2:1 ratio. Insets show detailed views of the 31P{1H} and 31P NMR resonances assigned to the Me3P- (left) and -P(H)Cy (right) fragments in [Me3PP(H)Cy]1+. A list of coupling constants deduced from the combination of 31P and 1H NMR is also given. (b) Sections of the 31P/1H HSQC spectrum showing a 1JPH coupling between Pβ and Hβ. (c) Sections of the 31P/1H HMBC spectrum showing a 2JPH coupling between Pα and Hβ. | ||
The formation of [Me3PH]1+ and the phosphinophosphonium salts is understood in broad terms as a metathesis step followed by a reductive elimination step as outlined in Scheme 6. We speculate that coordination of Cy2PH to one of the antimony centres in [10(Me)][OTf]4 is followed by intramolecular deprotonation by PMe3 to yield the observed [Me3PH]1+ cation and a tricationic intermediate, [A]3+. This trication can undergo rapid intramolecular reductive elimination of the first equivalent of the phosphinophosphonium cation to give dication [B]2+. A second round of coordination, deprotonation and reductive elimination completes the reduction of antimony to its elemental form and furnishes the observed distribution of products. Unfortunately, the partially reduced species were not observed and appear to be fleeting intermediates. Nevertheless, formation of [Me3PP(H)R]1+ from reactions involving primary phosphines (Scheme 7) is consistent with the proposed mechanism, although it is unclear why the second deprotonation does not occur to yield the corresponding dication [(Me3P)2PR]2+. As before, Raman analysis of the black precipitate matches the amorphous α-phase of metallic antimony rather than pyramidal Sb4.
The observation that [10(Me)][OTf]4 serves as a source of PMe3, which deprotonates added primary and secondary phosphines, implies a labile and polarized P–Sb bond that undergoes facile heterolytic cleavage. Consistently, addition of Cy2PCl or CyPCl2 to a solution of [10(Me)][OTf]4 results in quantitative formation of [Me3PPCy2]1+ or [(Me3P)2PCy]2+,52 respectively, concomitant with deposition of elemental antimony (Scheme 8). In these cases, [10(Me)]4+ behaves overall as a chloride abstractor and phosphine donor. We tentatively propose formation of chloroantimony species as transients that undergo loss of chlorine gas to yield elemental antimony as there is no evidence of Sb–Cl bond stretching modes in the Raman spectra of the insoluble black solid isolated from these reactions. However, since no products expected from reactions of dissolved Cl2 could be detected, the fate of the chlorine atoms cannot yet be definitively described. When intermediate stoichiometries of CyPCl2 are employed, formation of the known [Me3PP(Cl)Cy]1+ cation53 is also observed, indicating a single chloride abstraction event, that is analogous to the formation of [Me3PP(H)Cy]1+ in reactions with CyPH2. 31P NMR data for [Me3PPCy2][OTf], [Me3PP(H)Cy][OTf], [(Me3P)2PCy][OTf]2, and [Me3PP(Cl)Cy][OTf] are given in Table 3.
 | ||
| Scheme 8 Formation of [Me3PPCy2]1+, [Me3PP(Cl)Cy]1+, and [(Me3P)2PCy]2+ from the reaction of [10(Me)][OTf]4 with Cy2PCl or CyPCl2. | ||
Reaction of [10(Me)][OTf]4 with PMe3
The 31P NMR spectrum of a reaction mixture containing 15 mol% of PMe3 and [10(Me)][OTf]4 shows slow disappearance of the signal due to the latter and evolution of broadened signals due to [11(Me)]2+ and free PMe3. Concomitantly, a mirror of antimony is deposited in the reaction vessel. Within 12 hours at 298 K, there is no evidence of [10(Me)]4+, while signals due to [11(Me)]2+ and free PMe3 persist, consistent with complete decomposition of the tetracation, catalyzed by PMe3. The proposed mechanisms (Scheme 9) involve nucleophilic attack by the added phosphine at either the antimony or the phosphorus centres. Attack at a stibine should yield intermediate [A]4+ (Scheme 9), featuring a hypercoordinate antimony centre. Several examples of such hypervalent P–Sb complexes have been reported.32,3 Due to its high charge concentration, this complex is predicted to be strongly oxidizing, and, in a process analogous to reductive elimination from [8(Me)]3+, an equivalent each of [11(Me)]2+ and intermediate [B]2+ (Scheme 9) can be generated, enabling dissociation of PMe3. Alternatively, attack at one of the phosphorus centres of [10(Me)]4+ directly generates intermediate [B]2+ together with [11(Me)]2+ and PMe3. The liberated phosphine can further reduce [B]2+ by a second nucleophilic attack either at Sb or P to evolve the second equivalent of [11(Me)]2+ and yield fully reduced antimony. Nucleophilic attack by a neutral two-electron ligand at tetracoordinate trimethylchlorophosphonium, trimethylphosphonium and dimethylthiophosphonium cations has been demonstrated previously.54–56 The broadness of signals for [11(Me)]2+ and PMe3 in these reaction mixtures is attributed to an exchange process that is also detected when free PMe3 is added to a solution of [11(Me)][OTf]2.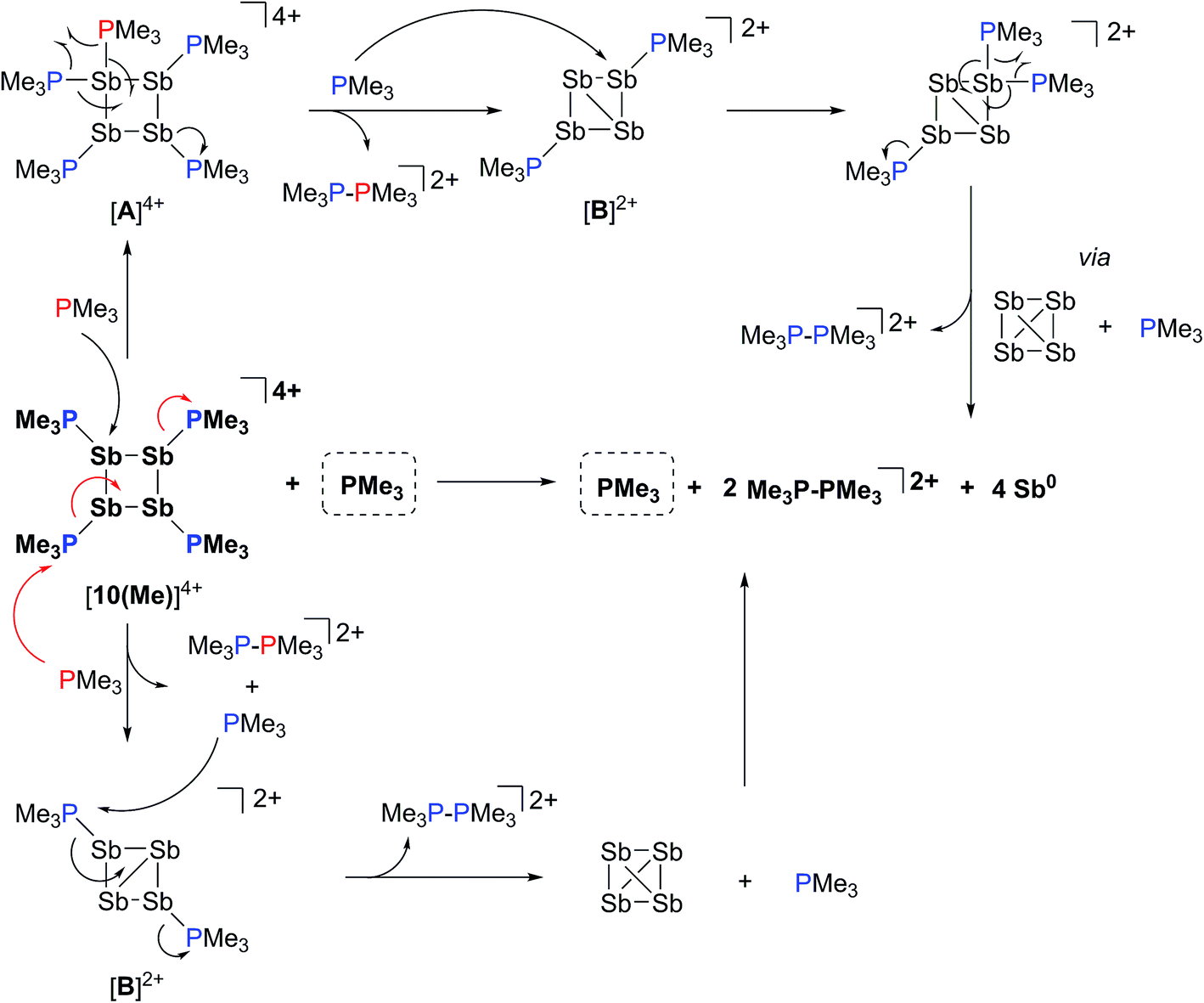 | ||
| Scheme 9 Catalytic decomposition of [10(Me)][OTf]4 by PMe3via nucleophilic attack at Sb (upper half) or P (lower half). | ||
The catalytic decomposition of [10(Me)][OTf]4 in the presence of PMe3 explains the difficulties encountered during synthesis of this salt. For instance, if the addition rate of PMe3 to FSb(OTf)2 is too high, a dark orange solution is obtained which rapidly deposits elemental antimony (see note in Experimental section). However, if a dynamic vacuum is applied to the dark orange solution to remove the volatile PMe3 (b.p. = 38 °C), the solution maintains a yellow colour, leading to the formation of [10(Me)][OTf]4. Moreover reactions with Lewis bases that displace PMe3 must be carried out with explicit steps to remove the liberated phosphine in order to avoid decomposition (vide infra).
Reaction of [10(Me)][OTf]4 with [Li][nacnac(dipp)]
In contrast to the sterically unhindered and neutral base PMe3, a bulky and anionic base is expected to yield products arising from ligand substitution rather than from addition. Consistently, the 31P{1H} NMR spectra of equimolar reaction mixtures of [10(Me)][OTf]4 and Li[nacnac(dipp)] (dipp = 2,6-diisopropylphenyl), indicate quantitative formation of [15(Me)][OTf]3 (Scheme 10). The 1,3-diketiminate anion [nacnac(dipp)]1−, abbreviated as nacnac, displaces one PMe3 ligand from [10(Me)]4+ to give [(Me3P)3Sb4(nacnac)]3+ ([15(Me)]3+), which is an analogue of [(Me3P)3Sb4(PCy2)]3+ (intermediate [A]3+ in Scheme 6). The 31P{1H} NMR spectrum (Fig. 6) of [15(Me)][OTf]3 shows the expected AX2 spin system [−26.6 ppm (triplet), −33.6 ppm (doublet), 3JPP = 32 Hz] and a corresponding AX2 spin system [−6.3 ppm (triplet), −2.5 ppm (doublet), 3JPP = 23 Hz] is also observed for [15(Et)]3+, prepared from the reaction of [10(Et)]4+ with [Li][nacnac(dipp)]. Isolation of [15(Me)][OTf]3 is only possible when the reaction is performed under a mild dynamic vacuum to remove the displaced phosphine, which effects redox decomposition at high concentrations, presumably via similar mechanisms as described above for [10(Me)]4+.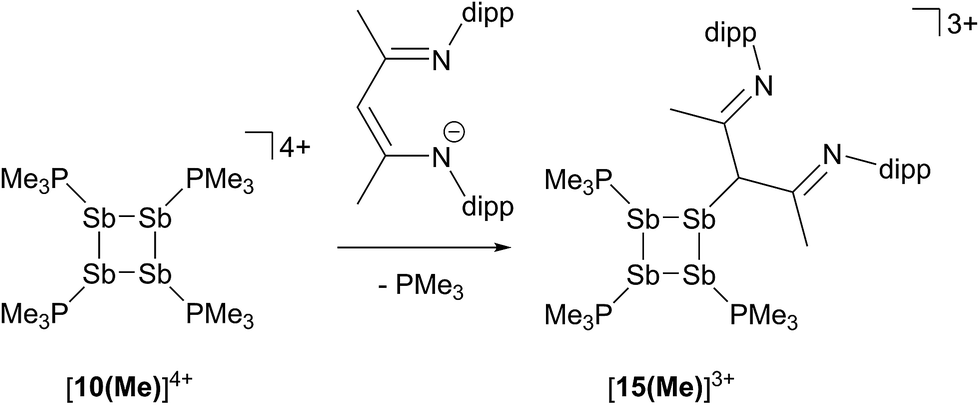 | ||
| Scheme 10 Formation of [15(Me)]3+ by nucleophilic displacement of PMe3 from [10(Me)]4+. | ||
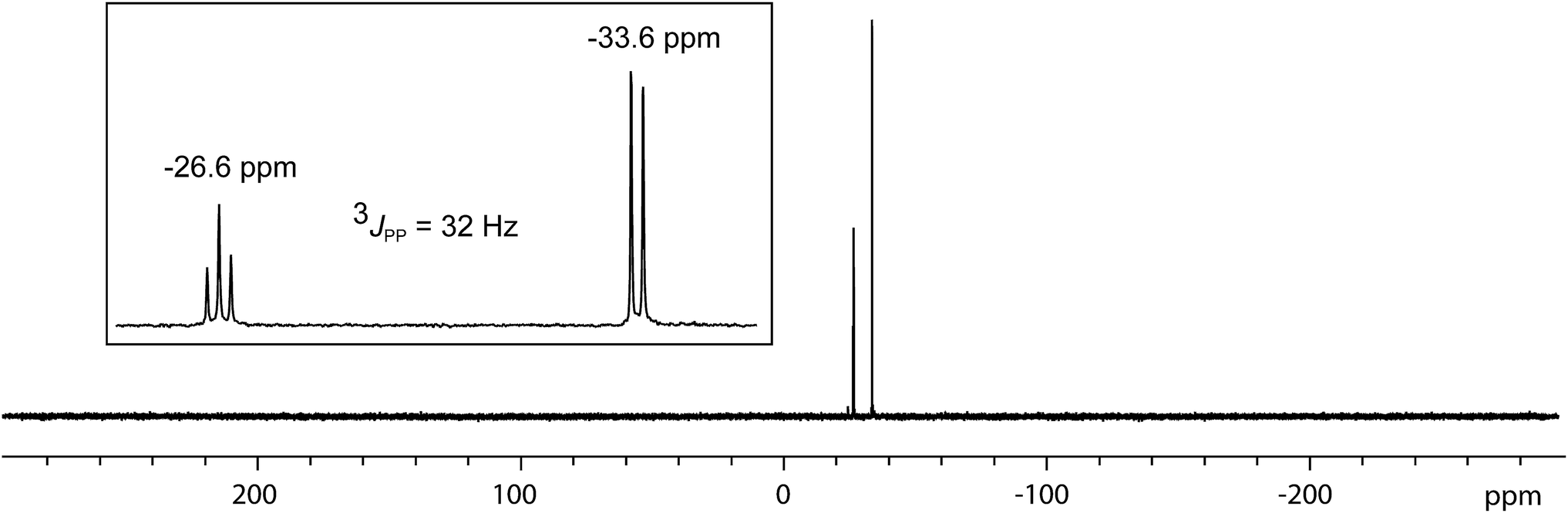 | ||
| Fig. 6 31P{1H} NMR spectrum of [15(Me)][OTf]3 at 298 K in CD3CN. Inset shows the AX2 spin system due to 3JPP coupling between two equivalent and one unique phosphorus environment in [15(Me)]3+. | ||
The solid-state structure of the cation in [15(Me)][OTf]3·MeCN (Fig. 7) shows three phosphine ligands and the rare γ-coordination mode for the nacnac substituent,57 which, to the best of our knowledge, has not been observed for haloantimony centres bound to this substituent.58 Heteroleptic substitution is very rare in antimony homocycles59 and examples for cationic systems have not been reported. The range of Sb–Sb [2.8209(5)–2.8612(5) Å] and Sb–P [2.538(5)–2.604(9) Å] distances are similar to those in [10(Me)]4+ indicating minimal distortion of the Sb4 ring upon displacement of PMe3 with nacnac. While [15(Me)][OTf]3 is stable in the solid state under inert atmosphere, 31P{1H} NMR spectra of MeCN solutions show decomposition over five days at 20 °C to elemental antimony, [10(Me)][OTf]4 and [11(Me)][OTf]2 (Fig. S9, ESI†).
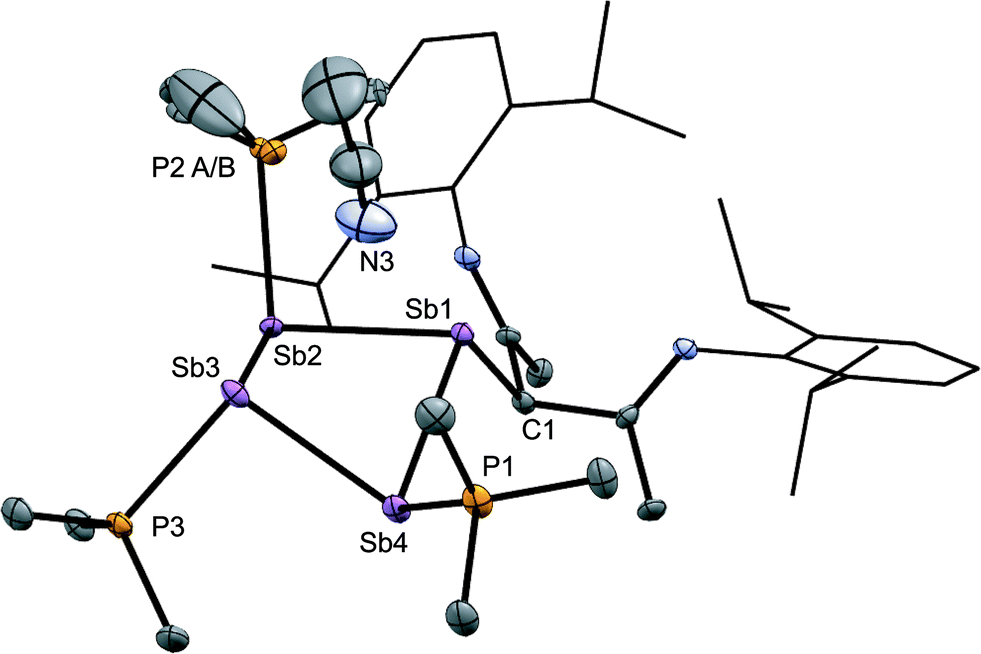 | ||
| Fig. 7 Molecular structure of the cation in [15(Me)][OTf]3·MeCN in the solid state. Hydrogen atoms and triflate anions have been omitted for clarity. Thermal ellipsoids are drawn at 30% probability level. Bond lengths (Å) and angles (°) are as follows: Sb1–Sb2 = 2.8209(5), Sb2–Sb3 = 2.8457(5), Sb3–Sb4 = 2.8501(5), Sb1–Sb4 = 2.8612(5), P1–Sb4 = 2.538(2), P2A–Sb2 = 2.548(5), P2B–Sb2 = 2.604(9), P3–Sb3 = 2.541(1), C1–Sb1 = 2.209(5), Sb1–Sb3 = 3.7344(5), Sb2–Sb4 = 3.7003(5), Sb1–N3 = 3.42(1), Sb3–N3 = 3.19(1), Sb1–Sb2–Sb3 = 82.45(1), Sb2–Sb3–Sb4 = 81.03(1), Sb3–Sb4–Sb1 = 81.67(2), Sb4–Sb1–Sb2 = 81.26(1), Sb1–Sb2–Sb3–Sb4 = −42.10(2). | ||
To assess whether or not bonding via the γ carbon of nacnac is a general feature of antimony compounds and because nacnac functionalized antimony centers are rare in the literature, we also prepared (nacnac)Sb(OTf)2 by salt metathesis between an equimolar mixture of in situ generated Sb(OTf)3 and [Li][nacnac(dipp)]. Upon removal of LiOTf, the compound was isolated as a pure substance and comprehensively characterized. The molecular structure of (nacnac)Sb(OTf)2, determined by X-ray diffraction, shows a see-saw geometry around antimony with two strongly-interacting triflate anions in axial positions (Fig. S10, ESI†). In contrast to γ-coordination observed for [15(Me)]3+, N,N′-chelation is observed for (nacnac)Sb(OTf)2, and we attribute the difference in bonding modes to the different steric environments around antimony in the two compounds, rather than intrinsic features of the nacnac-Sb interaction.
Interestingly, the 19F resonances for the two triflate CF3 groups in (nacnac)Sb(OTf)2 are different (−78.3 and −78.4 ppm), implying a rigid ring system with non-equivalent positions above and below the plane of the ring. Consistently, the isopropyl substituents show two unique resonances for the Cipso protons. Furthermore, there is restricted rotation around the Cipso–Cphenyl bond giving rise to four unique signals for the methyl groups in the 1H NMR spectrum of the compound. We speculate that this is due to solution-phase persistence of the weak hydrogen bonding interactions between the nitrogen atoms and the isopropyl Cipso protons, detected as short contacts in the solid state molecular structure (Fig. S10, ESI†).
Reaction of [10(Me)][OTf]4 with dmap
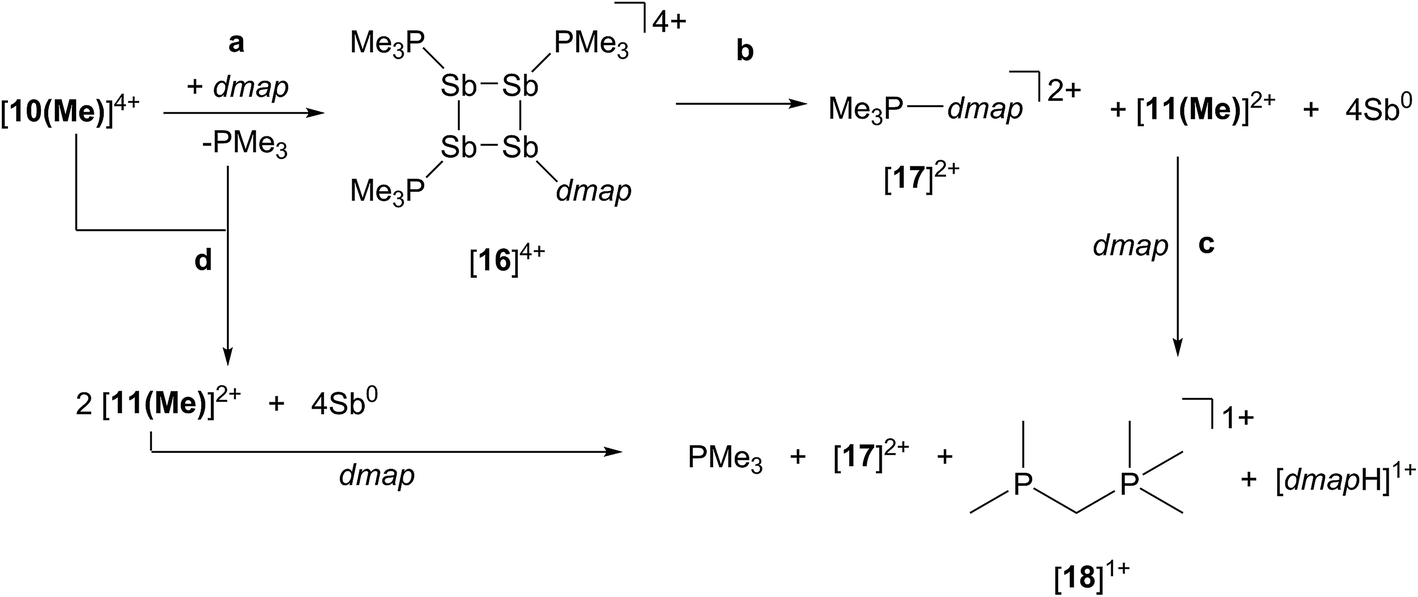
The reaction of [10(Me)][OTf]4 with 4-dimethylaminopyridine (dmap) has been examined by 31P NMR (Fig. 8) and shows displacement of one phosphine ligand by dmap (Scheme 11). It was not possible to isolate the resulting products. Following filtration of the reaction mixture (black suspension), the yellow-green filtrate shows the expected AX2 spin system (triplet at +66.9 ppm, doublet at +42.5 ppm, 3JPP = 24 Hz), tentatively assigned to [(Me3P)3Sb4(dmap)]4+ ([16]4+), and broad signals due to PMe3 (−62 ppm), [Me3P(dmap)]2+ ([17]2+, +89.0 ppm)54 and [11(Me)]2+. Within hours, signals due to [Me3PCH2PMe2]1+ ([18]1+, doublet at −53.9 ppm, doublet at +26.0 ppm, 2JPP = 58 Hz)60 appear in the 31P NMR spectrum and a significant amount of [dmapH]+ is observed by 1H NMR spectroscopy. We propose that the latter two species arise from deprotonation of the slightly acidic protons of [11(Me)]2+ by dmap and the subsequent rearrangement of [Me3PPMe2CH2]1+ (Scheme 11). Consistently, a 1:1 control reaction of dmap and [11(Me)]2+ initially shows broad signals for [17]2+ and free PMe3 as the kinetic products, but within 4 hours signals due to [18]1+ and [dmapH]1+ are observed, revealing them to be the thermodynamic products (Scheme 12, Fig. S11 in ESI†). In addition to [18]1+, four unique and mutually coupled phosphorus environments (by 31P NMR spectroscopy) are also observed which could not been assigned definitively.
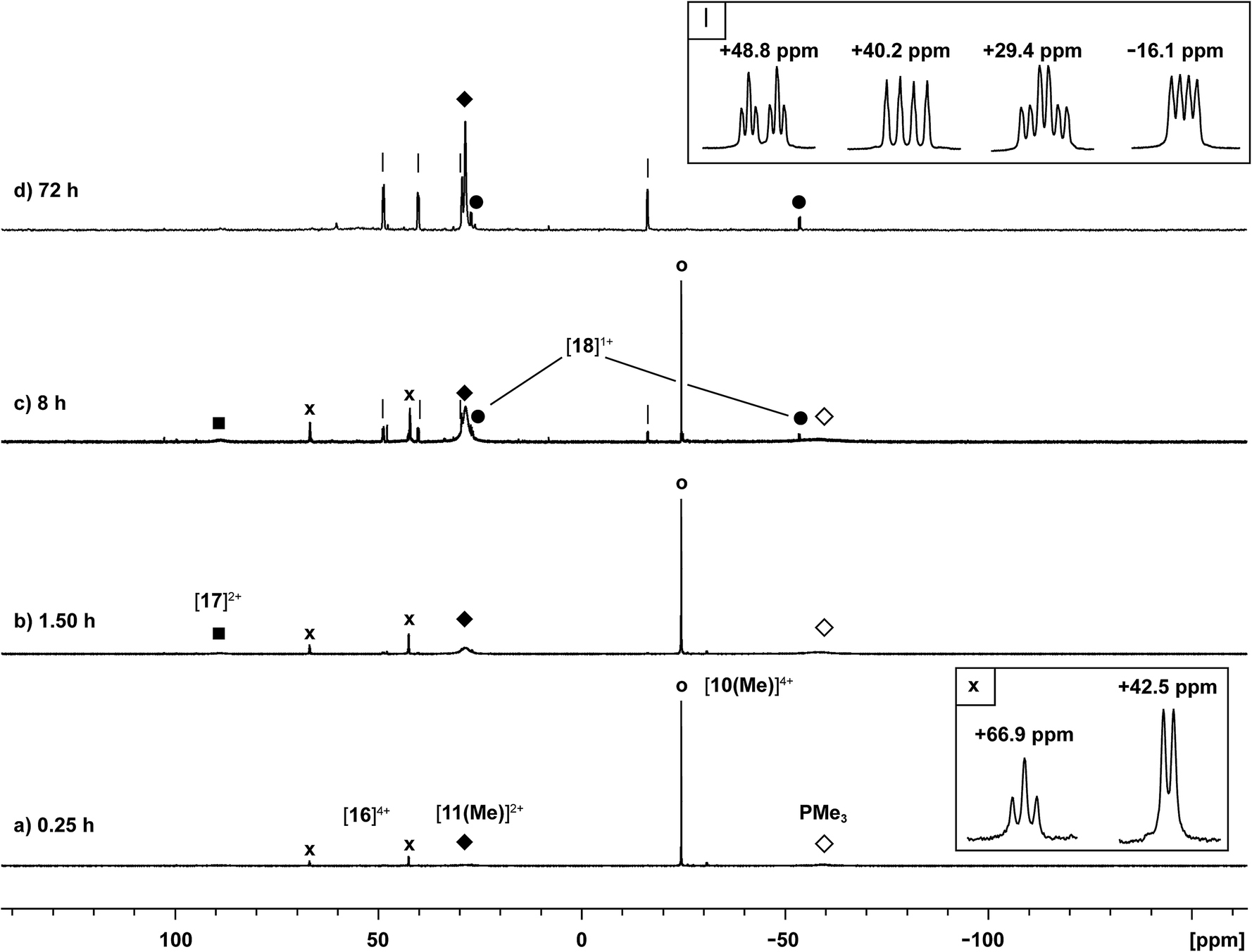 | ||
| Fig. 8 Formation of [11(Me)]2+ (♦), [16]4+ (×), [17]2+ (■), [18]1+ (●), and PMe3 (◊) in the equimolar reaction of dmap with [10(Me)][OTf]4 (○). Peaks labelled with a vertical line (|) correspond to an unidentified product. Insets show the spin systems observed for [16]4+ (×) and the unidentified product (|). | ||
 | ||
| Scheme 11 Proposed pathways to formation of [16]4+ (a), [17]2+, [11(Me)]2+, elemental antimony (b and d), and [18]1+ (c) in reaction mixtures containing [10(Me)][OTf]4 and dmap in a 1:1 stoichiometry. | ||
 | ||
| Scheme 12 Kinetic and thermodynamic pathways in the reaction between [11(Me)]2+ and dmap. | ||
The 3JPP coupling constant for the signal assigned to [16]4+ (24 Hz) is comparable to the values in [15(Me)]3+ (32 Hz) and [15(Et)]3+ (23 Hz). However, the 31P{1H} NMR chemical shifts observed for [16]4+ (A: +66.9, X2: +42.5) are significantly downfield from those of [15(Me)]3+ and [15(Et)]3+ (Table 4), and this cannot be attributed solely to the different formal charges in the species as the PMe3 groups in tetracationic [10(Me)]4+ resonate at −24.5 ppm. We propose that dmap-stabilized main-group cations generally show 31P NMR chemical shifts that are substantially downfield from their PMe3-stabilized homologues (Table 4) due to the greater electronegativity of nitrogen relative to phosphorus, supporting the assignment for [16]4+.
| 31P NMR | Reference | |
|---|---|---|
| [Me2PPMe3]1+ | +18 (−59) | 27 |
| [Me2P(dmap)]1+ | +91 | 54 |
| [Ph2P(PMe3)]1+ | −23 (+15) | 51 |
| [Ph2P(dmap)]1+ | +88 | 55 |
| [Me2(S)PPMe3]1+ | +16 (+38) | 56 |
| [Me2(S)P(dmap)]1+ | +88 | 56 |
| [11(Me)]2+ | (+28.4) | 27 |
| [17]2+ | (+89.0) | 54 |
| [10(Me)]4+ | (−24.5) | This work |
| [15(Me)]3+ | (−26.6), (−33.6) | This work |
| [15(Et)]3+ | (−2.5), (−6.3) | This work |
| [16]4+ | (+66.9), (+42.5) | This work |
| [18]1+ | (+26.0), −53.9 | 60 |
Reaction of [10(Me)][OTf]4 with [10(Et)][OTf]4
Neutral catena-antimony rings are known to participate in ring–ring equilibria unless bulky substituents or dilute solutions are employed. For instance, solutions of hexaphenylcyclohexastibine (Ph6Sb6) equilibrate to give a mixture of four-, five-, and six-membered rings suggesting labile Sb–Sb bonds.61To assess the possibility of preparing heteroleptic derivatives of [10(R)][OTf]4, pure samples of [10(Me)][OTf]4 and [10(Et)][OTf]4 were combined in a 1:1 stoichiometry. The 31P{1H} NMR spectrum (Fig. 9c) of the resulting mixture suggests formation of multiple constitutional isomers of [(PMe3)x(PEt3)(4−x)Sb4]4+, implicating a scrambling process in the two ring systems via Sb–Sb or P–Sb bond cleavage. A scrambling process involving Sb–Sb cleavage has been described previously for distibines.62 However, a control experiment, where free PEt3 was added to [10(Me)][OTf]4, also showed (Fig. 9d) formation of these isomers. Therefore a nucleophilic displacement pathway, where a bound PR3 ligand is displaced by an added PR′3 ligand, cannot be precluded. However this displacement route to heteroleptically substituted derivatives also yields significant amounts of [11(Me)]2+ and elemental antimony, presumably due to free PMe3 catalyzed decomposition of [10(Me)][OTf]4 as described earlier. Although, it has not yet been possible to purify these reaction mixtures and isolate the first examples of heteroleptically substituted catena-antimony rings, signal multiplicities consistent with AM2X, A2X2, and AA′XX′ spin systems are observed, as expected from a mixture of [10(Me)3(Et)]4+, cis/trans-[10(Me)2(Et)2]4+, and [10(Me)(Et)3]4+ (Scheme 13). Moreover, the coupling constants lie in the 21–26 Hz range and are comparable to 3JPP coupling constants detected in [15(Me)]3+ (32 Hz), [15(Et)]3+ (23 Hz), and [16]4+ (24 Hz). Collectively, these data enable a tentative assignment of the spectral features observed in Fig. 9.
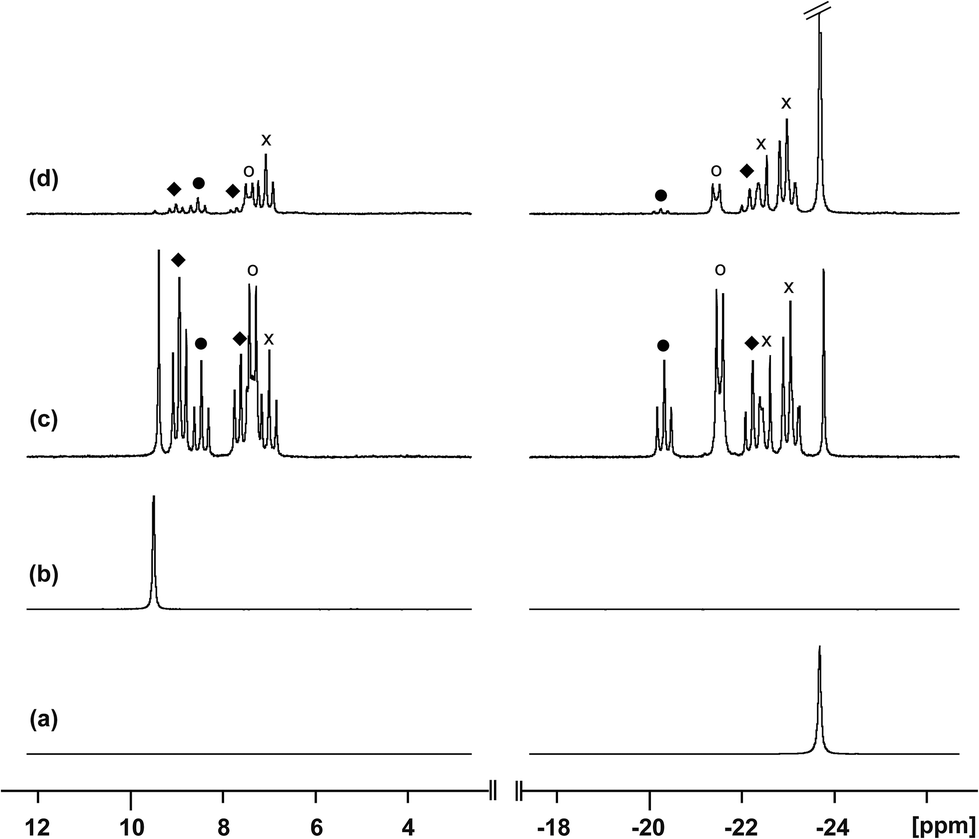 | ||
| Fig. 9
31P{1H} NMR spectra (CD3CN, 298 K) of (a) [10(Me)]4+, (b) [10(Et)]4+, (c) 1:1 mixture of [10(Me)]4+ and [10(Et)]4+, and (d) 1:1 mixture of [10(Me)]4+ and PEt3. Symbols denote tentative assignments for [10(Me)3(Et)]4+ (×), cis-[10(Me)2(Et)2]4+ (○), trans-[10(Me)2(Et)2]4+ (●), and [10(Me) (Et)3]4+ (♦). | ||
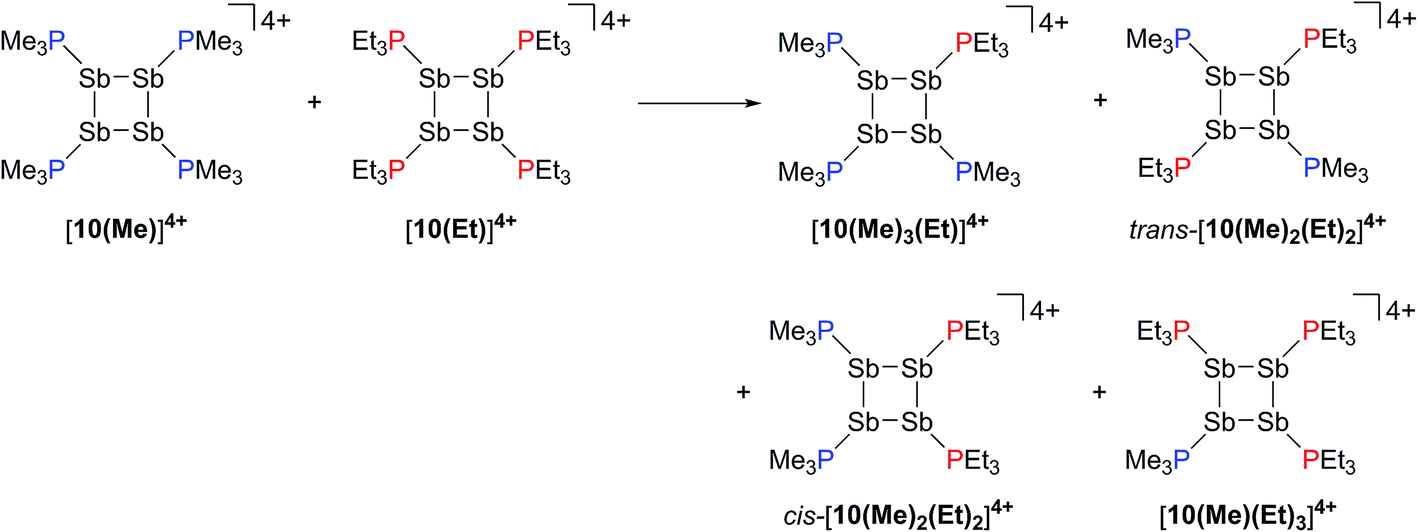 | ||
| Scheme 13 Proposed formation of constitutional isomers from the equimolar reaction of [10(Me)]4+ and [10(Et)]4+. | ||
Conclusions
The reductive elimination of diphosphonium dications [11(R)]2+ from trialkylphosphine complexes of highly electrophilic antimony(III) centres is reported. The reduced antimony(I) fragments cyclize into frameworks identified as cyclo-tetra(stibinophosphonium) tetracations, [10(R)]4+. As outlined in Scheme 3, a phosphine catalyzed mechanism is proposed for fluoroantimony complexes, and isolation or spectroscopic characterization of key mechanistic intermediates is presented. The scope of this reductive assembly is dependent upon the steric bulk of the phosphine employed as demonstrated by non-productive reactions involving PiPr3. Formation of cyclic (R-Pn)n or [L-Pn]n(n+) species (R = aryl group, L = alkylphosphine ligand, E = heavy pnictogen) appears to be the general fate of low-valent (R-Pn) or [L-Pn]1+ monomers, respectively. A multi-gram scale synthesis for the triflate salt of a prototypical cyclo-tetra(stibinophosphonium) tetracation, [10(Me)][OTf]4, has enabled reactivity studies that are summarized in Scheme 14.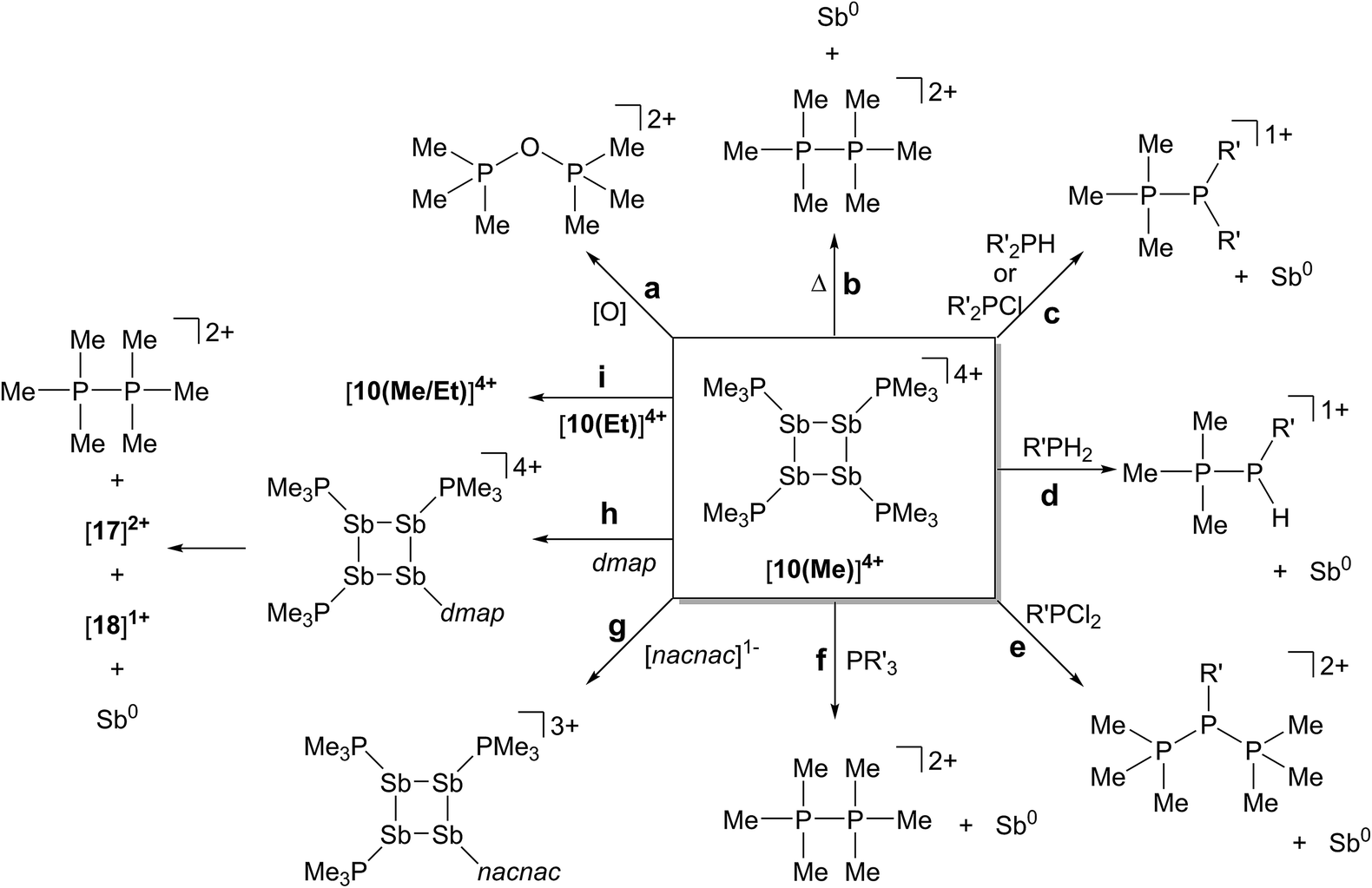 | ||
| Scheme 14 Reactivity of a prototypical cyclo-tetra(stibinophosphonium) tetracation, [10(Me)]4+. See text for descriptions of a–i. | ||
In broad terms, the reactivity of catena-antimony(I) cation [10(Me)]4+ is directed by two features: (i) high charge concentration, and (ii) the presence of strongly polarized P–Sb bonds. The former explains the electrophilicity of cation [10(Me)]4+, its thermolysis to extrude [11(Me)]2+, and the observed facility for reductive elimination to yield elemental antimony (Scheme 14, reactions a–f). The significant polarization of the P–Sb bonds enables activation of a wide spectrum of bonds with the unusual outcome of yielding the same products via reaction with oppositely polarized substrates (e.g. P–Cl and P–H containing reagents) (Scheme 14, reactions c–f). This unique feature has led to the spectroscopic detection of the an H-phosphino-phosphonium cation, [Me3PP(H)Cy]1+, examples of which have not been reported previously. The high P–Sb bond polarization also supports a coordinate bonding model, consistent with ligand displacement reactivity demonstrated for cation [10(Me)]4+ (Scheme 14, reactions g–i). Ligand displacement has permitted functionalization of the four-membered Sb ring with substituents such as [nacnac]1− or dmap (transiently). A heteroleptic phosphine substitution pattern around the Sb4 is feasible, but multiple isomers are observed on a relatively shallow potential energy surface hindering the isolation of a single derivative.
Within the broader context of phosphines as ubiquitous ligands in coordination chemistry, evidence of a novel ligand activation pathway has been presented and the associated reactants and products characterized. Taken together with previous, albeit less definitive, detection of such reactivity,10,42 the observation of this reductive elimination pathway confirms that these prototypical ligands can behave simultaneously as reducing agents and stabilizing ligands, a feature that may be generally applicable for phosphine complexes of highly electrophilic acceptors across the periodic table. Diversification of this synthetic protocol may therefore provide access to more extensively catenated systems for antimony as well as other elements. As demonstrated for [10(Me)]4+, a unique and rich reaction chemistry can be expected, in addition to the potential for valuable emergent properties such as σ-bond conjugation and cooperative catalysis due to metal catenation.
Acknowledgements
We thank the Natural Sciences and Engineering Research Council (NSERC) of Canada and the Vanier Canada Graduate Scholarships Program for funding. We gratefully acknowledge financial support from the ERC (SynPhos 307616) for a six month research stipend for S.S.C. at the TU Dresden. We also thank Dipl.-Chem. Kai Schwedtmann for experimental assistance and Prof. Lisa Rosenberg for valuable discussion.References
- W. Levason and C. A. McAuliffe, Coord. Chem. Rev., 1976, 19, 173–185 CrossRef CAS.
- N. C. Norman and N. L. Pickett, Coord. Chem. Rev., 1995, 145, 27–54 CrossRef CAS.
- J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65–115 CrossRef CAS PubMed.
- S. S. Chitnis and N. Burford, Dalton Trans., 2015, 44, 17–29 RSC.
- W. Levason, G. Reid and W. Zhang, Coord. Chem. Rev., 2011, 255, 1319–1341 CrossRef CAS PubMed.
- M. L. H. Green, J. Organomet. Chem., 1995, 500, 127–148 CrossRef CAS.
- For select examples, see: D. Marcoux and A. B. Charette, J. Org. Chem., 2008, 73, 590–593 CrossRef CAS PubMed; F. E. Goodson, T. I. Wallow and B. M. Novak, J. Am. Chem. Soc., 1997, 119, 12441–12453 CrossRef; L. V. Rybin, E. A. Petrovskaya, M. I. Rubinskaya, L. G. Kuz'mina, Y. T. Struchkov, V. V. Kaverin and N. Y. Koneva, J. Organomet. Chem., 1985, 288, 119–129 CrossRef; A. C. Filippou, E. O. Fischer and H. G. Alt, J. Organomet. Chem., 1988, 215–225 CrossRef; G. Bellachioma, G. Cardaci, A. Macchioni, C. Venturi and C. Zuccaccia, J. Organomet. Chem., 2006, 691, 3881–3888 CrossRef PubMed.
- See for example: A. Takaoka, A. Mendiratta and J. C. Peters, Organometallics, 2009, 28, 3744–3753 CrossRef CAS; R. B. Bedford, M. Betham, C. P. Butts, S. J. Coles, M. Cutajar, T. Gelbrich, M. B. Hursthouse, P. N. Scully and S. Wimperis, Dalton Trans., 2007, 459–466 RSC; F. A. Cotton, F. Barceló, P. Lahuerta, R. Llusar, J. Payá and M. A. Ubeda, Inorg. Chem., 1988, 27, 1010–1013 CrossRef; M. W. Avis, K. Vrieze, J. M. Ernsting, C. J. Elsevier, N. Veldman, A. L. Spek, K. V. Katti and C. L. Barnes, Organometallics, 1996, 15, 2376–2392 CrossRef.
- See for example: D. K. Morita, J. K. Stille and J. R. Norton, J. Am. Chem. Soc., 1994, 117, 8576–8581 CrossRef; K. C. Kong and C. H. Cheng, J. Am. Chem. Soc., 1995, 117, 6313–6315 Search PubMed.
- R. M. Siddique and J. M. Winfield, Can. J. Chem., 1989, 67, 1780–1784 CrossRef CAS.
- M. D. Fryzuk, Can. J. Chem., 1992, 70, 2839–2845 CrossRef CAS.
- L. Liang, Coord. Chem. Rev., 2006, 250, 1152–1177 CrossRef CAS PubMed.
- M. D. Fryzuk, T. S. Haddad and D. J. Berg, Coord. Chem. Rev., 1990, 99, 137–212 CrossRef CAS.
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815–7880 CrossRef CAS PubMed; V. Y. Lee and A. Sekiguchi, Acc. Chem. Res., 2007, 40, 410–419 CrossRef PubMed.
- S. Brownridge, I. Krossing, J. Passmore, H. D. B. Jenkins and H. K. Roobottom, Coord. Chem. Rev., 2000, 197, 397–481 CrossRef CAS.
- S. Inoue, M. Ichinohe, T. Yamaguchi and A. Sekiguchi, Organometallics, 2008, 27, 6056–6058 CrossRef CAS; K. Takanashi, V. Y. Lee, T. Matsuno, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2005, 127, 9978–9979 CrossRef PubMed; A. Sekiguchi, T. Matsuno and M. Ichinohe, J. Am. Chem. Soc., 2000, 122, 11250–11251 CrossRef.
- A. Sekiguchi, M. Tsukamoto and M. Ichinohe, Science, 1997, 275, 60–61 CrossRef CAS.
- A. Althaus, H. J. Breunig and E. Lork, Chem. Commun., 1999, 1971–1972 RSC.
- C. Hering, M. Lehmann, A. Schulz and A. Villinger, Inorg. Chem., 2012, 51, 8212–8224 CrossRef CAS PubMed.
- H. J. Breunig, M. Denker and E. Lork, Angew. Chem., Int. Ed., 1996, 35, 1005–1006 CrossRef CAS.
- R. Minkwitz and C. Hirsch, Z. Anorg. Allg. Chem., 1999, 625, 1674–1682 CrossRef CAS.
- E. Conrad, N. Burford, U. Werner-Zwanziger, R. McDonald and M. J. Ferguson, Chem. Commun., 2010, 46, 2465–2467 RSC.
- M. Lindsjö, A. Fischer and L. Kloo, Angew. Chem., Int. Ed., 2004, 43, 2540–2543 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- S. S. Chitnis, Y. Carpenter, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., Int. Ed., 2013, 52, 4863–4866 CrossRef CAS PubMed.
- J. J. Weigand, S. D. Riegel, N. Burford and A. Decken, J. Am. Chem. Soc., 2007, 129, 7969–7976 CrossRef CAS PubMed.
- V. G. Nenadjenko, N. E. Schevchenko and E. S. Balenkova, Chem. Rev., 2003, 103, 229–282 CrossRef PubMed.
- A. J. Roering, J. J. Davidson, S. N. MacMillan, J. M. Tanski and R. Waterman, Dalton Trans., 2008, 4488–4498 RSC.
- R. Waterman and T. D. Tilley, Angew. Chem., Int. Ed., 2006, 45, 2926–2929 CrossRef CAS PubMed.
- E. V. Nikitin, A. S. Romakhin, V. A. Zagumennov and Y. A. Babkin, Electrochim. Acta, 1997, 42, 2217–2224 CrossRef CAS.
- S. S. Chitnis, N. Burford, R. McDonald and M. J. Ferguson, Inorg. Chem., 2014, 53, 5359–5372 CrossRef CAS PubMed.
- S. S. Chitnis, N. Burford and M. J. Ferguson, Angew. Chem., Int. Ed., 2013, 52, 2042–2045 CrossRef CAS PubMed.
- S. L. Benjamin, J. Burt, W. Levason, G. Reid and M. Webster, J. Fluorine Chem., 2012, 135, 108–113 CrossRef CAS PubMed.
- M. Schaefer, J. Pebler, B. Borgsen, F. Weller and K. Dehnicke, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 1243–1250 CAS.
- I. Becker, M. Windhaus and R. Mattes, Z. Naturforsch., B: J. Chem. Sci., 1994, 49, 870–876 CAS.
- J. Lipkowski, M. S. Fonari, V. C. Kravtsov, Y. A. Simonov, E. V. Ganin and V. O. Gelmboldt, J. Chem. Crystallogr., 1996, 26, 823–833 CrossRef CAS.
- M. S. Fonari, E. V. Ganin, V. O. Gelmboldt, J. A. Lipkowski, S. A. Kotlyar and G. L. Kamalov, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m1021–m1023 CAS.
- W. Levason, M. E. Light, S. Maheshwari, G. Reid and W. Zhang, Dalton Trans., 2011, 40, 5291–5297 RSC.
- P. P. Man, Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, John Wiley & Sons Ltd, Chichester, 2000, pp. 12224–12265 Search PubMed.
- A. P. M. Robertson, S. S. Chitnis, H. A. Jenkins, R. McDonald, M. J. Ferguson and N. Burford, Chem. Eur. J., 2015 Search PubMed , In Press.
- A. P. M. Robertson, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., Int. Ed., 2014, 126, 3548–3551 CrossRef.
- H. Yow and L. S. Bartell, J. Mol. Struct., 1973, 15, 209–215 CrossRef CAS.
- T. M. Bernhardt, B. Stegemann, B. Kaiser and K. Rademann, Angew. Chem., Int. Ed., 2003, 42, 199–202 CrossRef CAS PubMed.
- X. Wang, K. Kunc, I. Loa, U. Schwarz and K. Syassen, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 134305–134310 CrossRef.
- A. J. Kornath, A. Kaufmann and S. Cappellacci, J. Mol. Spectrosc., 2009, 255, 189–193 CrossRef CAS PubMed.
- J. Mühlbach, P. Pfau, E. Recknagel and K. Sattler, Surf. Sci., 1981, 106, 18–26 CrossRef.
- C. Schwarzmaier, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 858–861 CrossRef CAS PubMed.
- J. B. Hendrickson and S. M. Schartzman, Tetrahedron Lett., 1975, 16, 277–280 CrossRef.
- C. A. Dyker and N. Burford, Chem.–Asian J., 2008, 3, 28–36 CrossRef CAS PubMed.
- N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404–14410 CrossRef CAS PubMed.
- Y. Carpenter, Investigations in the Reactivity and Structure of Phosphinophosphonium Cations and Related Species, Ph. D. dissertation, Dalhousie University, Halifax, 2010.
- Y. Carpenter, C. A. Dyker, N. Burford, M. D. Lumsden and A. Decken, J. Am. Chem. Soc., 2008, 130, 15732–15741 CrossRef CAS PubMed.
- J. J. Weigand, N. Burford, A. Decken and A. Schulz, Eur. J. Inorg. Chem., 2007, 4868–4872 CrossRef CAS.
- N. Burford, P. Losier, A. D. Phillips, P. J. Ragogna and T. S. Cameron, Inorg. Chem., 2003, 42, 1087–1091 CrossRef CAS PubMed.
- J. J. Weigand, N. Burford, D. Mahnke and A. Decken, Inorg. Chem., 2007, 46, 7689–7691 CrossRef CAS PubMed.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3065 CrossRef CAS PubMed.
- L. A. Lesikar and A. F. Richards, J. Organomet. Chem., 2006, 691, 4250–4256 CrossRef CAS PubMed.
- Selected examples include: G. Balazs, H. J. Breunig, E. Lork and S. Mason, Organometallics, 2003, 22, 576–585 CrossRef CAS; G. Balázs, H. J. Breunig and E. Lork, Z. Anorg. Allg. Chem., 2003, 629, 1937–1942 CrossRef; H. J. Breunig, R. Roesler and E. Lork, Z. Anorg. Allg. Chem., 1999, 625, 1619–1623 CrossRef; M. Westerhausen, S. Weinrich and P. Mayer, Z. Anorg. Allg. Chem., 2003, 629, 1153–1156 CrossRef.
- H. H. Karsch, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1979, 34, 31–43 Search PubMed.
- H. J. Breunig, K. H. Ebert, S. Gülec and J. Probst, Chem. Ber., 1995, 128, 599–603 CrossRef CAS.
- G. Balázs, H. J. Breunig, E. Lork and S. Mason, Organometallics, 2003, 22, 576–585 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03939d |
| This journal is © The Royal Society of Chemistry 2015 |