
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled trinuclear arsenic and antimony macrobicycles†
Mary S.
Collins
a,
Robert Y.
Choi
a,
Lev N.
Zakharov
b,
Lori A.
Watson
c,
Benjamin P.
Hay
d and
Darren W.
Johnson
*a
aDepartment of Chemistry & Biochemistry and Materials Science Institute, University of Oregon, Eugene, Oregon, USA 97403-1253. E-mail: dwj@uoregon.edu; Fax: +1-541-346-4643; Tel: +1-541-346-1695
bCAMCOR – Center for Advanced Materials Characterization in Oregon, University of Oregon, Eugene, OR 97403-1443, USA
cDepartment of Chemistry, Earlham College, Richmond, IN 47374-4095, USA
dSupramolecular Design Institute, Oak Ridge, TN 37830-7185, USA
First published on 18th February 2015
Abstract
The synthesis of six trinuclear Pn3L2 macrobicycles (Pn = As, Sb) was achieved by self-assembly of a pnictogen trichloride and a 2,4,6-trisubstituted-1,3,5-benzenetrimethanethiol ligand. 1H-NMR spectroscopy reveals self-assembly in 1,1,2,2-tetrachloroethane is dynamic in solution producing two structural isomers. The symmetric and the asymmetric isomers (in which a single chloride ligand is cast in an opposing direction from other chlorides) of the As3L2 complexes exist in a ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 distribution, whereas only the symmetric isomer is observed in solution for Sb3L2. Solvent effects appear to influence conformational isomerism and conversion to the final products. Macrobicycles were confirmed by 1H-NMR spectroscopy and X-ray crystallography and further studied by MP2/LANL2DZ optimizations.
:1 distribution, whereas only the symmetric isomer is observed in solution for Sb3L2. Solvent effects appear to influence conformational isomerism and conversion to the final products. Macrobicycles were confirmed by 1H-NMR spectroscopy and X-ray crystallography and further studied by MP2/LANL2DZ optimizations.
Introduction
Supramolecular systems rely on multiple weak interactions to achieve stable, specific coordination to their hosts. Through creative ligand design, one can incorporate binding elements that enable directional self-assembly. Historically, a diversity of self-assembled systems incorporate the predictable coordination geometry of transition metals as a directional tool.1–6 Only recently has more effort been made toward understanding the supramolecular self-assembly of main group elements, including Group 15.7–10 We utilize mercaptomethyl substituents as a preorganizing binding motif to accommodate the pnictogen's trigonal pyramidal geometry and thiophilicity. In addition to this predictable geometric configuration, electron density from the ligand provides a supporting non-covalent stabilizing force with the Lewis acidic pnictogen, known as a Pn⋯π interaction.11,12 Further studies by our laboratory have investigated how self-assembly is dictated by the structure of the dithiolate ligand by altering its length, width or the placement of steric bulk (Chart 1).13–16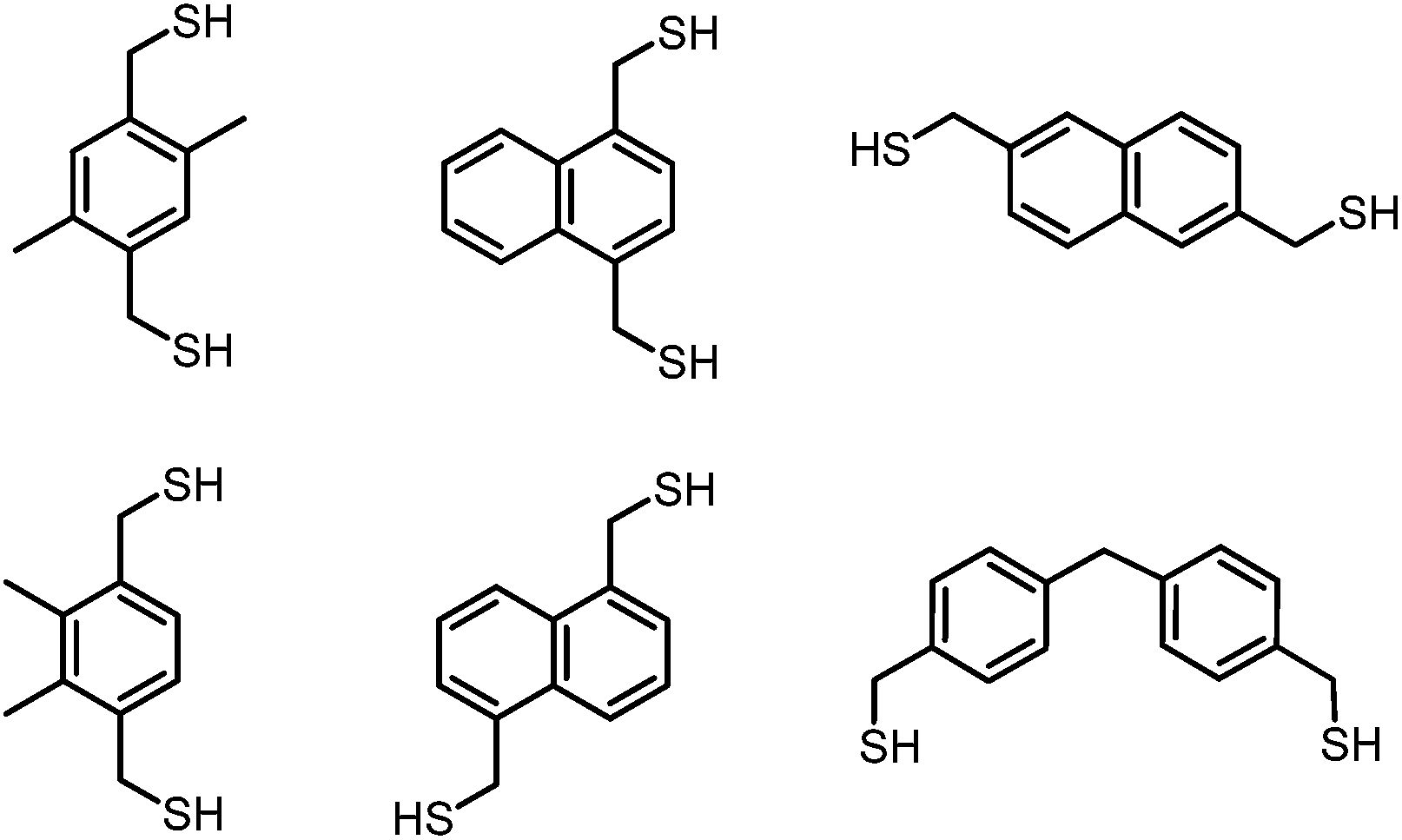 | ||
| Chart 1 Selection of dithiol ligands used for Pn–thiolate self-assembly. | ||
The 1,3,5-trisubstituted-2,4,6-triethyl benzene scaffold has been exploited in numerous supramolecular hosts since Mislow first demonstrated steric gearing of adjacent substituents, where each substituent is positioned in an alternating up-down arrangement.17 While this preorganization technique has now been readily utilized to form supramolecular structures,18 its use with Group 15 metalloids (As, Sb) has not yet been explored, and led to some surprising structures. In this study, a series of six trinuclear arsenic and antimony supramolecular macrobicycles [Pn3L2Cl3] were synthesized using a trisubstituted, trithiol ligand (Fig. 1). In addition to the syntheses, we address some unusual dynamic solution behaviour that arises from these new ligands that feature this steric gearing component. Typically, steric gearing serves as an effective preorganizational tool for metal–ligand-based supramolecular systems, providing enhanced thermodynamic stability of a complex and/or faster kinetics of formation. In this system we observed a surprising deviation where this gearing effect does not influence the rate of self-assembly in mediating thiol reactivity with a pnictogen. Rather, interaction with the solvent and/or other external additives (such as chloride and tetrafluoroborate salts) more directly influences the course of the self-assembly reaction. For instance, a subtle change in solvent or the addition of a salt accelerate formation of macrobicycles in solution and additionally facilitates or prohibits a change in conformation to favour an unexpected, slightly higher energy asymmetric macrobicycle in tandem with the more stable, symmetric macrobicycle. Furthermore, close Pn⋯πaryl contacts suggest a Pn⋯πaryl interaction acts as a stabilizing force during assembly formation. These structures represent the first known trinuclear supramolecular arsenic– or antimony–thiolate assemblies.8
 | ||
| Fig. 1 Ball and stick representations (hydrogen atoms and solvent molecules are omitted for clarity) of the symmetric conformations of all new macrobicyclic structures reported: (a) [As3LH2Cl3] (b) [As3LMe2Cl3] (c) [As3LEt2Cl3] (d) [Sb3LH2Cl3] (e) [Sb3LMe2Cl3] (f) [Sb3LEt2Cl3]. | ||
Results and discussion
Symmetric and asymmetric Pn3L2Cl3 macrobicycles
To study the steric effect imparted by a trisubstituted benzene in arsenic–thiolate self-assembly, three bridging tridentate 2,4,6-trisubstituted-1,3,5-tris(mercaptomethyl)benzene ligand derivatives H3LR (R = H, Me, Et) were synthesized (see ESI† for complete experimental details). When treated with a stoichiometric amount of AsCl3 in 1,1,2,2-tetrachloroethane (TCE), a discrete trinuclear As3L2Cl3 macrobicycle emerges (Scheme 1). The unsubstituted metallacyclophane As3LH2Cl3 can alternatively be synthesized in a more facile method through the addition of tetrabutylammonium chloride at 0 °C in chloroform. The presence of a chloride guest was initially used to catalyze or template formation of a tetrahedral cage; however, TBA+Cl− instead induced a rapid precipitation of pure macrobicycles in the form of colorless needles (45% isolated crystalline yield after filtration, see ESI†).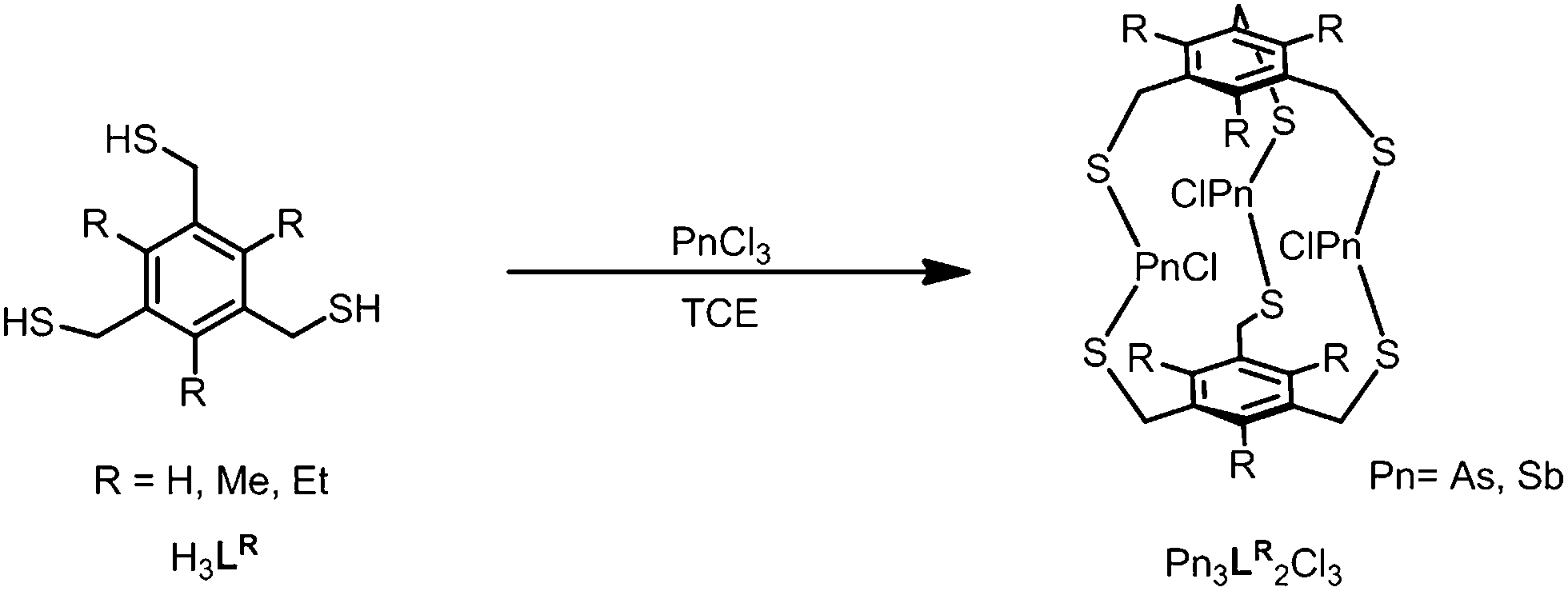 | ||
| Scheme 1 Self-assembly of Pn3L2Cl3 macrobicycles. | ||
Each prepared ligand derivative (H3LH,Me,Et) provides quantitative conversion to As3L2Cl3 upon treatment with AsCl3 as observed by 1H-NMR spectroscopy. Layering benzene over a solution of H3LMe,Et and AsCl3 in TCE gave crystals suitable for X-ray diffraction in a 25% yield for [As3LMe2Cl3] and 15% for [As3LEt2Cl3], respectively, within one week. Antimony macrobicycles were isolated from a mixture of the corresponding H3L species and an excess of SbCl3 and crystallized by slow evaporation of TCE. The formation of Sb3LMe,Et2Cl3 complexes under these conditions could not be observed by 1H-NMR spectroscopy,20 and only partial conversion to [Sb3LH2Cl3] from SbCl3 and H3LH was observed when heating the solution to 70 °C (Fig. S18†). Nevertheless, crystallization of these solutions appears to induce formation of these macrocbicycles, and the structures of each [Sb3L2Cl3] complex were determined by single crystal X-ray diffraction (ESI Table 1†). These Sb-macrobicycles are then stable and can be redissolved TCE-d2 to verify their structure in solution (see Fig. S18, S21 and S24†).
By incorporating the 2,4,6-triethyl-substituted benzene scaffold, we envisioned the rate of self-assembly for both arsenic and antimony complexes to be favored over ligands that do not have such steric gearing components. While this preorganizing element has been successfully utilized to overcome high energy binding conformations or potential unfavorable binding entropies in relation to free host, guest or solvated host–guest complexes,21 we did not observe steric gearing as the primary driving force by 1H-NMR in the formation of pnictogen–thiolate macrobicycles as expected. The unsubstituted analogue [As3LH2Cl3], bearing no steric gearing component, forms faster in solution (1 day) by 1H-NMR than its ethyl-substituted counterpart (4 days) in TCE-d2 (Fig. S6 and S16†).
However, evidence for steric gearing was observed in the crystalline state (single crystal structure data and parameters are shown in ESI Table 1, see ESI† for complete details). As anticipated, none of the 2,4,6-triethyl substituents are directed toward the arsenic atoms in the crystal structure of [As3LEt2Cl3], since the 1,3,5-trimercaptomethyl groups of the ligand are all pointed in that direction in order to converge on the three arsenic ions (Fig. 1C and 2). The electron-rich chlorine atoms coordinated to arsenic would also sterically disfavour the presentations of ethyl groups toward the center of the assembly. Interestingly, the increased steric bulk of the ethyl groups appears to affect the flexibility of the molecule to adopt a position that maintains the shortest As⋯πaryl contact. This trend is apparent in both the As and Sb complexes, in which the Pn⋯πaryl contact increases in distance from [Pn3LH2Cl3] to [Pn3LMe2Cl3] to [Pn3LEt2Cl3] (Table 1). As shown in Fig. 2A, each pnictogen atom is positioned endo relative to the aromatic ligand backbone, allowing for the stereochemically active pnictogen lone pairs to be directed inside the cavity of the macrobicycle. This arrangement facilitates an attractive Pn⋯πaryl interaction, which we have observed in many other structures of this type and use as a key design feature.11,16 The smallest As⋯πaryl contacts in the As3L2Cl3 macrobicycles range between 3.17 and 3.27 Å, which is shorter than the sum of the van der waals radii for C and As (3.7 Å),10,11,22–24 suggesting a stabilizing As⋯π interaction (Table 1). The close As⋯πaryl contacts for the unsubstituted macrobicycle and the faster self-assembly of these congeners suggest that providing for As⋯π interactions in the ligand design may play a more significant directing role in the self-assembly than steric gearing. Moreover, the less restricted rotation around the C–aryl bonds in the less sterically hindered ligand may accelerate self-assembly.21
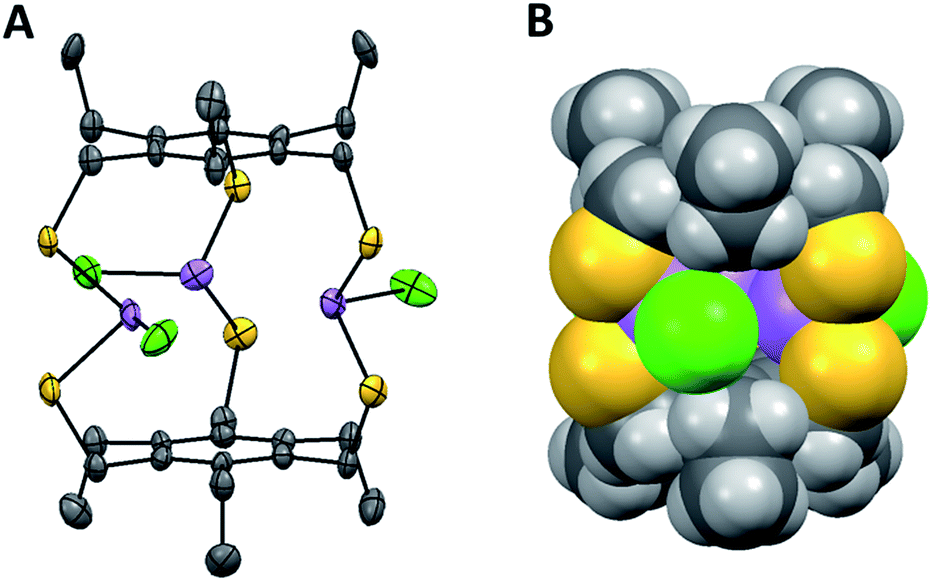 | ||
| Fig. 2 ORTEP representation (50% probability ellipsoids, hydrogen atoms and solvent molecules omitted for clarity) (A) and space filling representation (B) of the symmetric conformer of the As3LEt2Cl3 macrobicycle. Arsenic is shown in purple, chloride in green, sulfur in yellow, carbon in black and hydrogen in gray. | ||
| As3LH2Cl3 | As3LMe2Cl3 | As3LEt2Cl3 | Sb3LMe2Cl3 | Sb3LEt2Cl3 | |
|---|---|---|---|---|---|
| a Sb3LH2Cl3 not included due to significant disorder in structure. b Denotes an averaged value. c Value provided corresponds to shortest Pn⋯Caryl contact. | |||||
| Pn–S (Å)b | 2.209(9) | 2.197(9) | 2.217(2) | 2.400(4) | 2.403(2) |
| Pn⋯Pn (Å)b | 3.882(13) | 4.40(19) | 4.110(2) | 3.768(34) | 4.023(1) |
| S–Pn–S (deg)b | 88.1(9) | 88.4(3) | 85.73(7) | 88.2(2) | 81.92(2) |
| Pn⋯Caryl (Å)c | 3.169(8) | 3.211(8) | 3.269(6) | 3.223(7) | 3.386(5) |
1H-NMR characterization of As macrobicycles was performed in TCE-d2. The methylene protons in the structure are inequivalent, creating an AB splitting system due to geminal coupling. Interestingly, all As3L2Cl3 derivatives exhibit the presence of an unusual, asymmetric isomer that accompanies the dominant symmetric isomer (sym-As3LMe2Cl3 and asym-As3LMe2Cl3, Fig. 3). The asymmetric conformer exhibits a framework in which a single chloride atom is pointing in the opposite direction relative to the other As–Cl bonds. This results in inequivalent methyl groups giving rise to singlets at 2.58, 2.48, and 2.43 ppm in TCE-d2. A similar type of symmetric vs. asymmetric isomerism was previously observed in two-fold symmetric, naphthalene-based arsenic–thiolate As2L2Cl2 macrocycles, in which a single ligand “flips” causing the loss of C2 symmetry of the complex.25Fig. 4 shows the mixture of sym- and asym-As3LMe2Cl3 macrobicycles obtained from dissolving single crystals of As3LMe2Cl3 and shows the AB splitting pattern observed for the methylene protons (Ha in figure). From integrating the resonances of each structure, it was determined that the symmetric isomer is favoured over the asymmetric isomer by a 63:37 ratio. The relative stability of the two different isomers was also investigated with electronic structure calculations. MP2/LANL2DZ geometry optimizations revealed the symmetric, unsubstituted As macrobicycle to be 3.63 kcal mol−1 lower in energy than the asymmetric isomer (Fig. 3C and D).
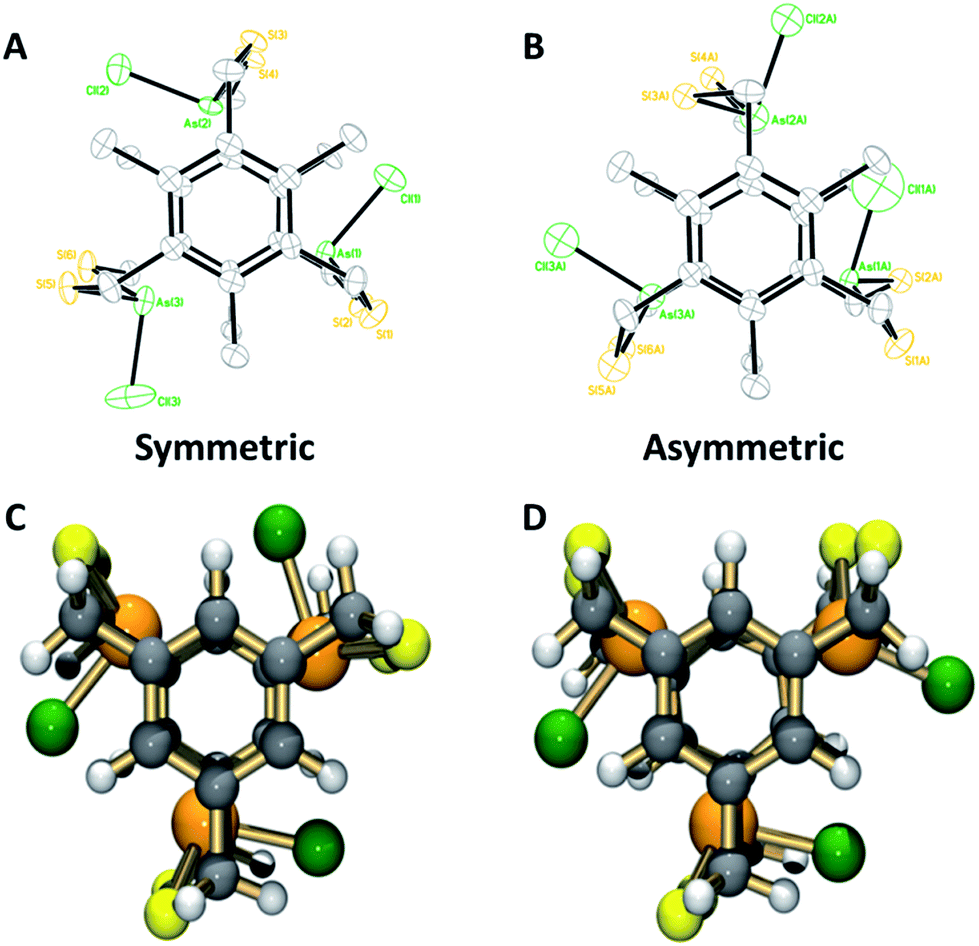 | ||
| Fig. 3 (Top) ORTEP representations comparing the symmetric and asymmetric conformations of (a) sym-As3LMe2Cl3 and (b) asym-As3LMe2Cl3 macrobicycles. (Bottom) Ball and stick representations of optimized geometries (MP2/LANL2DZ) of (c) sym-As3LH2Cl3, (d) asym-As3LH2Cl3 macrobicycles. | ||
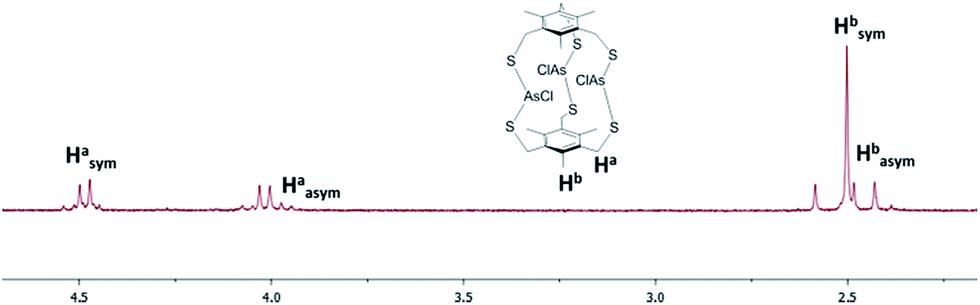 | ||
| Fig. 4 1H NMR spectrum for As3LMe2Cl3 macrobicycle. The signals from the symmetric and asymmetric macrobicycle are labeled. | ||
While a variation in the dithiolate ligand core appeared to dictate the isomer ratio in related studies on As2L2Cl2 macrocycles,26 in the case of the macrobicycles the substitution at the 2,4,6 position did not affect the sym:asym ratio between derivatives in solution as estimated by 1H NMR spectroscopy: 68:32 ([As3LH2Cl3]), 63:37 ([As3LMe2Cl3]), and 69:31 ([As3LEt2Cl3]). Given that the sym-isomers are favoured by this ∼2:1 ratio in solution, then at 25 °C these sym-isomers are ca. 0.4 kcal mol−1 more stable than the asym-isomers. The difference between the relative energy of the two isomers in solution versus that calculated in vacuum presumably reflects the influence of solvation. Additionally, 1H-NMR reveals a slow equilibrium between sym- and asym-As3L2Cl3: each isomer has a distinct NMR spectrum at room temperature, meaning the equilibration between the two species is slow on the NMR timescale. However, upon dissolving single crystals of As3L2Cl3 macrobicycles, the equilibrium mixture of sym- and asym-isomers is established within minutes, suggesting this equilibration is fast on the seconds to minutes timescale.
Structural characterization using single crystal X-ray diffraction is in agreement with solution state NMR studies, suggesting the symmetric conformers are again the dominant species. The preferred C3h-symmetric As3LMe2Cl3 conformer co-crystallizes with the Cs asymmetric complex in a 90:10 ratio (Fig. 3A and B). The opposing chloride in the asymmetric structure shows a significant amount of disorder. The crystal packing structures for ethyl- and methyl-substituted macrobicycles reveal a formation of columns in a herringbone fashion (Fig. S12 and S17†). Discernible stabilizing forces in each As-containing macrobicycle are intermolecular interactions seen in the form of short S⋯S contacts between 3.28–3.67 Å. In addition, a single benzene molecule co-crystallizes orthogonal to the 1D stacks in the crystal structure of the unsubstituted derivative, [As3LH2Cl3].
Effect of external additives and solvent dependence
Due to the low energy barrier of conversion from the symmetric macrobicycle to the asymmetric macrobicycle, the kinetic stability of the macrobicycles was tested by heating the solution to 120 °C over several days. Both -sym and -asym conformers were kinetically stable under these conditions and showed no apparent conversion from asym-As3L2Cl3 to the symmetric complex on the NMR timescale. Interestingly, the self-assembled isomerism was found to be strongly influenced by solvent effects as revealed by solution studies performed in deuterated chloroform. Reacting unsubstituted ligand H3LH with AsCl3 results in only partial conversion to the macrobicycle after more than a week in CDCl3; whereas 100% conversion to the macrobicycle in TCE-d2 is achieved in only one day, suggesting some unusual solvent effect is at play in TCE that facilitates macrobicycle formation. In benzene, insoluble kinetic intermediates rapidly precipitate from the reaction solution, resulting in limited conversion to macrobicycle. The degree of solubility for these oligomeric intermediates also appears to impact the formation of a single stereoisomer over another. This was most apparent in tetrahydrofuran where only the symmetric isomer is formed in solution; whereas, dichloromethane, benzene, and chloroform hinder the self-assembly of both sym/asym-macrobicycles. Supplementary to solvent effects, tetrabutylammonium tetrafluoroborate increases the relative rate of self-assembly in TCE giving complete conversion to As3LH2Cl3 in 8 minutes without subsequent precipitation of product seen with TBA+ chloride.19 This change in reactivity when subjected to an external agent is another unusual example of a self-assembly reaction driven by a small molecule that facilitates the organization process,27,28 and augurs well for the formation of even greater, more intricate self-assembled systems using related thiol ligands and main group ion building blocks.C 3h-Symmetric Sb3L2Cl3 macrobicycles appear to be more static in solution when compared to the arsenic-based congeners as only symmetric macrobicycles are present in TCE-d2 suggesting the asymmetric isomers are much more stable relatively. Similar to its As counterpart, the Sb3LH2Cl3-sym is only 4.46 kcal mol−1 lower in energy than the asymmetric conformer calculated via MP2/LANL2DZ. Single crystal X-ray diffraction of [Sb3LMe,Et2Cl3] reveal columnar packing in a herringbone pattern with short S⋯S contacts of 3.62 Å and intramolecular Sb⋯Caryl distances ranging between 3.24–3.39 Å (VDW radii is 3.85 Å for Sb). Similar to As macrobicycles, the sulfur–sulfur interaction poses as the prominent stabilizing factor in crystal packing. However, the solid state structure of Sb3LEt2Cl3 adopts more sulfur contacts between the layers of columns than [As3LEt2Cl3] (Fig. S26†). A similar trend in Pn⋯π interaction is observed, with the distance in this stabilizing force increasing with added steric bulk at the ligand core. Surprisingly, no evidence for asym-Sb3L2Cl3 conformers was observed in solution, although under certain conditions the asym-conformer does contribute to a minor disordered component in single crystal X-ray structures. These disordered crystals still lead to NMR spectra indicating only the presence of sym-Sb3L2Cl3 macrobicycles. This is in stark conflict with previous studies on dinuclear Pn2L3 cryptands, which revealed added stability in Sb (and Bi) complexes over the As derivatives.25
Conclusion
In conclusion, this is the first report of a series of self-assembled trinuclear As(III) and Sb(III) coordination complexes, and we report the syntheses of six such macrobicyclic assemblies. The As-derivatives exist in two different conformations: a slightly more stable C3h-symmetric structure and an asymmetric conformer that lacks the three-fold symmetry. Steric gearing at the tripodal core of the 1,3,5-trisubstituted benzene ring does not appear to influence the rate of self-assembly in solution nor the equilibrium between different macrobicyclic conformers, negating the presence of any apparent steric gearing effects during self-assembly and dynamic motion in solution. However, X-ray crystallography suggests pnictogen⋯πaryl interactions may act as a strong directing force in self-assembly complementary to steric gearing. Additionally, the self-assembly of these macrobicycles is sometimes enhanced and/or suppressed by different organic solvents as well as the addition of chloride and tetrafluoroborate salts.Acknowledgements
The authors thank the University of Oregon for partial support of this work. D.W.J. is a Scialog Fellow of Research Corporation for Science Advancement (Award ID 20317). We thank Dr Charlie Peck and the Earlham College Cluster Computing Group for providing computational resources used in this work (http://cluster.earlham.edu).Notes and references
- A. Schmidt, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS.
- R. A. Bilbeisi, J.-C. Olsen, L. J. Charbonnière and A. Trabolsi, Inorg. Chim. Acta, 2014, 417, 79–108 CrossRef CAS.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- D. L. Caulder and K. N. Raymond, J. Chem. Soc., Dalton Trans., 1999, 1185–1200 RSC.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354–1379 RSC.
- M. M. Watt, M. S. Collins and D. W. Johnson, Acc. Chem. Res., 2013, 46, 955–966 CrossRef CAS PubMed.
- N. R. Lindquist, T. G. Carter, V. M. Cangelosi, L. N. Zakharov and D. W. Johnson, Chem. Commun., 2010, 46, 3505–3507 RSC.
- M. E. Carnes, N. R. Lindquist and D. W. Johnson, Main Group Supramolecular Coordination Chemistry in Encyclopedia of Supramolecular Chemistry, ed. J. L. Atwood, J. W. Steed and K. J. Wallace, Taylor and Francis, New York, 2012 Search PubMed.
- M. A. Pitt and D. W. Johnson, Chem. Soc. Rev., 2007, 36, 1441–1453 RSC.
- H. Schmidbaur and A. Schier, Organometallics, 2008, 27, 2361–2395 CrossRef CAS.
- J. Zukerman-Schpector, A. Otero-de-la-Roza, V. Luaña and E. R. T. Tiekink, Chem. Commun., 2011, 47, 7608–7610 RSC.
- V. M. Cangelosi, L. N. Zakharov, S. A. Fontenot, M. A. Pitt and D. W. Johnson, Dalton Trans., 2008, 3447–3453 RSC.
- V. M. Cangelosi, L. N. Zakharov, J. L. Crossland, B. C. Franklin and D. W. Johnson, Cryst. Growth Des., 2010, 10, 1471–1473 CAS.
- M. A. Pitt, L. N. Zakharov, K. Vanka, W. H. Thompson, B. B. Laird and D. W. Johnson, Chem. Commun., 2008, 3936–3938 RSC.
- V. M. Cangelosi, M. A. Pitt, W. J. Vickaryous, C. A. Allen, L. N. Zakharov and D. W. Johnson, Cryst. Growth Des., 2010, 10, 3531–3536 CAS.
- D. J. Iverson, G. Hunter, J. F. Blount, J. R. Damewood and K. Mislow, J. Am. Chem. Soc., 1981, 103, 6073–6083 CrossRef CAS.
- G. Hennrich and E. V. Anslyn, Chem.–Eur. J., 2002, 8, 2218 CrossRef CAS.
- M. E. Carnes, M. S. Collins, N. R. Lindquist, E. Guzmán-Percástegui, M. D. Pluth and D. W. Johnson, Chem. Commun., 2014, 50, 73–75 RSC.
- W. J. Vickaryous, L. N. Zakharov and D. W. Johnson, Main Group Chem., 2006, 5, 51–59 CrossRef CAS.
- X. Wang and F. Hof, Beilstein J. Org. Chem., 2012, 8, 1–10 CrossRef CAS PubMed.
- T. Probst, O. Steigelmann, J. Riede and H. Schmidbaur, Chem. Ber., 1991, 124, 1089–1093 CrossRef CAS.
- H. Schmidbaur, W. Bublak, B. Huber and G. Müller, Angew. Chem., Int. Ed. Engl., 1987, 26, 234–236 CrossRef.
- H. Schmidbaur, R. Nowak, O. Steigelmann and G. Müller, Chem. Ber., 1990, 123, 1221–1226 CrossRef CAS.
- V. M. Cangelosi, T. G. Carter, J. L. Crossland, L. N. Zakharov and D. W. Johnson, Inorg. Chem., 2010, 49, 9985–9992 CrossRef CAS PubMed.
- V. M. Cangelosi, A. C. Sather, L. N. Zakharov, O. B. Berryman and D. W. Johnson, Inorg. Chem., 2007, 46, 9278–9284 CrossRef CAS PubMed.
- T. Hasell, X. Wu, J. T. A. Jones, J. Bacsa, A. Steiner, T. Mitra, A. Trewin, D. J. Adams and A. I. Cooper, Nat. Chem., 2010, 2, 750–755 CrossRef CAS PubMed.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; NMR spectra; crystallographic information; and computational details, absolute energies and atomic coordinates of the discussed macrobicycles. CCDC 1041514–1041519. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc04042b |
| This journal is © The Royal Society of Chemistry 2015 |