
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold(I)-catalyzed [2 + 2 + 2] cycloaddition of allenamides, alkenes and aldehydes: a straightforward approach to tetrahydropyrans†
Hélio
Faustino‡
a,
Iván
Varela‡
a,
José L.
Mascareñas
*a and
Fernando
López
*ab
aCentro Singular de Investigación en Química Biológica y Materiales Moleculares (CIQUS) and Departamento de Química Orgánica, Universidad de Santiago de Compostela, C/ Jenaro de la Fuente s/n, 15782, Santiago de Compostela, Spain. E-mail: joseluis.mascarenas@usc.es; fernando.lopez@csic.es
bInstituto de Química Orgánica General (CSIC), Juan de la Cierva 3, 28006, Madrid, Spain
First published on 2nd March 2015
Abstract
Allenamides participate as two-carbon components in an intermolecular [2 + 2 + 2] cycloaddition with alkenes and aldehydes when treated with catalytic amounts of a phosphite gold complex. The reaction is highly regio- and chemoselective, and works with different types of alkenes, including styrenes, enol ethers or enamides, as well as with aromatic and aliphatic aldehydes. Accordingly, different types of 2,6-disubstituted tetrahydropyrans can be stereoselectively assembled in a single step from commercial or very accessible starting materials.
Introduction
Transition metal catalyzed [2 + 2 + 2] cycloadditions constitute one of the most attractive methodologies for the construction of six-membered cyclic systems.1 Despite the significant achievements reported in this field, intermolecular examples involving three different cycloaddition partners are extremely scarce, most probably because of the chemo- and regioselectivity issues associated with these multicomponent annulations.2 The few examples reported so far involve the use of Rh, Ru, Nb or Ni catalysts and at least one alkyne as cycloaddition component.2 Curiously, and despite the fact that gold catalysis has proven to be very efficient for unveiling novel types of cycloadditions,3 fully intermolecular [2 + 2 + 2] examples are almost unknown4 and, to the best of our knowledge, those of three different two-atom components are unprecedented.5Herein, we are pleased to report a fully intermolecular gold-catalyzed [2 + 2 + 2] cycloaddition involving three different π-unsaturated components, namely an allene, an alkene and an aldehyde. The reaction takes place with excellent chemo- and regioselectivity and provides a straightforward and atom-economical entry to tetrahydropyrans (THPs). THPs, and in particular their 2,6-disubstituted counterparts, are privileged scaffolds that are present in a myriad of biologically active molecules (Fig. 1).6 Although many elegant methods have been developed to construct these motifs,6,7 none of them encompass the coupling of three readily available components in a single catalytic annulation step.8
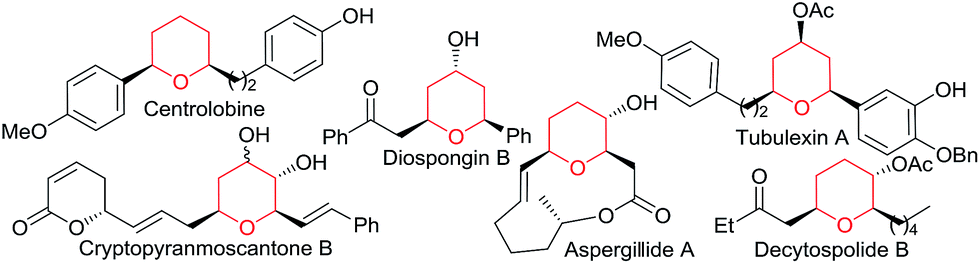 | ||
| Fig. 1 Tetrahydropyran frameworks in biologically active products. | ||
Over the past few years, we have developed different types of Au-catalyzed annulations,9 including a cycloaddition between allenamides and oxoalkenes that affords oxabridged medium-sized carbocycles (Scheme 1, eqn (1)).10,11 This annulation was proposed to proceed through the intermediate I,12 which evolves to the product by the sequential formation of species II and III. On this basis, we then wondered whether it would be possible to achieve an annulation between the allenamide, alkene and carbonyl units in a fully intermolecular way, a process that would directly afford 2,6-disubstituted THPs like 4 (Scheme 1, eqn (2)). Despite the fact that the process could be viewed as an intermolecular version of the previous annulation, the timely assembly of three different components in a programmed manner is extremely challenging. Indeed, the feasibility of the reaction could be seriously compromised since more simple [2 + 2] adducts of type 5 and 6,9c acyclic products like 7, or alternative [2 + 2 + 2] adducts (8/9) could be likewise expected.13
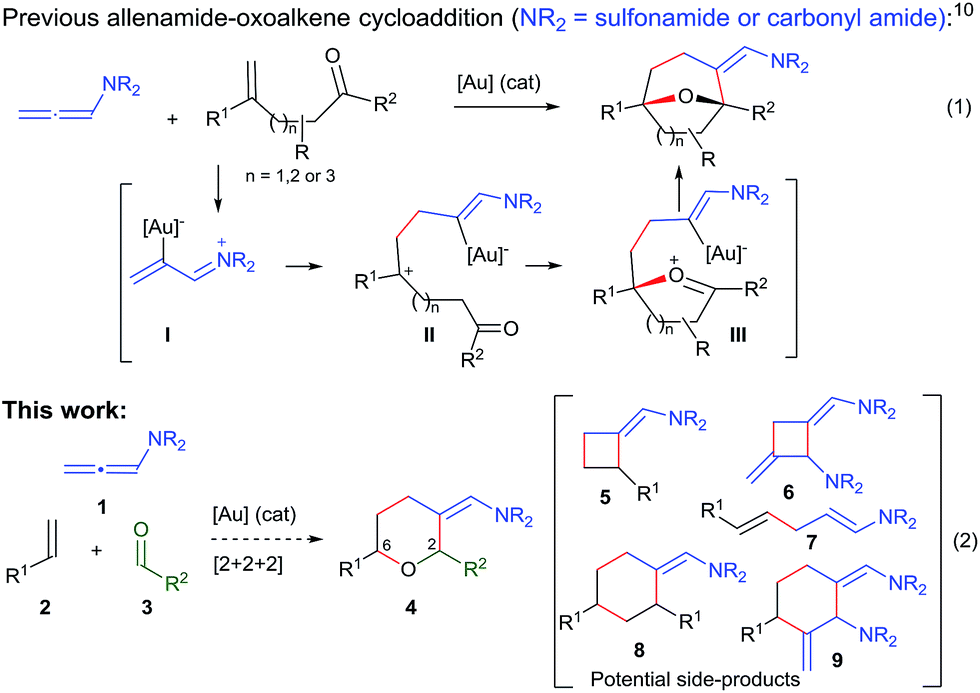 | ||
| Scheme 1 Previous gold-catalyzed cascade cycloadditions and current proposal. | ||
Results and discussion
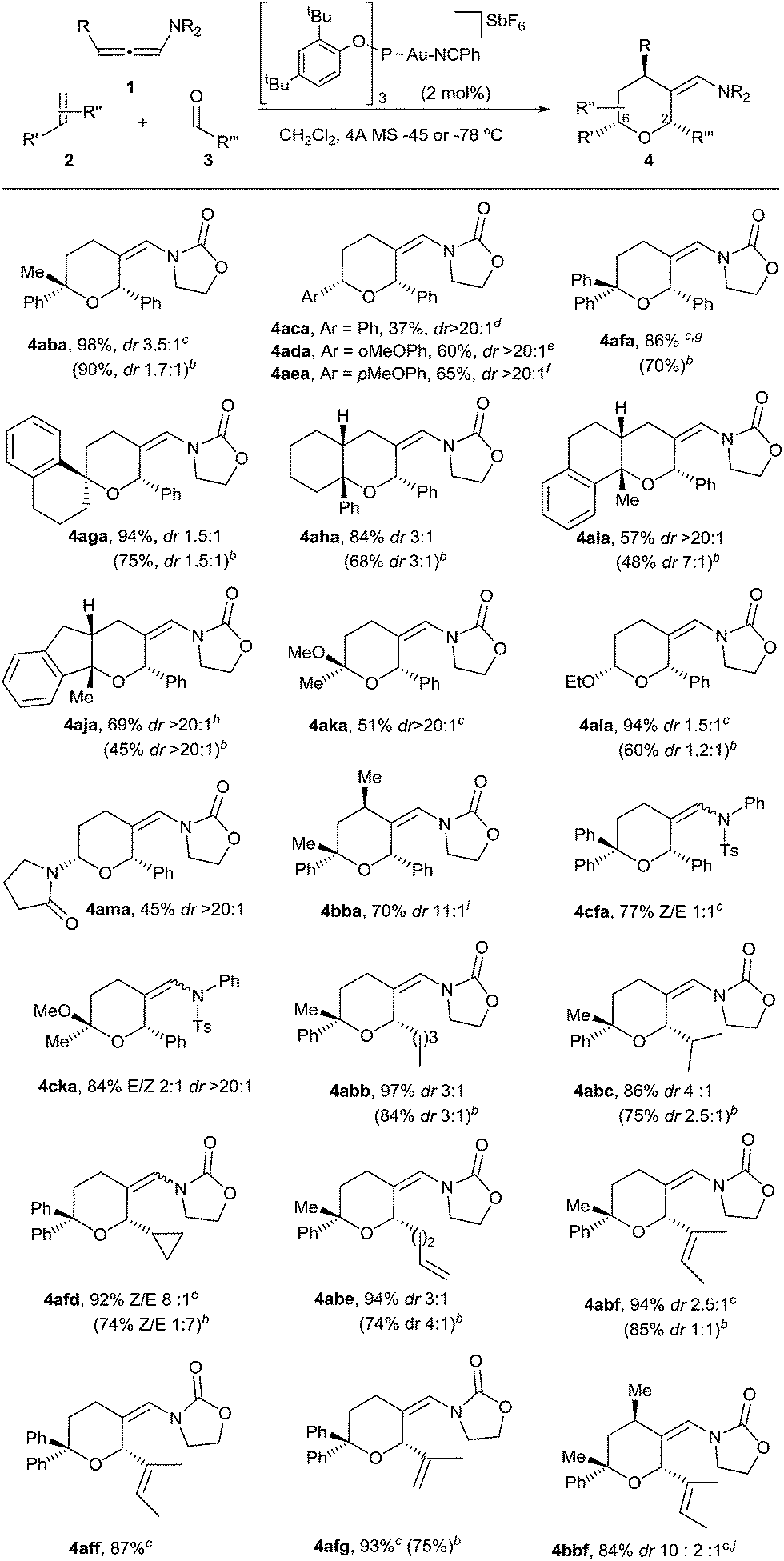
We began our studies by analyzing the reactivity of allenamide 1a with (E)-β-methylstyrene (2a) and benzaldehyde (3a) (Table 1). Initial assays confirmed the expected difficulties for controlling the chemoselectivity of the process. Indeed, despite using an excess of the aldehyde (10 equiv.), and adding the allenamide over 2 hours, the gold complex Au1 induced the formation of the [2 + 2] allenamide dimerization adduct 6a in 44% yield, together with a minor amount of the cyclobutane 5aa,9c resulting from the [2 + 2] cycloaddition between 1a and 2a (entry 1). A [2 + 2 + 2] adduct, eventually identified as the 2,6-cis THP 4aaa, was also detected, but only in trace amounts. Similarly, other frequently used gold catalysts such as Ph3PAuNTf2 or the NHC–gold complex Au2 provided very low yields of the [2 + 2 + 2] adduct 4aaa (entries 2 and 3), with poor mass recovery balances in all these cases. Interestingly, when using the phosphite-gold complex Au3, we observed a significant increase in the global yield of the reaction, which provided 5aa in 60% yield along with the [2 + 2 + 2] adduct 4aaa in 21% yield (entry 4). This last yield could be further improved up to 35% by carrying out the reaction at −45 °C (entry 5).14|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | [Au] (mol%) | 2 | R1 | R2 | Conv. | 4 (%) | 5 (%) | 6 (%) |
a
1a (1 equiv.) added over 2 h to a solution of 2 (2 equiv.), 3a (10 equiv.), [Au] (X mol%) and 4 Å MS, in CH2Cl2 at −15 °C, unless otherwise noted.
b Conversion of 1a and yields of 4–6 determined by 1H-NMR of the crude mixture using 1,3,5-(MeO)3C6H3 as internal standard (IS).
c Carried out at −45 °C, (1 h).
d Overall yield for the mixture of 2,6-cis (4aba) and trans (4aba′); dr = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The major isomer is that drawn.
e
1a added in one portion.
f Overall yield. dr 1.5:1.
g Carried out at −78 °C, (1 h).
h 90% overall isolated yield, dr 3.5:1 (4aba:4aba′).
i Carried out in F3C–Ph at −25 °C (4 h).
j 86% overall isolated yield, dr 4.5:1. :1. The major isomer is that drawn.
e
1a added in one portion.
f Overall yield. dr 1.5:1.
g Carried out at −78 °C, (1 h).
h 90% overall isolated yield, dr 3.5:1 (4aba:4aba′).
i Carried out in F3C–Ph at −25 °C (4 h).
j 86% overall isolated yield, dr 4.5:1. 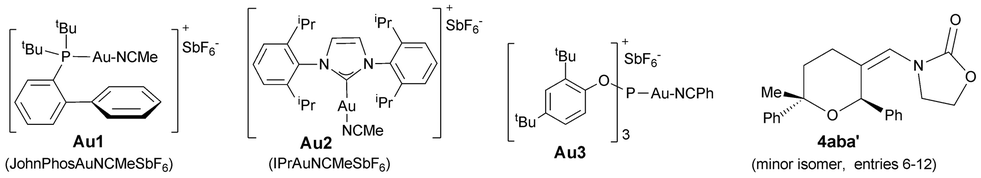
|
||||||||
| 1 | Au1 (5%) | 2a | H | Me | 99% | 4aaa, 2 | 5aa, 4 | 6a, 44 |
| 2 | Ph3PAuNTf2 (5%) | 2a | H | Me | 60% | 4aaa, 2 | 5aa, 0 | 6a, 7 |
| 3 | Au2 (5%) | 2a | H | Me | 99% | 4aaa, 15 | 5aa, 7 | 6a, 22 |
| 4 | Au3 (2%) | 2a | H | Me | 99% | 4aaa, 21 | 5aa, 60 | 6a, 8 |
| 5c | Au3 (2%) | 2a | H | Me | 99% | 4aaa, 35 | 5aa, 37 | — |
| 6 | Au3 (2%) | 2b | Me | H | 99% | 4aba, 98d | — | — |
| 7e | Au3 (2%) | 2b | Me | H | 99% | 4aba, 99d | — | — |
| 8 | Au1 (2%) | 2b | Me | H | 99% | 4aba, 51d | 5ab, 17 | — |
| 9 | Ph3PAuNTf2 (2%) | 2b | Me | H | 99% | 4aba, 77f | 5ab, 14 | — |
| 10 | Au2 (2%) | 2b | Me | H | 99% | 4aba, 80d | 5ab, 6 | — |
| 11e,g | Au3 (2%) | 2b | Me | H | 99% | 4aba, 98h | — | — |
| 12e,i | Au3 (2%) | 2b | Me | H | 99% | 4aba, 98j | — | — |
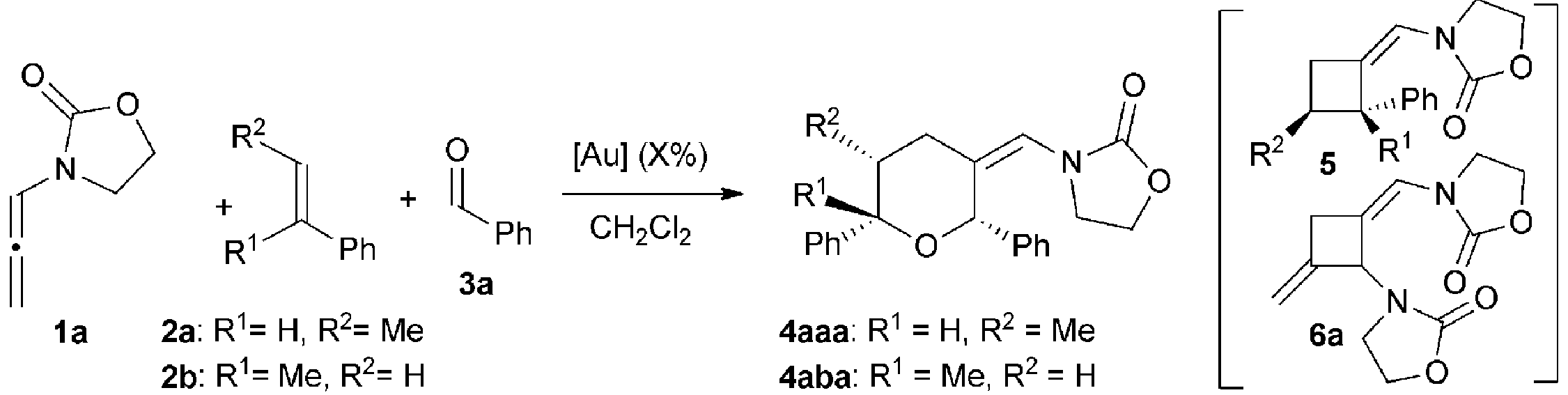
At this point, we envisioned that an additional stabilization of the putative carbocationic species of type II, resulting from the addition of the alkene to intermediate I (Scheme 1), could eventually facilitate its intermolecular capture by the aldehyde.
In consonance with this hypothesis, we were pleased to find that the use of α-methylstyrene (2b) instead of β-methylstyrene (2a) provided, under otherwise identical conditions, the desired THP in an excellent 98% yield, as a 2:1 mixture of 2,6-cis (4aba) and 2,6-trans (4aba′) diastereoisomers (entry 6).15 The same result was obtained when 1a was added in one portion (entry 7). Gold catalysts such as JohnPhosAuNCMeSbF6 (Au1), Ph3PAuNTf2 or IPrAuNCMeSbF6 (Au2), also provided the desired [2 + 2 + 2] cycloadduct 4aba as the major adduct; however, yields and chemoselectivities were significantly lower than those obtained with the phosphite–gold catalyst Au3 (entry 7 vs. 8–10). Moreover, with this latter catalyst the diastereoselectivity could be improved by either performing the reaction at −78 °C (dr 3.5:1, 90% isolated yield, entry 11) or by using α,α,α-trifluorotoluene as solvent (dr 4.5:1, 86% yield, entry 12).
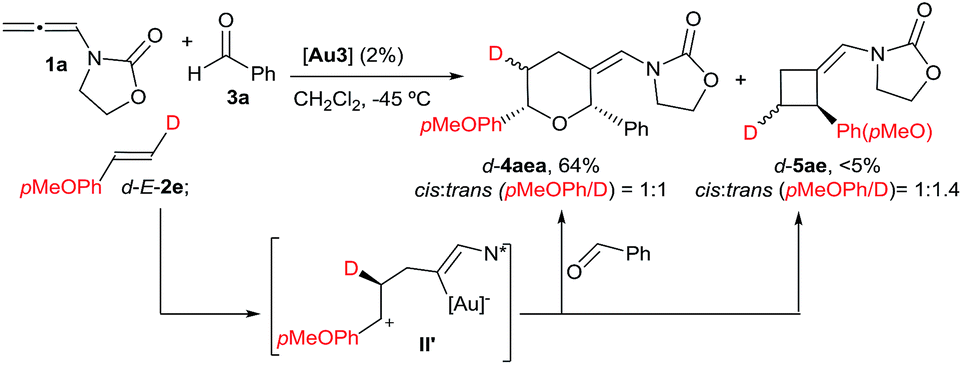
With these results in hand, we next analyzed the scope of the process (Table 2). In consonance with the performance of β-methylstyrene (2a, Table 1, entry 5), the cycloaddition of styrene (2c) with 1a and benzaldehyde provided the desired 2,6-disubstituted THP (4aca) in a moderate 37% yield, but with complete 2,6-cis selectivity (5ac was also isolated in 45% yield). Gratifyingly, use of styrenes with electron-donating groups (e.g. p-MeO or o-MeO) allowed significant improvement of the chemoselectivity, so the corresponding THPs, 4ada and 4aea, were isolated in good yields (60–65% yield) and with complete 2,6-cis diastereoselectivity.
a
1 (1 equiv.) added to a solution of 2 (2 equiv.), aldehyde (10 equiv.), [Au3] (2 mol%) and 4 Å MS, in CH2Cl2 at −45 °C, unless otherwise noted. Conversions >99% (1H-NMR). When a mixture of 2,6-isomers is formed, the major is that drawn.
b Carried out at −45 °C with a 1/2/3 molar ratio of 1/1.25/2.
c Carried out at −78 °C.
d 45% of 5ac was also isolated.
e 21% of 5ad was also isolated.
f Traces of 5ae and 8ae (5% yield) were also isolated.
g Traces of 7af (5% yield) were also isolated.
h Traces of 7aj (5% yield) were also isolated.
i 17% yield of 5bb was also isolated.
j For the structure of the minor isomers, see the ESI. 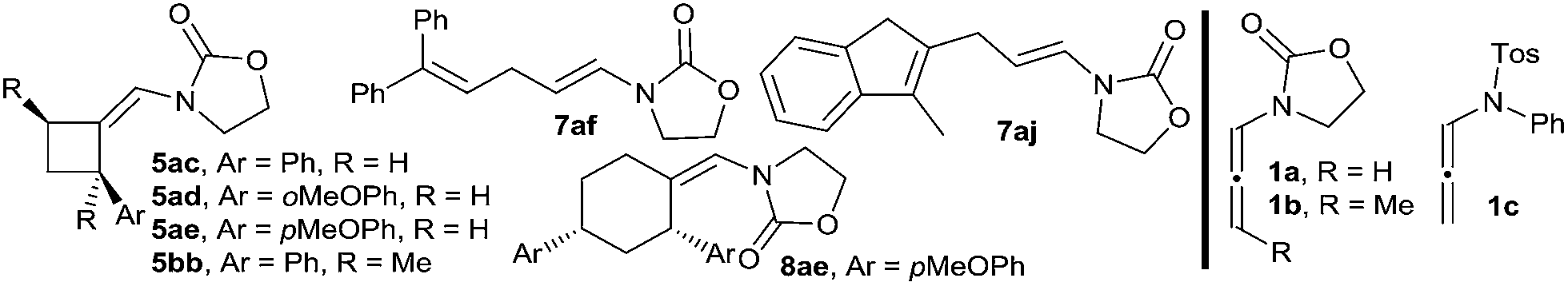
|
|---|
|
On the other hand, the cycloaddition with α-phenylstyrene provided the desired THP (4afa) in an excellent 86% yield, whereas the use of exo-methylenes such as 1-methylene-tetrahydronaphthalene allowed an efficient access to spirotetrahydropyran derivatives like 4aga, which was isolated in an excellent 94% yield (dr 1.5:1).16
Remarkably, cyclic alkene derivatives were also excellent partners for this process. Thus, the cycloadditions of allenamide 1a and benzaldehyde with 1-phenylcyclohexene, 4-methyl-1,2-dihydronaphthalene or 3-methyl-1H-indene provided the corresponding THPs (4aha–4aja) in good yields (57–84% yield) and moderate (4aha) or complete (4aia–aja) stereoselectivity.17 X-ray analysis of crystals of 4aha and 4aia unambiguously confirmed their structures and relative stereochemistry (Fig. 2).14
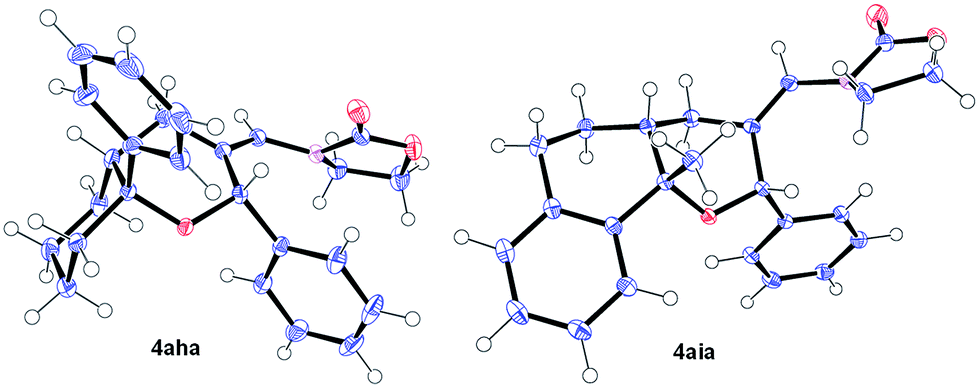 | ||
| Fig. 2 X-ray structures of 4aha (left, major isomer) and 4aia (right).14 | ||
We next explored the use of alternative electron-rich alkenes. Gratifyingly, the cycloaddition could also be performed with enol ethers such as 2-methoxyprop-1-ene or ethoxyethene, to obtain the corresponding cyclic acetals (4aka–4ala) with moderate to good yields. Similarly, the cycloaddition between 1a, 3a and 1-vinylpyrrolidin-2-one was also feasible, providing the cyclic hemiaminal ether 4ama in 45% yield and with complete diastereoselectivity.
These annulations are also feasible with other allenamides. For instance, the reaction of γ-methyl-substituted allenamide 1b (see Table 2, footnote) with α-methylstyrene and benzaldehyde provided the [2 + 2 + 2] adduct 4bba, featuring three new stereogenic centers, in 70% yield and with excellent diastereoselectivity (dr 11:1).18 On the other hand, N-tosylphenyl allenamides such as 1c were also suitable partners. Thus, the [2 + 2 + 2] adduct 4cfa, resulting from the cycloaddition of 1c, benzaldehyde and α-phenylstyrene was obtained in 77% yield, whereas the adduct 4cka, from 2-methoxyprop-1-ene, was obtained in 84% yield and, importantly, with complete stereoselectivity.
Remarkably, the scope of the method is not limited to benzaldehyde. Indeed, the reaction of α-methylstyrene, allenamide 1a and an aliphatic aldehyde such as pentanal led to the desired adduct, 4abb, in 97% yield (dr 3:1). Other aldehydes such as isobutyraldehyde, cyclopropanecarbaldehyde or pent-4-enal also gave the THPs 4abc–4abe in excellent yields. α,β-Unsaturated aldehydes such as 2-methylbut-2-enal or methacrolein also participated in the annulation yielding the desired THPs (4abf, 4aff, 4afg) in yields above 90%. Moreover, the cycloaddition of the γ-substituted allenamide 1b with an aliphatic aldehyde such as 2-methylbut-2-enal was also feasible, providing 4bbf in 84% yield (dr 10:2:1).14
Overall, it is important to highlight that the current method constitutes one of the very few catalytic approaches that affords THPs featuring fully substituted carbons at the oxygen-adjacent position (e.g. C6).19 On the other hand, while the above reactions were carried out using a relatively large excess of the aldehyde, gratifyingly, we found that in most of the cases the reaction can be efficiently performed using an allenamide (1)/alkene (2)/aldehyde (3) molar ratio of 1/1.2/2 (Table 2, footnote b, results in parentheses). Thus, using these conditions, THPs 4aba, 4afa, 4aga, 4ala, 4abb, 4abc, 4afd, 4abe, 4abf or 4afg were obtained in yields varying from 60% to 90% (Table 2).20 Additionally, more complex polycyclic systems like 4aha–4aja could also be obtained in yields from 45% to 68%.14
We next explored some manipulations of the exo-enamide moiety of the products (Scheme 2). Thus, THPs the like 4afd or 4aea can be dihydroxylated to afford the α-hydroxo aldehydes 11 and 12 in excellent yields and with very good or complete diastereoselectivity (Scheme 2, eqn (1)). Moreover, both types of enamides (e.g.4afd and 4cfa) could be easily converted into their corresponding ketones upon ozonolysis (Scheme 2, eqn (2)).
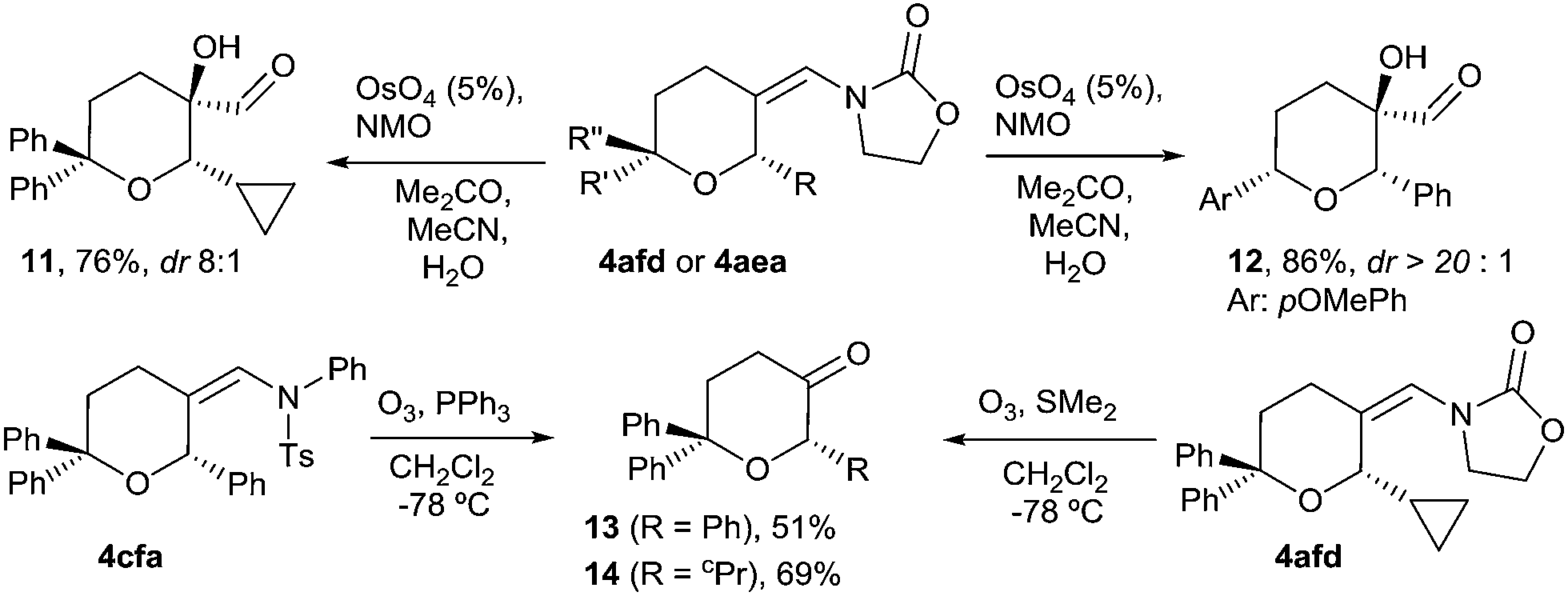 | ||
| Scheme 2 Functionalization of the exo-enamide moiety. | ||
With regard to the mechanism of the annulation, the general proposal indicated in Scheme 1 could also apply for this intermolecular process; however, we found some results that were indicative of a more complex scenario. In particular, it is curious that while the [2 + 2] product (5aa) obtained from 1a and E-β-methylstyrene retains the trans configuration of the alkene, the [2 + 2 + 2] adduct 4aaa displays these groups in a cis disposition (Table 1, entry 5). On the contrary, polycyclic [2 + 2 + 2] adducts like 4aha, 4aia or 4aja retained the configuration of the parent alkene. To shed light on this divergence, we carried out the cycloaddition of 1a and pentanal with the trans-deuterated styrene d-E-2c (Scheme 3). As expected, the reaction provided a mixture of the [2 + 2 + 2] and [2 + 2] adducts d-4acb (38% yield) and d-5ac (43% yield), respectively. Interestingly, d-5ac incorporates the Ph and the deuterium atom in a trans disposition, whereas the [2 + 2 + 2] adduct, d-4acb, holds these groups in a cis arrangement. These results strongly suggest the formation of an intermediate of type II that preserves the stereochemical information of the alkene due to an stabilizing electrostatic interaction between the gold atom and the benzylic carbocation (Scheme 3).21 A subsequent nucleophilic anti attack of the carbonyl moiety would lead to intermediate III and, eventually, to the product d-4acb. The preferential formation of this THP with the C2 and C6 substituents in cis is in agreement with a transition state that places these groups in equatorial disposition (Prins-like cyclization from III to 4). On the other hand, if species II collapses to render a [2 + 2] adduct, the Ph and the D atom would retain the initial trans arrangement, as observed in d-5ac.
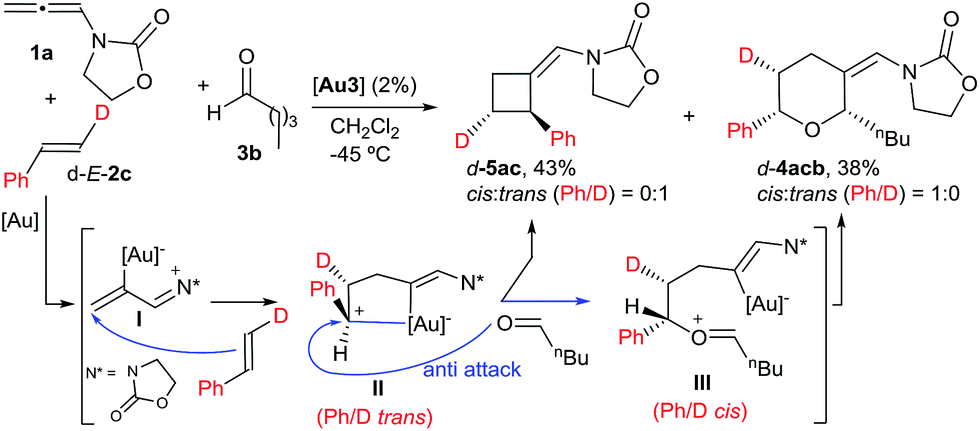 | ||
| Scheme 3 Cycloaddition of d-E-2c and the proposed key intermediate II. | ||
We also analysed the cycloaddition with deuterated α-methylstyrene (d-2b) as a model for α-substituted alkenes (Scheme 4). Curiously, the expected [2 + 2 + 2] isomeric adducts d-4abb and d-4abb′ were obtained as mixtures of cis/trans (Ph/D) isomers. Accordingly, an acyclic carbocation species like II′ or, alternatively, a fast equilibrium between the Ph/D-trans and cis intermediates II and II′′, could account for this result.22,23 Considering this proposal, the exclusive formation of the cis-fused polycyclic THPs 4aha–4aja from cyclic alkene precursors can be also understood.
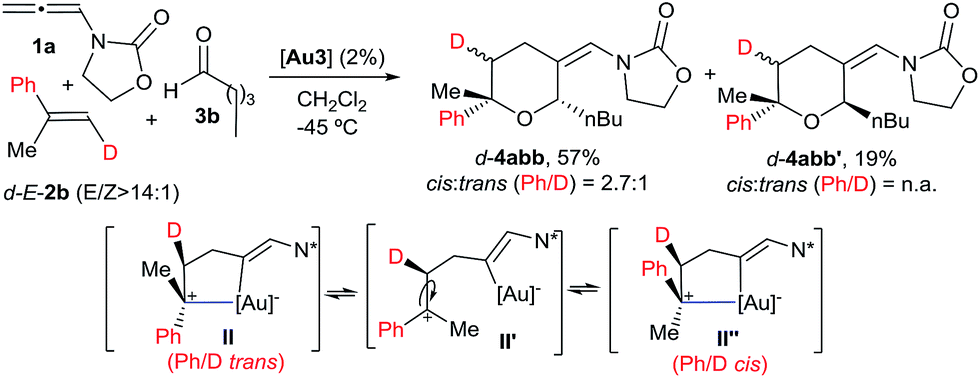 | ||
| Scheme 4 Cycloaddition of d-E-2b and the proposed key intermediate II′. | ||
Finally, we carried out the above cycloadditions of Schemes 3 and 4 using the NHC–gold catalyst Au2, instead of Au3. Not unexpectedly, lower chemoselectivities and yields of the corresponding [2 + 2 + 2] adducts were obtained in both cases but, interestingly, the stereochemistry of each deuterated cycloadduct (d-4acb, d-5ac and d-4abb), turned out to be identical to that obtained with Au3.14 Thus, the σ-donor or π-acceptor characteristics of the ligand at the gold atom do not seem to significantly affect the nature of the intermediate of type II.
Conclusions
In summary, we have developed a gold-catalyzed fully intermolecular [2 + 2 + 2] cycloaddition that constitutes one of the few transition metal catalyzed annulations involving three different π-unsaturated components. The process shows a broad scope with regard to the alkenes and aldehydes that can be used, and provides an efficient, atom-economical and stereoselective access to a variety of 2,6-disubstituted THPs from easily accessible or even commercially available materials.Acknowledgements
This work was supported by the Spanish MINECO (SAF2013-41943-R, SAF2010-20822-C02), the ERDF, the European Research Council (Adv. Grant no. 340055) and the Xunta de Galicia (GRC2013-041). HF acknowledges the Fundação para a Ciência e Tecnologia (Portugal) and POPH/FSE for a PhD grant (SFRH/BD/60214/2009).Notes and references
- (a) D. L. J. Broere and E. Ruijter, Synthesis, 2012, 44, 2639 CrossRef CAS PubMed; (b) G. Dominguez and J. Pérez-Castells, Chem. Soc. Rev., 2011, 40, 3430 RSC.
- (a) J. Hara, M. Ishida, M. Kobayashi, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2014, 53, 2956 CrossRef CAS PubMed; (b) M. Kobayashi, T. Suda, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2011, 50, 1664 CrossRef CAS PubMed; (c) Y. Satoh and Y. Obora, Org. Lett., 2011, 13, 2568 CrossRef CAS PubMed; (d) S. Ogoshi, A. Nishimura and M. Ohashi, Org. Lett., 2010, 12, 3450 CrossRef CAS PubMed; (e) N. Mori, S. Ikeda and Y. Sato, J. Am. Chem. Soc., 1999, 121, 2722 CrossRef CAS.
- For selected recent reviews, see (a) D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462 CrossRef CAS; (b) I. D. G. Watson and F. D. Toste, Chem. Sci., 2012, 3, 2899 RSC; (c) F. López and J. L. Mascareñas, Beilstein J. Org. Chem., 2011, 7, 1075 CrossRef PubMed; (d) G. Abbiati and E. Rossi, Beilstein J. Org. Chem., 2014, 10, 481 CrossRef CAS PubMed.
- For fully intermolecular (trimolecular) Au-catalyzed [2 + 2 + 2] cycloadditions of two different components, see (a) S. N. Karad and R.-S. Liu, Angew. Chem., Int. Ed., 2014, 53, 9072 CrossRef CAS PubMed; (b) R. B. Dateer, B. S. Shaibu and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 113 CrossRef CAS PubMed.
- For selected examples of partially intermolecular (bimolecular) Au-catalyzed [2 + 2 + 2] cycloadditions of three different components, see (a) C. Obradors and A. M. Echavarren, Chem.–Eur. J., 2013, 19, 3547 CrossRef CAS PubMed; (b) D. B. Huple and R.-S. Liu, Chem. Commun., 2012, 48, 10975 RSC; (c) M. Schelwies, R. Moser, A. L. Dempwolff, F. Rominger and G. Helmchen, Chem.–Eur. J., 2009, 15, 10888 CrossRef CAS PubMed; (d) T.-M. Teng and R.-S. Liu, J. Am. Chem. Soc., 2010, 132, 9298 CrossRef CAS PubMed . For a review highlighting the challenges of Au-catalyzed intermolecular annulations, see: ; (e) M. E. Muratore, A. Homs, C. Obradors and A. M. Echavarren, Chem.–Asian J., 2014, 9, 3066 CrossRef CAS PubMed.
- For recent reviews, see (a) M. A. Perry, S. D. Rychnovsky and N. Sizemore, Synthesis of Saturated Tetrahydropyrans, in Synthesis of Saturated Oxygenated Heterocycles, ed. J. Cossy, Topics in Heterocyclic Chemistry, Springer-Verlag, Berlin, 2014 Search PubMed; (b) X. Han, G. Peh and P. E. Floreancig, Eur. J. Org. Chem., 2013, 1193 CrossRef CAS.
- For selected recent methods for THP synthesis, see (a) Y. Xie and P. E. Floreancig, Angew. Chem., Int. Ed., 2014, 53, 4926 CrossRef CAS PubMed; (b) J. Zeng, Y. J. Tan, J. Ma, M. L. Leow, D. Tirtorahardjo and X. W. Liu, Chem.–Eur. J., 2014, 20, 405 CrossRef CAS PubMed; (c) I. Shin, G. Wang and M. J. Krische, Chem.–Eur. J., 2014, 20, 13382 CrossRef CAS PubMed.
- For a tandem [[2 + 2] + 2] cycloaddition, see: A. T. Parsons and J. S. Johnson, J. Am. Chem. Soc., 2009, 131, 14202 CrossRef CAS PubMed.
- (a) F. López and J. L. Mascareñas, Chem. Soc. Rev., 2014, 43, 2904 RSC; (b) F. López and J. L. Mascareñas, Beilstein J. Org. Chem., 2013, 9, 2250 CrossRef PubMed; (c) H. Faustino, P. Bernal, L. Castedo, F. López and J. L. Mascareñas, Adv. Synth. Catal., 2012, 354, 1658 CrossRef CAS; (d) H. Faustino, F. López, L. Castedo and J. L. Mascareñas, Chem. Sci., 2011, 2, 633 RSC; (e) I. Alonso, H. Faustino, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2011, 50, 11496 CrossRef CAS PubMed.
- H. Faustino, I. Alonso, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2013, 52, 6526 CrossRef CAS PubMed.
- For a review on allenamides, see: T. Lu, Z. Lu, Z. X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862 CrossRef CAS PubMed.
- (a) S. Montserrat, H. Faustino, A. Lledós, J. L. Mascareñas, F. López and G. Ujaque, Chem.–Eur. J., 2013, 19, 15248 CrossRef CAS PubMed . See also: ; (b) Y. Horino, Y. Takata, K. Hashimoto, S. Kuroda, M. Kimura and Y. Tamaru, Org. Biomol. Chem., 2008, 6, 4105 RSC; (c) M. C. Kimber, Org. Lett., 2010, 12, 1128 CrossRef CAS PubMed.
- (a) [2 + 2] adducts of type 5 and 6 were previously reported, see ref. 9c; For addition products related to 7, see ; (b) A. W. Hill, M. R. Elsegood and M. C. Kimber, J. Org. Chem., 2010, 75, 5406 CrossRef CAS PubMed.
- See the ESI† for further details.
- (a) The [2 + 2] adducts 5ab, 6a, or other side-products, were not detected in the crude mixture (1H-NMR); ; (b) The structure and relative stereochemistry of both THP isomers (4aba/4aba′) were established by NMR and, additionally, those of the major isomer (4aba), with the Ph groups in cis, were further confirmed by X-ray ESI.†14.
- When a diastereoisomeric mixture is formed in the reactions of Table 2, the 2,6-cis and trans isomers could usually be separated by standard chromatography. ESI.†.
- The ring fusion was exclusively cis in all these polycyclic systems. Therefore, dr refers to the substituents at the 2,6-THP positions.
- An α-alkyl-substituted allenamides such as 3-(buta-2,3-dien-2-yl)oxazolidin-2-one provided a complex mixture of products.
- (a) Previously reported approaches are essentially limited to the formation of THPs with monosubstituted C2 or C6 carbons,6a something that significantly facilitates a high stereoselection; For isolated catalytic examples yielding products with fully substituted-C2 or C6 carbons, see: ; (b) M. Jacolot, M. Jean, N. Levoin and P. van de Weghe, Org. Lett., 2012, 14, 58 CrossRef CAS PubMed; (c) M. P. Castaldi, D. M. Troast and J. A. Porco Jr, Org. Lett., 2009, 11, 3362 CrossRef CAS PubMed; (d) J. S. Yadav, B. V. Subba Reddy, G. G. K. S. Narayana Kumar and S. Aravind, Synthesis, 2008, 395 CrossRef CAS PubMed.
-
4aba can even be obtained using a 1:1:1 ratio (86% yield, dr 1.8:1).
- (a) A. Z. Gonzalez, D. Benitez, E. Tkatchouk, W. A. Goddard and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 5500 CrossRef CAS PubMed; (b) See also ref. 12a.
- The preferential formation of the THP 4abb, is also in agreement with a preferred Prins-like transition state that holds the bulkier groups at C2 and C6 in equatorial disposition.
- Similarly, the cycloaddition of an electron-rich styrene such as trans-deuterated p-methoxystyrene (d-E-2e) with 1a and benzaldehyde provided d-4aea (64% yield) and traces of the [2 + 2] adduct d-5ae, both as almost equimolar mixtures of cis and trans (pMeOPh/D) isomers.14 Thus, an intermediate of type II′ (or an equilibrium between the cis and trans cyclic isomers II, Scheme 4) might also operate in this case.
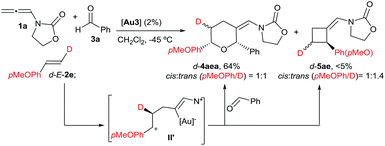 .
.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data and experimental procedures. CCDC 1038447–1038449. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00295h |
| ‡ HF and IV equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2015 |