
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterconvertible multiple photoluminescence color of a gold(I) isocyanide complex in the solid state: solvent-induced blue-shifted and mechano-responsive red-shifted photoluminescence†
Tomohiro
Seki
a,
Taichi
Ozaki
a,
Takuma
Okura
a,
Kiyotaka
Asakura
b,
Aya
Sakon
c,
Hidehiro
Uekusa
*c and
Hajime
Ito
*a
aDivision of Chemical Process Engineering and Frontier Chemistry Center, Faculty of Engineering, Hokkaido University, Sapporo, Hokkaido 060-8628, Japan. E-mail: hajito@eng.hokudai.ac.jp
bCatalysis Research Center, Hokkaido University, Sapporo, Hokkaido 001-0021, Japan
cDepartment of Chemistry and Materials Science, Graduate School of Tokyo Institute of Technology, Meguro-ku, Tokyo, 152-8551, Japan
First published on 21st January 2015
Abstract
In this study, we report the interconvertible tetracolored solid state photoluminescence of gold(I) isocyanide complex 2 upon various external stimuli through solid state structural changes. Soaking complex 2 in acetone yields blue emission as a result of the formation of 2B. The subsequent removal of acetone yields 2G through a crystal-to-crystal phase transition, which exhibits green emission. This green-emitting solid 2G exhibits stepwise emission color changes to yellow and then to orange upon mechanical stimulation by ball-milling, which corresponds to the formation of 2Y and 2O, respectively. 2B could be recovered upon the addition of acetone to 2G, 2Y, and 2O. Thus, these four emitting solid states of 2 can be switched between repeatedly by means of acetone soaking and the application of mechanical stimulation. Importantly, single crystal and powder X-ray diffraction (PXRD) studies fully show the detailed molecular arrangements of 2B, 2G, and 2Y. This is the first mechanochromic compound to show interconvertible four color emission in the solid state. We also present the first example of using PXRD measurements and the Rietveld refinement technique for the structural analysis of a ground powder in a luminescence mechanochromism study. We obtained complete molecular-level structural information of the crystalline states of 2B, 2G, 2Y, and 2O. In comparison with a more solvophobic analogue 1, we suggest that the weak interaction of 2 with acetone in the solid state would allow a solvent inclusion/release mode, which is an important structural factor for the unprecedented multicolor mechanochromic luminescence.
Introduction
Solid compounds that show a visible response upon mechanical stimulation have recently emerged as interesting smart materials. A mechanical stimulus, typically grinding or shearing, induces a luminescence or color change of the solid compounds, which is referred to as mechanochromism.1 These materials are promising in sensory and recording applications. Further development of smarter functions, such as multicolor mechanochromism with a detailed understanding of the relationship between structural and optical properties, is desirable. Most luminescent mechanochromic compounds show a single-step phase change upon application of a mechanical force, and there have only been a few reports of materials exhibiting dual emission color changes upon mechanical stimulation. Kato reported a brightly tricolored mechanochromic anthracene derivative which took advantage of the phase change of the liquid crystalline phase.2 Jia reported a mechanical force strength-dependent mechanochromic molecule containing two different chromophore units, which exhibited two individual emission color changes.3 The group of Yamaguchi and Saito found that tetrathiazolyl thiophene exhibits distinct emission color changes with a change of pressure and shearing force.4 More recently, the Yagai and Ito group reported the rational design of a mechanochromic compound with dipolar and amphiphilic character that showed a two-step emission color change by mechanical stimulation.5 However, solid materials that show multiple luminescence color changes are still rare. To the best of our knowledge, no mechanochromic materials that show tetracolored luminescence changes have been reported.The luminescence properties of molecules that show luminescent mechanochromism are very sensitive to environmental changes around the molecules and are strongly related to their solid state structures. In a typical case of luminescent mechanochromism, the crystalline structure of the mechanochromic compound is changed to an amorphous phase, which exhibits a different luminescence color from that observed in its crystalline state.3,6 The amorphous phase can revert to the original crystalline structure through recrystallization by solvent fuming or heating. We previously reported the first demonstration of the reversible luminescent mechanochromism of 1 based on this mechanism (Fig. 1 and 2a).6a Mechanical grinding or shearing is non-coherent and exerts random stimulation on the solid. Thus, these mechanical stresses tend to induce a crystalline-to-amorphous phase transition, in which the latter phase has a random structure when compared with the former. Approximately 80% of reported crystalline mechanochromic luminescent materials show this type of phase transition.3,6 Some mechanochromic compounds cocrystallize with a solvent and yield an amorphous phase after grinding, accompanied by solvent release.7 Luminescent mechanochromism caused by the conversion of one crystal structure to another with a different molecular arrangement constitutes only 10% of mechanochromic luminescent materials.4,8 More remarkable mechanisms, such as mechano-triggered single-crystal-to-single-crystal phase transition of mechanochromic gold(I) isocyanide complexes, have been reported by our group.9 In most examples, luminescent mechanochromism compounds can only show a single phase transition upon mechanical stimulation. Multiple phase transitions can realize multiple responses; however, such materials have been rarely reported.
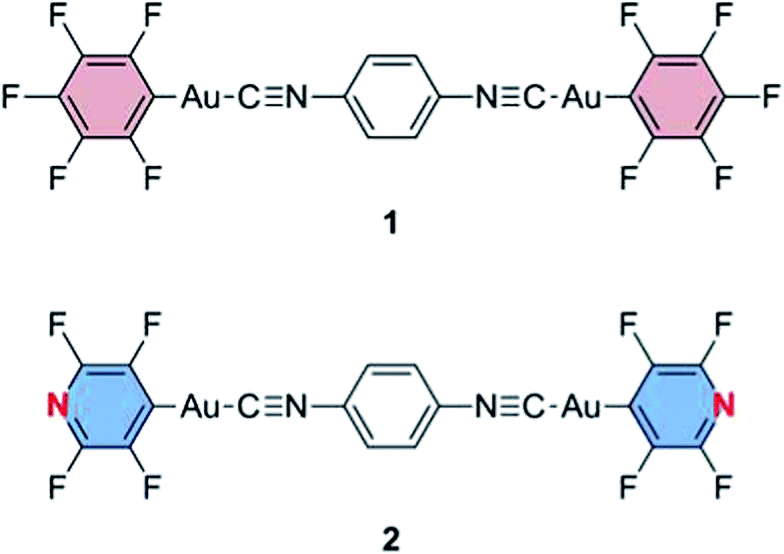 | ||
| Fig. 1 Structures of gold(I) isocyanide complexes 1 and 2. | ||
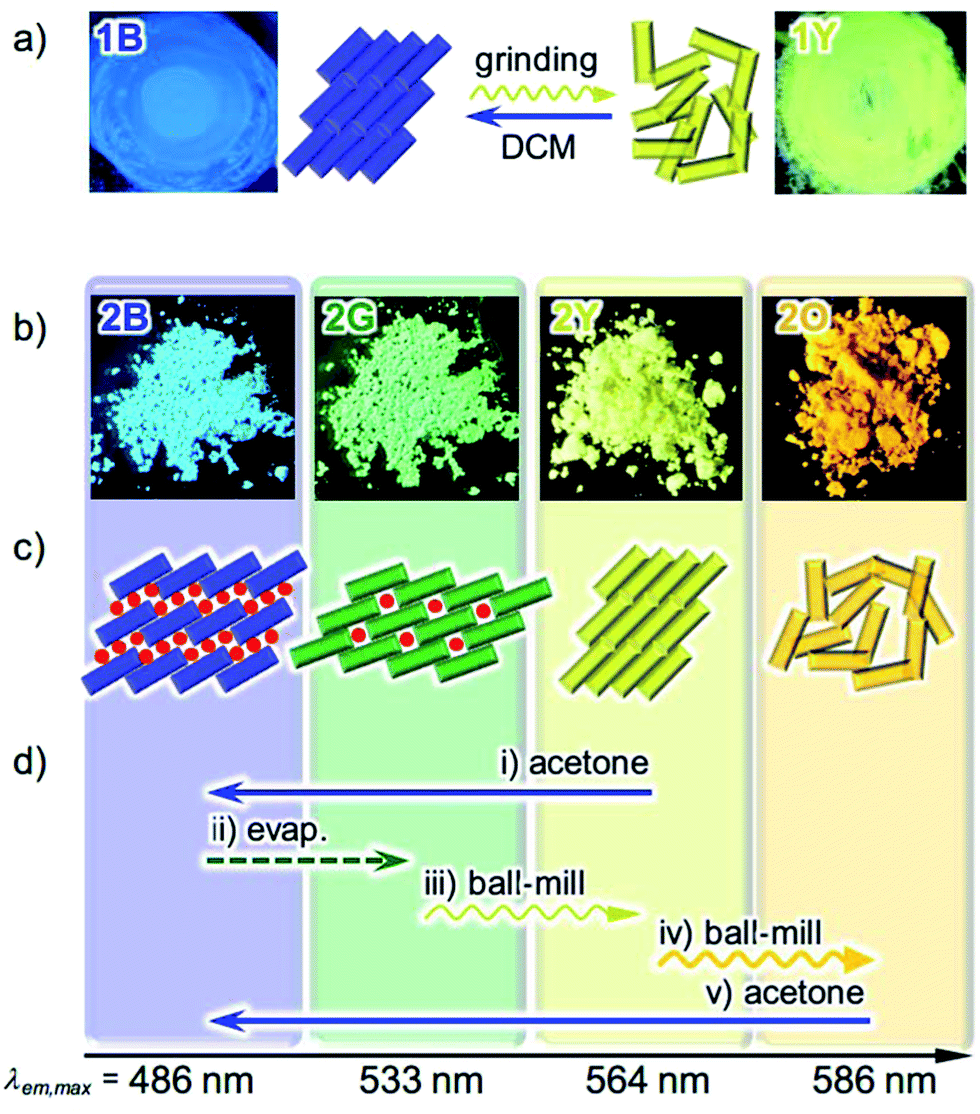 | ||
| Fig. 2 (a) Photographs and schematic representation of the molecular arrangements of 1 showing different photoluminescence upon applying external stimuli. A molecule of 1 is denoted as a rectangle. (b) Photographs of the powder forms of 2 showing different photoluminescence under UV light at 365 nm. (c) Schematic representation of the solid state molecular arrangements of 2, in which a molecule of 2 is denoted as a rectangle, with the colors of the corresponding emission. Solvent molecules are denoted as red circles. (d) Specific procedures for the interconversion of the four emitting states of 2. | ||
One obstacle facing researchers in the study of mechanochromism is the difficulty of the structural analysis of the powdery solid samples obtained after the mechanical process. This is in contrast to solid structure analyses before mechanical stimulation of the sample, in which the crystalline material can be analyzed by single crystal X-ray analysis. Powder X-ray diffraction (PXRD) measurements and Rietveld refinement techniques are known to be powerful methods for crystalline structure analyses of powdered materials;10 however, there have been no reports of this method being applied to mechanochromic materials.
In this paper, we report a new mechanochromic luminescent material 2 (Fig. 1), showing crystal-to-crystal-to-amorphous phase changes upon mechanical grinding and solvent-related structural modifications. As a result, compound 2 shows four individual solid state emissions, which are interconvertible by the addition of solvent and application of mechanical force (Fig. 2b–d). We studied the optical properties and single crystal and powder X-ray diffraction analyses of the new compound 2. For the first time, we solved the crystalline structures of ground powders of the luminescent mechanochromic compound by PXRD measurements and Rietveld refinement. Thermal analyses of 2 provide the unique profile features of its crystalline structure change upon mechanical stimulation. Compound 2 is shown to be the first mechanochromic compound incorporating four interconvertible structures with different emission properties. The structural analysis reveals that weak interactions between the solvent and 2 created the solvent releasing crystal-to-crystal conversion, which is an unprecedented mechanochromic structural change. The combination of two crystal-to-crystal phase changes and one crystal-to-amorphous phase change results in the novel interconversion between four different colors.
Results and discussion
Stimuli-responsive emission color changes
We previously reported the reversible two-colored luminescent mechanochromism of gold(I) isocyanide complex 1, exhibiting a phase transition from blue-emitting 1B to yellow-emitting 1Y (Fig. 2a).6a This emission color change arises from the crystalline-to-amorphous phase transition as revealed by the PXRD pattern. We suggested that the yellow emission in amorphous ground powder 1Y was responsible for the formation of aurophilic interactions.1d,e,11,12 The original blue emission was recovered upon the addition of dichloromethane to 1Y, owing to partial dissolution followed by recrystallization. The role of dichloromethane is to facilitate the recrystallization of amorphous 1Y to crystalline 1B. Both 1B and 1Y are solvophobic and do not contain any solvent in their crystal lattice.The new complex 2 shows four individual emission colors which are interconvertible by treatment with acetone and mechanical stimulation (Fig. 2b–d). The as-synthesized yellow solid of 2 exhibits yellow emission under UV light irradiation at 365 nm and thus is referred to as 2Y (Fig. 2b). The emission color of 2Y immediately turns into blue upon soaking the powder in acetone (step (i) in Fig. 2d and S1†), and the resulting polymorph is referred to as 2B. It should be noted that complex 2 is scarcely soluble in acetone (cmax = 0.2 mg mL−1) and its acetone solution is not emissive in the visible region.13 Upon air drying, the polymorph 2B immediately transforms to a green-emitting polymorph 2G (step (ii) in Fig. 2d and S1†). 2G may contain acetone molecules in the crystal lattice, but did not show any further emission color changes even under reduced pressure for weeks. When 2G was ground using a pestle, yellow and orange emissions were observed upon gentle and hard grinding, respectively (Fig. S2a†). After several experiments, we determined that two different solid states with distinct emissions emerge in a stepwise fashion by ball-milling over a short and long duration (Fig. S2b†). When 2G was mechanically stimulated in a ball-mill at 4600 rpm for 10 min (short grinding), the emission color of the powder was yellow (step (iii) in Fig. 2d), indicating the recovery of 2Y. When 2Y is ground by ball-milling for an additional 5 min (long grinding), orange colored emission was observed, corresponding to the formation of 2O (step (iv) in Fig. 2d). Similar mechano-responsive stepwise emission color changes have rarely been reported in the literature.3,5 After further grinding of 2O for 1 h, no subsequent changes in the orange emission are observed. The reversion of 2O to 2B occurs by soaking the powder in acetone (step (v) in Fig. 2d), indicating the interconvertibility between the four emission colors of 2.14 The following solid state spectroscopic studies of 2 indicate that treatment with acetone induces crystalline structure changes with blue-shifted emission (steps (i) and (v) in Fig. 2d), whereas mechanical grinding results in those with red-shifted emission (steps (iii) and (iv) in Fig. 2d).
The optical properties of the four emitting solid states of 2 were investigated by steady-state spectroscopy (Fig. 3). Under excitation at 365 nm, all of the solid materials of 2 show broad emission bands that are devoid of vibrational structures (solid lines in Fig. 3). The emission spectra of 2B, 2G, 2Y, and 2O have maxima at 486, 533, 564, and 586 nm, respectively, confirming the wide coverage of the visible spectral region. This is in contrast to the THF solution of 2 which is not emissive [absolute emission quantum yield (Φem) is 0%, Fig. S3†]. This indicates that the solid state emission properties of 2 are dependent on aggregation in the solid phase. The excitation spectra of 2B, 2G, 2Y, and 2O detected at the emission maxima show broad bands with peaks at 385, 415, 397, and 459 nm, respectively (dashed lines, Fig. 3). The UV-vis absorption spectrum of the THF solution of 2 showed an absorption band in the range of 200–300 nm (Fig. S4†), shorter than the regions of the excitation bands observed for 2B, 2G, 2Y, and 2O. The different excitation spectra suggest that the ground state structures of 2B, 2G, 2Y, and 2O in the solid phase are distinct from each other.
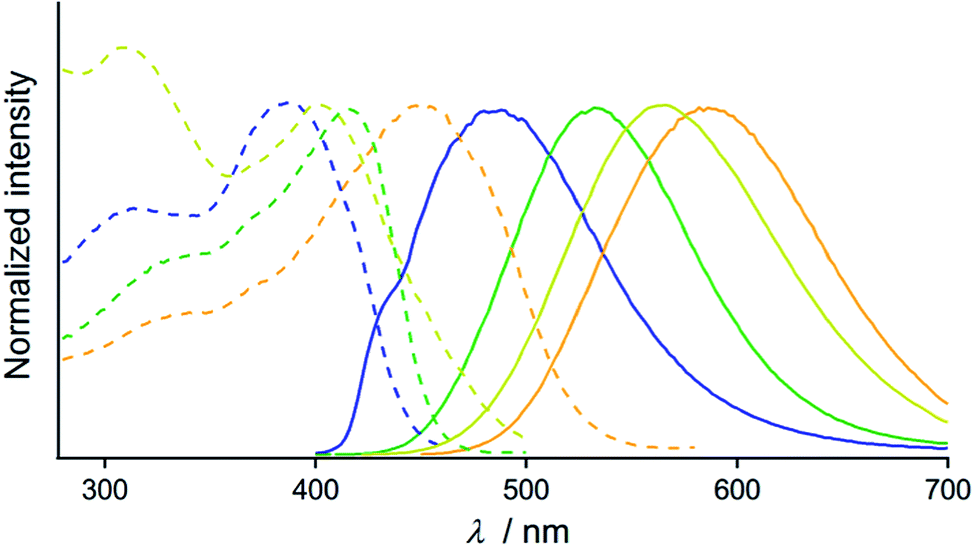 | ||
| Fig. 3 Normalized excitation (dashed lines, monitored at the emission maxima) and emission spectra (solid lines, λem = 365 nm) of 2B (blue lines), 2G (green lines), 2Y (yellow lines), and 2O (orange lines). | ||
Photophysical properties were investigated for the powders of 2 with different emission properties and the results are summarized in Table S1.† The Φem of 2B and 2G are both 10%, whereas Φem of 2Y and 2O, both obtained by ball-milling, are 27 and 30%, respectively (Table S1†). Mechanical force-induced emission color change with increased Φem was reported for organometallic complexes.6a,9b Photoluminescence lifetime spectroscopy was also carried out and the emission decay profiles are presented in Fig. S5†. The emission decay profiles of 2B, 2G, 2Y, and 2O were all fitted to a biexponential curve. The average lifetime τav [=(∑Aiτi)/(∑Ai)] of 2G, 2Y, and 2O are almost the same, within the range of 0.4–0.7 μs (Table S1†). However, a longer τav value of 2.55 μs was observed for 2B.
Stimuli-responsive crystalline structure changes
PXRD measurements indicate that 2B, 2G, 2Y, and 2O have distinct packing arrangements. It has been reported that the emission properties of solid materials are significantly related to their crystalline structures and the intermolecular interaction patterns.15 The powders of 2B, 2G, and 2Y showed several intense diffractions (Fig. 4), indicating their crystalline nature. The diffraction of 2Y, obtained by short grinding (ball-milling for 10 min), is principally similar to that of the as-synthesized powder (Fig. S6†), consistent with the similarity in their emission. Moreover, the PXRD patterns indicate that crystalline-to-crystalline phase transition from 2G to 2Y occurs upon mechanical stimulation.4,8 Unlike the above three patterns, the PXRD pattern of 2O showed diffractions with very small intensity (orange line in Fig. 4), indicating that 2O is in an amorphous phase. The crystalline-to-amorphous phase transition from 2Y to 2O is likely a result of the long grinding (ball-milling for 15 min) disrupting the ordered molecular arrangements as is commonly observed for mechanochromic compounds.3,6 The four different diffraction patterns of 2B, 2G, 2Y, and 2O indicate that each of their crystalline arrangements are distinct. As multicolored luminescent mechanochromic compounds have rarely been reported,2–5 the relationship between their detailed crystalline structures and emission properties should give useful insight.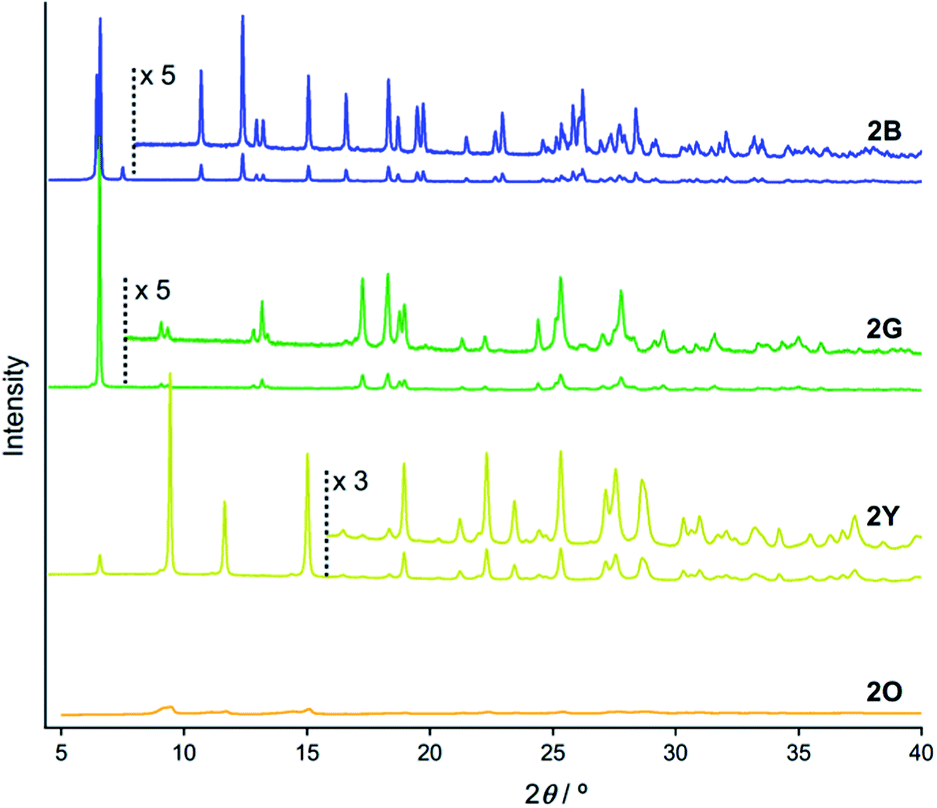 | ||
| Fig. 4 PXRD patterns of 2B (blue line), 2G (green line), 2Y (yellow line), and 2O (orange line). | ||
To gain a better understanding of the structure–property relationship of 2B, we performed single crystal X-ray diffraction analysis. The blue-emitting single crystal was prepared from a saturated acetone solution of 2. 2B crystallized in the triclinic system P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 5, Table 1 and S2†). The central isocyanide benzene ring of the molecule is on the inversion center. The simulated powder pattern obtained from the single crystal structure is identical to the PXRD pattern of 2B (Fig. S7a†). Molecules in 2B form a layer-like structure with an interlayer spacing, d, of 13.22 Å in which the molecular tilt angle is 28.61°. For each molecule, the dihedral angle θdihedral between the central benzenes and lateral pyridines is 34.03°. Within the layer, the molecules form four CH⋯F intermolecular interactions with the adjacent two molecules (Fig. S8†) to construct a tape-like motif. 2B contains 2 equivalents of disordered acetone molecules which form sublayers between the tape-like structures of 2 (2B: [2] × 2 = [acetone]; Fig. 5b). The gold molecules in the tapes and the acetone molecules in the sublayers interact via CH⋯O and CH⋯F intermolecular interactions (Fig. S8†). The absence of defined intermolecular interactions between the tapes, owing to the presence of the acetone sublayers, is thought to make 2B unstable under acetone-free conditions. The molecules in the tapes stack on top of each other without a prominent longitudinal offset through π–π stacking interactions between both the benzene rings and between the pyridine rings with perpendicular distances of 3.314 and 3.334 Å, respectively. These intermolecular interactions in the tapes of 2B may play an important role in achieving a longer wavelength emission compared with that of the solution phase. However, the Au⋯Au distance of 3.5452(7) Å (Fig. 5a) is beyond the limit of aurophilic interactions, which indicates their negligible influence on the emission energy level of 2B.
(Fig. 5, Table 1 and S2†). The central isocyanide benzene ring of the molecule is on the inversion center. The simulated powder pattern obtained from the single crystal structure is identical to the PXRD pattern of 2B (Fig. S7a†). Molecules in 2B form a layer-like structure with an interlayer spacing, d, of 13.22 Å in which the molecular tilt angle is 28.61°. For each molecule, the dihedral angle θdihedral between the central benzenes and lateral pyridines is 34.03°. Within the layer, the molecules form four CH⋯F intermolecular interactions with the adjacent two molecules (Fig. S8†) to construct a tape-like motif. 2B contains 2 equivalents of disordered acetone molecules which form sublayers between the tape-like structures of 2 (2B: [2] × 2 = [acetone]; Fig. 5b). The gold molecules in the tapes and the acetone molecules in the sublayers interact via CH⋯O and CH⋯F intermolecular interactions (Fig. S8†). The absence of defined intermolecular interactions between the tapes, owing to the presence of the acetone sublayers, is thought to make 2B unstable under acetone-free conditions. The molecules in the tapes stack on top of each other without a prominent longitudinal offset through π–π stacking interactions between both the benzene rings and between the pyridine rings with perpendicular distances of 3.314 and 3.334 Å, respectively. These intermolecular interactions in the tapes of 2B may play an important role in achieving a longer wavelength emission compared with that of the solution phase. However, the Au⋯Au distance of 3.5452(7) Å (Fig. 5a) is beyond the limit of aurophilic interactions, which indicates their negligible influence on the emission energy level of 2B.
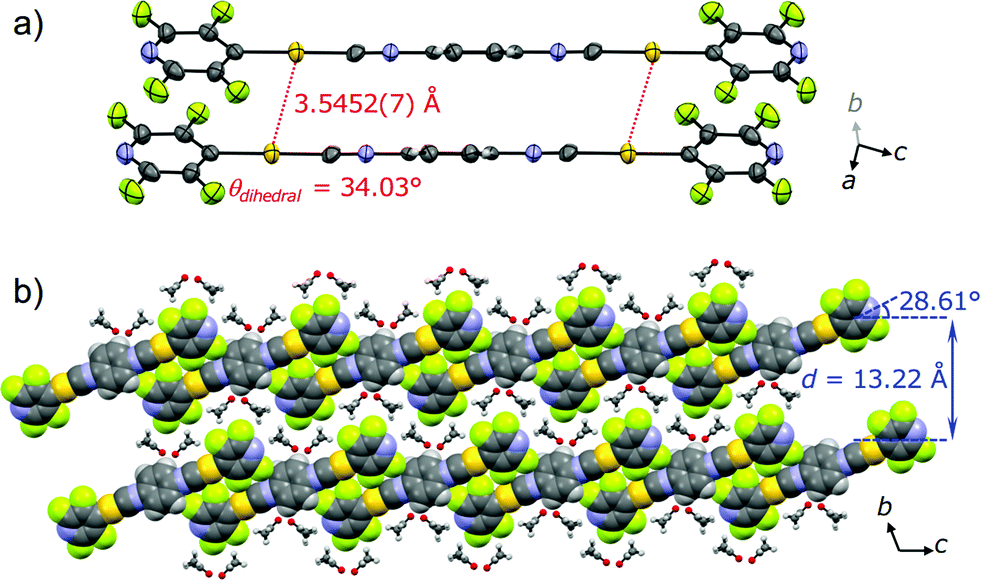 | ||
| Fig. 5 (a) ORTEP representation of the dimer and (b) the space-filling representation of the packing structure of 2B viewed along the direction of the a axis. Acetone molecules are depicted as ball-and-stick models in (b). | ||
| 2B | 2G | 2Y | |
|---|---|---|---|
| a For data with I > 2.00σ(I). b For all reflection data. c Goodness of fit. | |||
| CCDC number | 1035806 | 1035808 | 1035810 |
| Specimen | Single crystal | Single crystal | Powder |
| Crystal system | Triclinic | Triclinic | Monoclinic |
| Space group |
P (#2) |
P (#2) |
P21/c (#14) |
| a/Å | 3.5452(4) | 3.5707(3) | 7.79058(18) |
| b/Å | 14.0087(15) | 10.3087(10) | 18.7202(5) |
| c/Å | 14.2490(15) | 14.2742(11) | 6.74157(17) |
| α/° | 109.292(3) | 107.689(4) | 90 |
| β/° | 91.673(3) | 92.505(5) | 103.167(3) |
| γ/° | 91.322(2) | 100.205(5) | 90 |
| V/Å3 | 667.23(13) | 489.99(8) | 957.35(5) |
| Z value | 1 | 1 | 2 |
| D calc/g cm−3 | 2.335 | 2.786 | — |
| R 1 /% | 4.73 | 10.08 | — |
| wR2b/% | 10.55 | 25.46 | — |
| GOFc | 1.152 | 1.114 | — |
| R wp/% | — | — | 4.62 |
| R p/% | — | — | 3.60 |
| R F2/% | — | — | 2.74 |
The single crystal X-ray diffraction analysis of 2G provides information on the origin of the emission color change. For preparation of the single crystal 2G, single crystal 2B was exposed either to air or water vapor to remove the incorporated acetone molecules. The resulting green-emitting crystals obtained by both methods afforded similar packing structures and the latter method yielded better diffraction data. Both methods are observed to yield 2G because their simulated powder patterns are identical to the PXRD pattern of 2G (Fig. S7b†). 2G crystallized in the triclinic space group P (Fig. 6, Table 1 and S2†). The central isocyanide benzene ring of the molecule is on the inversion center. 2G forms layered structures (Fig. 6b) similar to 2B. The layer spacing, d, of 2G is 9.821 Å in which the molecular tilt angle is 21.66°. In 2G, a 1-D channel structure with some residual electron density along the a axis exists between the molecular tape structures (Fig. S9a†), indicating the inclusion of a small amount of acetone molecules. Based on the X-ray diffraction analyses and 1H NMR spectroscopy, the included acetone molecules in 2G are less than 0.5 equivalents (2G: [2] × n = [acetone], n < 0.5).16 Owing to the lack of an “acetone sublayer” in 2G, the gold complexes in the tape structure of 2G can interact with adjacent complexes through multipoint F⋯F and CH⋯F interactions (Fig. S9b†). As a result, flat 2-D sheets which extend along the bc-planes are formed. Between sheets, molecules stack on top of each other without offset through π–π stacking interactions with a perpendicular distance of 3.509 Å (benzene rings) and 3.330 Å (pyridine rings). 2G does not contain aurophilic interactions [Au⋯Au separation: 3.571(2) Å] (Fig. 6a). The smaller excitation energy of 2G compared with that of 2B may be caused either by smaller amounts of solvent inclusion, which may enhance chromophore–chromophore interactions, or by the rather flat conformations of the chromophore (θdihedral = 11.92°), which may lead to an effective intra and/or intermolecular conjugation.17
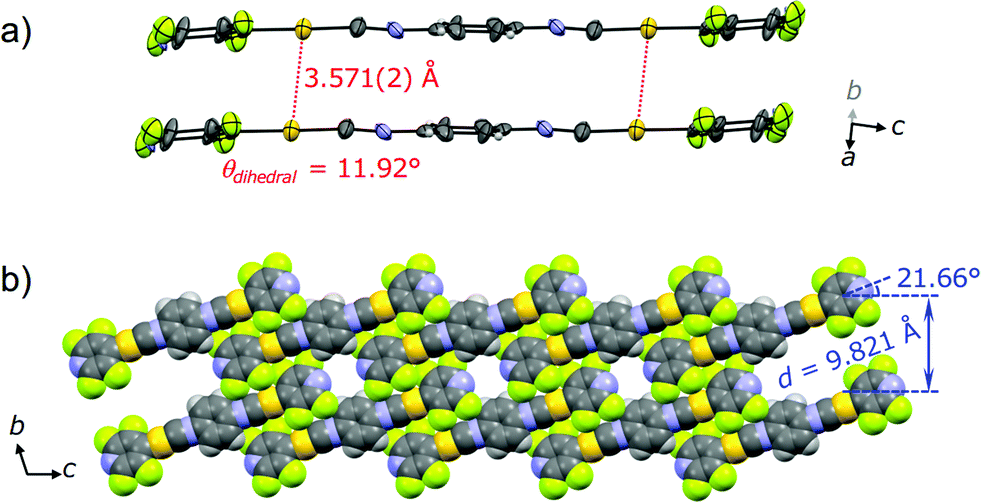 | ||
| Fig. 6 (a) ORTEP representation of the dimer viewed roughly along the direction of the b axis and (b) space-filling representation of the packing structure of 2G viewed along the direction of the a axis. | ||
PXRD measurements and Rietveld refinement disclosed the detailed crystalline structure of 2Y demonstrating that its yellow emission is caused by aurophilic interactions. The molecular packing arrangement of 2Y was determined with suitable quality from the PXRD data with a range of 2θ = 7–60° (Fig. S10†).18 It should be noted that the present study is the first example showing the great advantage of ab initio structural analysis of ground powders of mechanochromic compounds.19 Ground powder 2Y crystallizes in the monoclinic space group P21/c (Fig. 7 and Table 1 and S3†). The central isocyanide benzene ring of the molecule is on the inversion center. The molecular packing arrangement of 2Y is rather different from those of 2B and 2G. For example, no solvent molecules exist in the crystalline lattice of 2Y, which is also supported by thermal analysis, elemental analysis and 1H NMR spectroscopy (Fig. 8 and S13 and Table S4†). Moreover, face-to-face stacking of the molecules is absent. Instead, infinite chains of Au⋯Au interactions with a distance of 3.428(2) Å are formed along the direction of the c axis (Fig. 7a). This distance is within the range of aurophilic interactions, and is likely to be responsible for the emission properties of 2Y with a low excited energy level. Perpendicular to the aurophilic bond, molecules afford a flat sheet through multipoint F⋯F interactions between the tape-like motif (Fig. S11a†). Within the sheet, all the molecular long axes are oriented along the same direction, and in the adjacent sheet molecules are oriented at approximately 90° with respect to those in the next layer (Fig. S11b†). This is the first report of mechano-induced crystal-to-crystal phase conversion with solvent release.
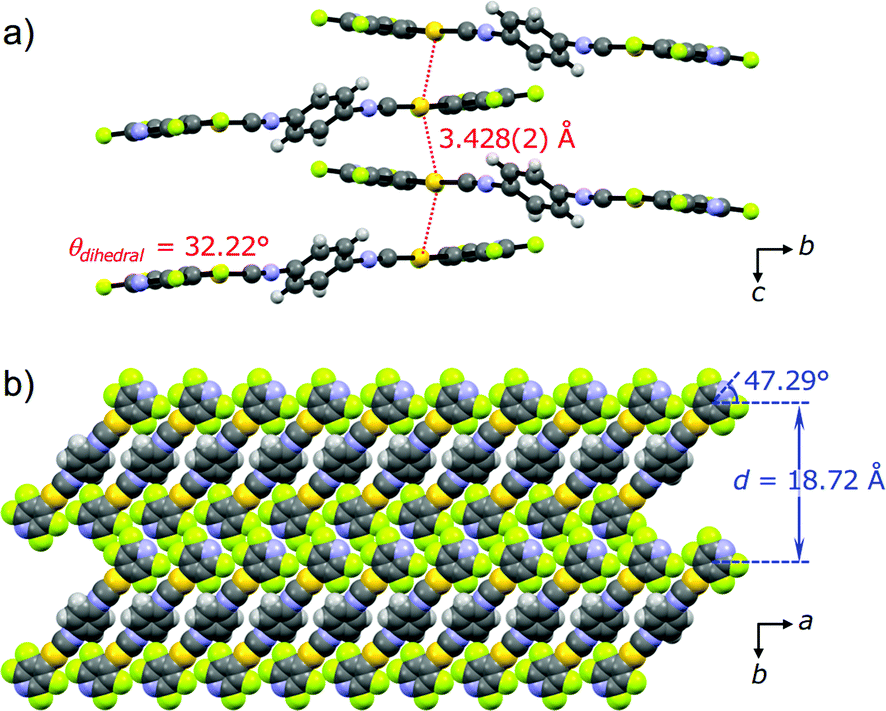 | ||
| Fig. 7 (a) Ball-and-stick representation of the tetramer viewed along the direction of the a axis, and (b) the space-filling representation of the packing structures of 2Y viewed along the direction of the c axis. | ||
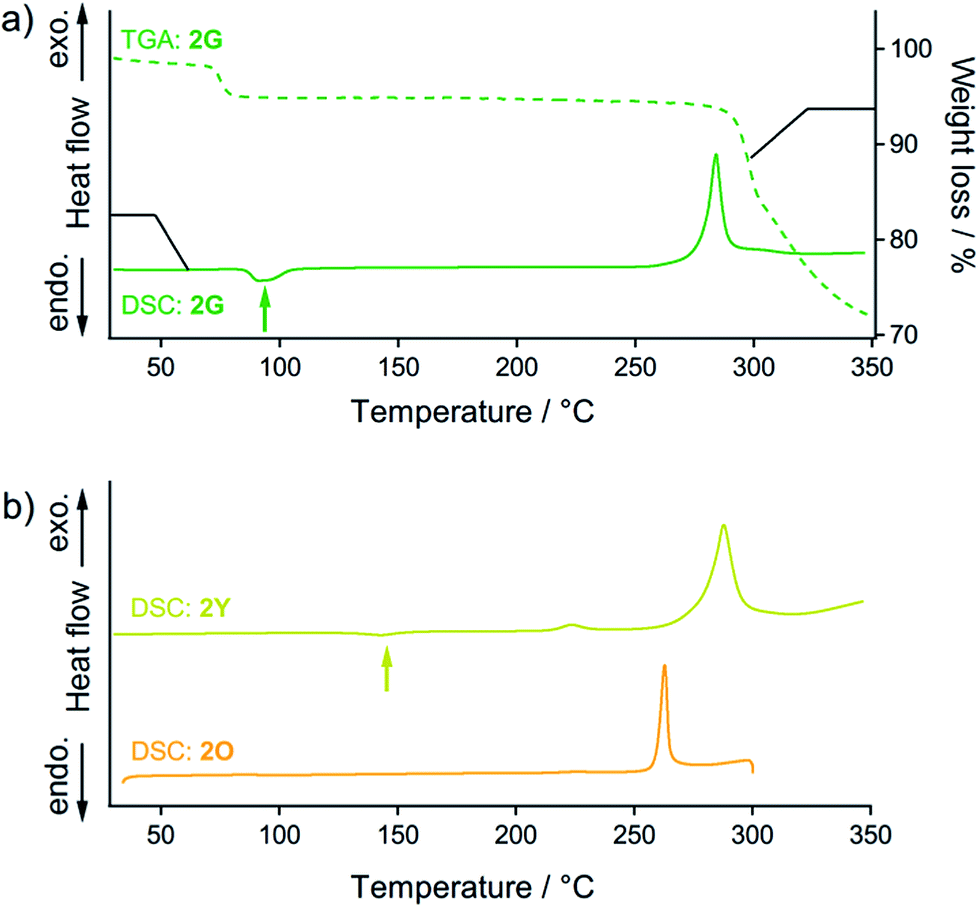 | ||
| Fig. 8 (a) DSC (solid line) and TGA (dashed line) profiles of 2G. The green arrow indicates the endothermic phase transition from 2G to 2Y. (b) DSC profiles of 2Y (yellow line) and 2O (orange line). The yellow arrow indicates the endothermic phase transition from 2Y to 2O. For the DSC and TGA profiles, the heating rate is 10 °C min−1. | ||
The orange emission of 2O, with the smallest excitation energy of the four different structures of 2, is attributed to the aurophilic bonds with the shortest Au⋯Au distance. Owing to the amorphous nature, the detailed intermolecular interaction patterns existing in 2O are unclear. TGA, elemental analysis and the 1H NMR spectrum revealed that 2O contains no solvent molecules (Fig. 8 and S14 and Table S4†), therefore chromophore–chromophore interactions, rather than chromophore–solvent interactions, must be involved. From the longest wavelength maxima in the excitation and emission spectra, it is more likely that aurophilic interactions with shorter Au⋯Au separation compared with those observed in 2Y should be present.6a In the amorphous ground phase of mechanochromic organometallic compounds, it is reported that metallophilic bonds are formed that effect their emission properties.6a,b,7e,g
The above discussion indicates that 2 can form two solvated (2B and 2G) and two non-solvated (2Y and 2O) solid states. This can be confirmed by the experimental results of the crystal structure analyses, thermal analyses, elemental analyses and 1H NMR spectroscopy as mentioned above (see the ESI†). This is further supported by IR spectroscopy: the IR spectra of 2B and 2G show carbonyl stretching vibrational bands at around 1715 cm−1, while those of 2Y and 2O do not (Fig. S15†). Therefore, crystalline structure changes with solvent inclusion/release mode are important for the interconvertible multiple photoluminescence color of 2.
Thermal analyses
Thermal analysis of 2G (Fig. 8a) reveals that its phase transition into the 2Y phase occurs with solvent release. DSC measurements of 2G show the endothermic peak at 95 °C (green arrow in Fig. 8a) before observation of the prominent exothermic peaks of decomposition above 250 °C. The endothermic peak at 95 °C can be attributed to the thermal phase transition from 2G to 2Y20 because the emission color of 2G changed from green to yellow at this temperature (Fig. S16†). This phase transition (2G → 2Y) occurs with solvent release: the TGA profile of 2G reveals a 5% weight loss at around at 90 °C (dashed line in Fig. 8a). This result suggests that the phase transition of 2G by short grinding also induces solvent release to form 2Y.21 Although mechanochromism with solvent release was reported previously,7 this is the first report of a crystal-to-crystal phase conversion.DSC analyses show the relative thermodynamic stability of the solvent-free forms of 2Y and 2O.22 DSC measurements of 2Y show the endothermic peak of the phase transitions at around 145 °C (yellow arrow in Fig. 8b). The emission color change of 2Y from yellow to orange was also observed at around 160 °C upon heating treatment (Fig. S16†). Thus, the endothermic peak of 2Y at 145 °C can be attributed to the thermal phase transition from 2Y to 2O. The TGA profile of 2Y does not show any discernible weight loss in this temperature range, because no solvent is included (Fig. S17†). However, DSC and TGA profiles of 2O do not show any peaks until decomposition above 250 °C (orange line in Fig. 8b and S17†), owing to the absence of the phase transition upon heating. This is supported by the fact that the thermal-induced emission color change of 2O is absent (Fig. S16†). From these results, 2Y can be considered as the solvent-free metastable polymorph of 2, while 2O is the solvent-free, thermodynamically more stable phase.23
The fact that the phase transition from 2G to 2Y requires solvent release and that 2Y is less thermodynamically stable than 2O is key to realize a unique two-step mechanochromism, 2G → 2Y → 2O (steps (iii) and (iv) in Fig. 2d). In the initial stage of grinding of 2G (step (iii) in Fig. 2d), included solvent molecules start to be released owing to decreasing the particle size and increasing the surface area of the solid sample. This initiates molecular rearrangement of 2G to give the solvent-free form. As a result, 2Y is initially formed as a kinetically trapped metastable intermediate. Additional mechanical stimulation provides the 2O phase (step (iv) in Fig. 2d), which is the more thermodynamically stable form of 2. This type of crystal-to-amorphous phase transition (2Y → 2O) upon mechanical stimulation in a solvent-free solid state condition is most commonly observed for mechanochromic compounds.3,6 As mentioned above, we confirm that 2B and 2G contain solvent molecules in their crystalline lattices while 2Y and 2O do not contain any solvent molecules based on crystal structure analyses, thermal analyses, elemental analyses and 1H NMR and IR spectroscopy (see the ESI†).
Role of tetrafluoropyridyl groups on the unique stimuli-responsivity of 2
It is surprising that substitution of the pentafluorophenyl group in 1 by a tetrafluoropyridyl group in 2 leads to a dramatic enhancement of its stimuli-responsivity (Fig. 2). It has been reported that subtle modifications of chemical structure can induce a significant change in solid state structures.15 As the overall crystalline structures are generally determined by accumulation of relatively weak intermolecular interactions, it is sometimes difficult to understand the relationships between molecular and crystalline structures. Indeed, at this stage, it is difficult to fully determine which molecular structural factor of 2 is important for its unique stimuli-responsivity.However, one possible explanation for the impact of the molecular structure on the mechanochromic properties concerns the relatively high polarity of 2 (Fig. S18†), which may help the formation of weak interactions with acetone. For the previously reported solvophobic complex 1 containing a C6F5 moiety, the molecules in the crystals were densely packed, and no solvent inclusion was observed.6a Complex 2, with a C5NF4 moiety, is more polar and less solvophobic compared to 1. This enables the solvent inclusion/release process owing to the weak interactions between the polar molecule 2 and acetone. In the presence of many solvent molecules, blue-emitting 2B was formed, then after evaporation of acetone, the less solvated 2G was formed. As the solvated acetone molecules were weakly absorbed in the crystal lattice, weak mechanical stimulation induces solvent release to produce non-solvated 2Y. The slight molecular structural change from 1 with the C6F5 moiety to less solvophobic 2 with the C5NF4 moiety is an important key factor to realize unprecedented crystal-to-crystal-to-amorphous phase transition with the different emission color changes.
Conclusions
We report complex 2 which can adopt four different solid state structures with different emission properties. The four different emissions of 2 in the solid state are reversible. Multiple switching of the solid state emission of 2 was caused by a combination of dual two-step emission color changes which are induced by solvent inclusion/release and short/long mechanical stimulation. In an acetone atmosphere, blue-emitting 2B was formed, and after acetone release, green-emitting 2G was formed. Upon applying a mechanical stimulus to 2G, two-step emission color changes to yellow (short ball-milling) and then to orange (long ball-milling) were observed, which correspond to phase changes into 2Y and 2O, respectively. Single crystal and powder X-ray diffraction analyses successfully revealed the detailed molecular arrangements of 2B, 2G, and 2Y. We found that both 2B and 2G formed solvated crystalline structures without defined aurophilic interactions. Conversely, the 2Y powder formed a solvent-free ordered molecular arrangement involving aurophilic interactions. The amorphous 2O phase is suggested to contain aurophilic interactions with the shortest Au⋯Au distance. Thermal analyses revealed that the phase transition from 2G to 2Y requires solvent release. Moreover, the amorphous 2O phase is thermodynamically more stable than the 2Y phase. Thus, this unique two-step mechanochromism (2G → 2Y → 2O) upon mechanical stimulation starts with solvent release from 2G followed by rearrangement into solvent-free 2Y (crystal-to-crystal phase transition), and then crystal-to-amorphous phase transition takes place to form the thermodynamically more stable 2O. Comparison between 1 with a C6F5 moiety and 2 with a C5NF4 moiety suggests that the more polar and less solvophobic nature of 2 may create the solvent inclusion/release structure transition. This is an important factor for the multiple structures and emission color transformation of 2. The present multicolored mechanochromic complex 2 with fully-solved structures can be considered as a new type of smart material.Experimental section
Synthesis of 2,3,5,6-tetrafluoropyridyl(tetrahydrothiophene) gold(I)
2,3,5,6-Tetrafluoropyridine (755.3 mg, 5.0 mmol) was dissolved in dry THF (20 mL) with stirring at −78 °C in a 100 mL round-bottomed flask, then n-BuLi (1.65 M hexane) was added dropwise for 15 min. After 1 h, chloro(tetrahydrothiophene)gold(I) (1.92 g, 6.0 mmol) was added to the reaction mixture. Then, the mixture was allowed to warm to room temperature and was stirred for 1 h. Addition of a small portion of water quenches the reaction. MgSO4 was added to the mixture and stirred for 20 min. MgSO4 was filtered off by filtration and the solvent was evaporated. The residue was quickly passed through a short silica gel column (CH2Cl2–hexane = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, 100 mL) to give a white-purple solid (1.72 g, 4.0 mmol, 79%). 1H NMR [400 MHz, CDCl3, δ (ppm)]: 2.24 (s, 4H), 3.46 (s, 4H). 13C NMR [100 MHz, CDCl3, δ (ppm)]: 30.7 (CH2), 38.7 (CH2), (CH), 141.7 (CH), 143.9 (CH), 144.3 (CH). HRMS-FAB (m/z): [M + H] calcd for C19H9AuNF4+, 436.0052; found, 436.0058. Anal. calcd for C19H8AuNF4: C, 24.84, H, 1.85; N, 3.22. Found: C, 24.87; H, 1.90; N, 3.05.
:50, 100 mL) to give a white-purple solid (1.72 g, 4.0 mmol, 79%). 1H NMR [400 MHz, CDCl3, δ (ppm)]: 2.24 (s, 4H), 3.46 (s, 4H). 13C NMR [100 MHz, CDCl3, δ (ppm)]: 30.7 (CH2), 38.7 (CH2), (CH), 141.7 (CH), 143.9 (CH), 144.3 (CH). HRMS-FAB (m/z): [M + H] calcd for C19H9AuNF4+, 436.0052; found, 436.0058. Anal. calcd for C19H8AuNF4: C, 24.84, H, 1.85; N, 3.22. Found: C, 24.87; H, 1.90; N, 3.05.
Synthesis of 2
A mixture of 1,4-diisocyanobenzene (64.1 mg, 0.5 mmol) and 2,3,5,6-tetrafluoropyridyl(tetrahydrothiophene)gold(I) (435.2 mg, 1.0 mmol) in CH2Cl2 (5 mL) was stirred for 30 min under a N2 atmosphere at room temperature. The precipitates were filtered and washed with CH2Cl2, and dried in vacuo to obtain 2 as a yellow solid (350.3 mg, 0.4 mmol, 85%). 1H NMR (400 MHz, THF-d8, δ (ppm)): 8.14 (s, 4H). The poor solubility of 2 in solvents hampered 13C NMR measurements. HRMS-FAB (m/z): [M + H] calcd for C18H5Au2N4F8+, 822.9712; found, 822.9711. Anal. calcd for C18H4Au2N4F8: C, 26.30, H, 0.49; N, 6.81. Found: C, 26.23; H, 0.64; N, 6.63.Preparation of 2B
The blue luminescent powder 2B was prepared from 2Y, 2G, and 2O immediately after they were soaked in acetone. IR (neat):![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 2218, 1704, 1628, 1543, 1421, 1214, 927, 832 cm−1.
= 2218, 1704, 1628, 1543, 1421, 1214, 927, 832 cm−1.
Preparation of 2G
Upon air drying a powder of 2B to evaporate acetone, green-emitting 2G was immediately formed. IR (neat): = 2209, 1628, 1453, 1207, 929, 838 cm−1. 1H NMR [400 MHz, THF-d8, Fig. S12,†δ (ppm)]: 8.14 (s, 4H). The methyl proton of included acetone was also observed at 2.11 ppm. Anal. calcd for C18H4Au2N4F8: C, 26.30, H, 0.49; N, 6.81. Found: C, 26.67; H, 0.89; N, 6.63.
Preparation of 2Y
As-prepared solid 2Y was analytically pure but, in terms of the crystalline arrangement, it is impure as shown by the PXRD pattern (Fig. S6†). For purification of as-synthesized 2Y, 2 was suspended in cyclohexanone (cmax = 1 mg mL−1) and filtered and washed with that solvent, then dried in vacuo to give the yellow-emitting powder 2Y. An alternative preparation method for 2Y is to grind 2G in a ball-mill at 4600 rpm for 10 min (Taitec Bead Crusher μT-01). Typically, 2G (50 mg) and a stainless bead (1/8 inch) were put into a micro tube (ϕ13 × 49 mm) with a screw cap and ball-milled for 10 min, during which time the container should be opened two or three times to facilitate solvent release. IR (neat): 2217, 1620, 1420, 1207, 921, 838 cm−1. 1H NMR [400 MHz, THF-d8, Fig. S13,†δ (ppm)]: 8.14 (s, 4H). Anal. calcd for C18H4Au2N4F8: C, 26.30, H, 0.49; N, 6.81. Found: C, 26.39; H, 0.60; N, 6.79.Preparation of 2O
For the preparation of 2O, 2Y was ground in a ball-mill at 4600 rpm for 5 min. IR (neat): = 2204, 1620, 1421, 1201, 916, 829 cm−1. 1H NMR [400 MHz, THF-d8, Fig. S14,†δ (ppm)]: 8.14 (s, 4H). Anal. calcd for C18H4Au2N4F8: C, 26.30, H, 0.49; N, 6.81. Found: C, 26.23; H, 0.64; N, 6.63.
Acknowledgements
This work was financially supported by the Funding Program for Next Generation World-Leading Researchers (NEXT Program, no. G002) from the Japan Society for the Promotion of Science (JSPS), the MEXT (Japan) program “Strategic Molecular and Materials Chemistry through Innovative Coupling Reactions” of Hokkaido University, and JSPS KAKENHI Grant-in-Aid for Challenging Exploratory Research, 26620053, and for Young Scientists (B), 26810042. We also thank the support of Frontier Chemistry Center Akira Suzuki “Laboratories for Future Creation” Project.Notes and references
- (a) Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC; (b) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC; (c) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed; (d) A. L. Balch, Angew. Chem., Int. Ed., 2009, 48, 2641–2644 CrossRef CAS PubMed; (e) C. Jobbágy and A. Deák, Eur. J. Inorg. Chem., 2014, 2014, 4434–4449 CrossRef.
- Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef CAS PubMed.
- Z. Ma, M. Teng, Z. Wang, S. Yang and X. Jia, Angew. Chem., Int. Ed., 2013, 52, 12268–12272 CrossRef CAS PubMed.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed.
- S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CAS.
- (a) H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044–10045 CrossRef CAS PubMed; (b) K. Kawaguchi, T. Seki, T. Karatsu, A. Kitamura, H. Ito and S. Yagai, Chem. Commun., 2013, 49, 11391–11393 RSC; (c) N. D. Nguyen, G. Zhang, J. Lu, A. E. Sherman and C. L. Fraser, J. Mater. Chem., 2011, 21, 8409–8415 RSC; (d) S. H. Lim, M. M. Olmstead and A. L. Balch, Chem. Sci., 2013, 4, 311–318 RSC; (e) H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 Search PubMed; (f) W. Li, L. Wang, J.-P. Zhang and H. Wang, J. Mater. Chem. C, 2014, 2, 1887–1892 RSC; (g) M. S. Kwon, J. Gierschner, J. Seo and S. Y. Park, J. Mater. Chem. C, 2014, 2, 2552–2557 RSC.
- (a) B.-C. Tzeng, T.-Y. Chang and H.-S. Sheu, Chem.–Eur. J., 2010, 16, 9990–9993 CrossRef CAS PubMed; (b) J. Ni, X. Zhang, N. Qiu, Y. H. Wu, L. Y. Zhang, J. Zhang and Z. N. Chen, Inorg. Chem., 2011, 50, 9090–9096 CrossRef CAS PubMed; (c) S. J. Choi, J. Kuwabara, Y. Nishimura, T. Arai and T. Kanbara, Chem. Lett., 2012, 41, 65–67 CrossRef CAS; (d) N. Zhao, Z. Yang, J. W. Lam, H. H. Sung, N. Xie, S. Chen, H. Su, M. Gao, I. D. Williams, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 8637–8639 RSC; (e) A. Han, P. Du, Z. Sun, H. Wu, H. Jia, R. Zhang, Z. Liang, R. Cao and R. Eisenberg, Inorg. Chem., 2014, 53, 3338–3344 CrossRef CAS PubMed; (f) C. Jobbágy, M. Molnár, P. Baranyai, A. Hamza, G. Pálinkás and A. Deák, CrystEngComm, 2014, 16, 3192–3202 RSC; (g) J. Ni, Y.-G. Wang, H.-H. Wang, Y.-Z. Pan, L. Xu, Y.-Q. Zhao, X.-Y. Liu and J.-J. Zhang, Eur. J. Inorg. Chem., 2014, 2014, 986–993 CrossRef CAS; (h) J. Ni, Y.-G. Wang, H.-H. Wang, L. Xu, Y.-Q. Zhao, Y.-Z. Pan and J.-J. Zhang, Dalton Trans., 2014, 43, 352–360 RSC; (i) X.-C. Shan, F.-L. Jiang, L. Chen, M.-Y. Wu, J. Pan, X.-Y. Wan and M.-C. Hong, J. Mater. Chem. C, 2013, 1, 4339–4349 RSC; (j) X. Zhou, H. Li, Z. Chi, X. Zhang, J. Zhang, B. Xu, Y. Zhang, S. Liu and J. Xu, New J. Chem., 2012, 36, 685–693 RSC.
- (a) N. Harada, Y. Abe, S. Karasawa and N. Koga, Org. Lett., 2012, 14, 6282–6285 CrossRef CAS PubMed; (b) N. Harada, S. Karasawa, T. Matsumoto and N. Koga, Cryst. Growth Des., 2013, 13, 4705–4713 CrossRef CAS; (c) S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed; (d) M.-S. Yuan, D.-E. Wang, P. Xue, W. Wang, J.-C. Wang, Q. Tu, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Chem. Mater., 2014, 26, 2467–2477 CrossRef CAS; (e) J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119–122 CrossRef CAS.
- (a) T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828–12832 CrossRef CAS PubMed; (b) H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 Search PubMed.
- (a) K. Fujii, Y. Ashida, H. Uekusa, F. Guo and K. D. M. Harris, Chem. Commun., 2010, 46, 4264–4266 RSC; (b) K. Fujii, H. Uekusa, N. Itoda, G. Hasegawa, E. Yonemochi, K. Terada, Z. Pan and K. D. M. Harris, J. Phys. Chem. C, 2010, 114, 580–586 CrossRef CAS; (c) K. Fujii, H. Uekusa, N. Itoda, E. Yonemochi and K. Terada, Cryst. Growth Des., 2012, 12, 6165–6172 CrossRef CAS; (d) K. Fujii, M. Aoki and H. Uekusa, Cryst. Growth Des., 2013, 13, 2060–2066 CrossRef CAS.
- (a) A. L. Balch, Gold Bull., 2004, 37, 45–50 CrossRef CAS; (b) P. Pyykko, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed; (c) V. W. Yam and E. C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC; (d) M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884–1895 RSC; (e) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC; (f) X. He and V. W.-W. Yam, Coord. Chem. Rev., 2011, 255, 2111–2123 CrossRef CAS; (g) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- Actually, direct evidence of the aurophilic interaction of 1Y has not been obtained even by EXAFS study measured at 4 K. The peak assignable to the Au⋯Au distance of 1Y was found around at 5 Å, which corresponded to the Au⋯Au distance of unground 1B. This result may be caused by only the small amount of aurophilic interactions in 1Y, so that even EXAFS studies could not observe the aurophilic interactions. A more detailed discussion will be reported elsewhere.
- Addition of other solvents, such as dichloromethane, chloroform, cyclohexanone and dimethoxyethane, induces various emission color changes of 2. For example, chloroform addition to 2G provided a red-emitting powder which shows emission spectrum peaked at 645 nm. Unfortunately, we can not determine, at present, detailed molecular arrangements of the resulting materials, and thus these behaviors will be discussed elsewhere. Moreover, recrystallization of 2 from cyclohexanone, THF, and dimethoxyethane (maximum solubility cmax > 0.1 mg mL−1) afforded a yellow-emitting solid (similar to 2Y). Since 2 can not solvate these solvents, it is proposed that non-solvated 2Y was formed under these conditions.
- 2Y is also transformed to 2B upon addition of acetone.
- (a) J. Bernstein, Polymorphism in molecular crystals, Clarendon Press/International Union of Crystallography, 2002, vol. 14 Search PubMed; (b) Y. Mnyukh, Fundamentals of Solid-State Phase Transitions: Ferromagnetism and Ferroelectricity, Authorhouse, 2001 Search PubMed.
- Residual electron density in 1-D channel is estimated to be seventeen electrons per unit cell. This is corresponding to 0.5 equivalents for acetone and/or 2 equivalents for water. 1H NMR spectrum of 2G confirms that both acetone and H2O could be present (Fig. S12†).
- D. Yan, G. Fan, Y. Guan, Q. Meng, C. Li and J. Wang, Phys. Chem. Chem. Phys., 2013, 15, 19845–19852 RSC.
- A diffraction peak at 6.6° is derived from the residual 2G phase contaminated within the 2Y phase (see also Fig. S7c†).
- For an example of PXRD measurements and the Rietveld refinement technique for the study of the crystalline size of the ground phase of mechanochromic compounds rather than full structure analysis, see: S. Perruchas, X. F. Le Goff, S. Maron, I. Maurin, F. Guillen, A. Garcia, T. Gacoin and J. P. Boilot, J. Am. Chem. Soc., 2010, 132, 10967–10969 CrossRef CAS PubMed.
- The 2Y phase thermally transformed from 2G can not further transform to the 2O phase upon phase transition; whereas mechanically prepared 2Y shows transformation into the 2O phase. This result indicates that the preparation method of the 2Y phase affects its phase transition behavior.
- As mentioned above, no solvent molecules exist in the crystalline lattice of 2Y based on thermal analyses, elemental analyses and 1H NMR and IR spectroscopy (Fig. 8, S13 and S17 and Table S4†).
- DSC analysis of 2B was not performed because it is unstable without acetone.
- This discussion agrees well with the fact that the as-synthesized form of 2 is principally similar to 2Y as shown in the PXRD pattern (Fig. S6†), likely because a metastable form of 2Y is kinetically trapped.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data, optical properties, characterization and interconversion of 2B, 2G, 2Y and 2O, and other additional information. CCDC 1035806, 1035808 and 1035810. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03960b |
| This journal is © The Royal Society of Chemistry 2015 |