DOI:
10.1039/C5SC00404G
(Edge Article)
Chem. Sci., 2015,
6, 2893-2902
Diverse reactivity of a tricoordinate organoboron L2PhB: (L = oxazol-2-ylidene) towards alkali metal, group 9 metal, and coinage metal precursors†
Received
3rd February 2015
, Accepted 22nd February 2015
First published on 23rd February 2015
Abstract
The reactivity of a tricoordinate organoboron L2PhB: (L = oxazol-2-ylidene) 1 towards metal precursors and its coordination chemistry were comprehensively studied. While the boron center in 1 is reluctant to coordinate to the alkali metals in their trifluoromethanesulfonate salts (MOTf) (M = Li, Na, K), the unprecedented compound 2 containing two L2PhB: units linked by a cyclic Li(OTf)2Li spacer was obtained from the reaction of 1 with LiOTf. Treatment of 1 with group 9 metal complexes [MCl(COD)]2 (M = Rh, Ir) afforded the first zwitterionic rhodium(I)–boronium complex 3 and the iridium(III)–borane complex 4, respectively. The reaction pathway may involve C–H activation followed by proton migration from the metals to the boron center, demonstrating the first example of the deprotonation of metal hydrides by a basic boron. In the reactions with coinage metals, 1 could act as a two-electron reducing agent towards the metal chlorides MCl (M = Cu, Ag, Au). Meanwhile, the reaction of 1 with gold chloride supported by a N-heterocyclic carbene (NHC) produced a heteroleptic cationic gold complex [(L2PhB)Au(NHC)]Cl (6) featuring both carbene and L2PhB: ligands on the gold atom. In contrast, an isolable gold chloride complex (L2PhB)AuCl (8) was obtained by direct complexation between 1 and triphenylphosphine-gold chloride via ligand exchange. X-ray diffraction analysis and computational studies revealed the nature of the B:→Au bonding interaction in complexes 6 and 8. Natural Population Analysis (NPA) and Natural Bond Orbital (NBO) analysis support the strong σ-donating property of the L2PhB: ligand. Moreover, preliminary studies showed that complex 8 can serve as an efficient precatalyst for the addition of X–H (X = N, O, C) to alkynes under ambient conditions, demonstrating the first application of a metal complex featuring a neutral boron-based ligand in catalysis.
Introduction
Transition metal complexes featuring boron-based ligands have attracted significant attention in boron chemistry because of the fundamental importance of understanding basic metal–boron bonding properties and their potential applications as catalysts in organic synthesis.1 Depending on the coordination number of boron and the metal–boron bonding mode, they are classified as boron cluster, borane, boryl, borylene, and boride complexes. In addition to several conventional strategies for the preparation of complexes featuring unique metal–boron bonding interactions, a new general methodology, namely direct transmetalation between isolable nucleophilic boron species and metal precursors, has been developed in recent years (Fig. 1). In 2007, Nozaki, Yamashita et al. reported the first nucleophilic attack of boryl lithium I2 on coinage metal chlorides supported by triphenylphosphine (Ph3P) or N-heterocyclic carbene (NHC) ligands which afforded the corresponding boryl complexes Ia possessing two-center two-electron M–B (M = Cu, Ag, Au) bonds.3 Since then, a large number of boryl complexes Ib have been synthesized by a similar approach, in which the metals varied extensively from rare earth metals and transition metals to main group metals.4 Similarly, Bertrand et al. illustrated that CAAC-stabilized boryl anion II can also be used as a nucleophile to synthesize the gold–boryl complex IIa.5 Braunschweig et al. revealed the radical reactivity of the boryl anion III towards heavier tetrel halides to produce IIIa possessing rare B–Sn and B–Pb bonds.6 They also showed that salt elimination between anionic dimetalloborylene IV and NHC-coinage metal chlorides afforded the trimetallic boride complexes IVa that contain delocalized Mn–B–M (M = Cu, Ag, Au) bonding,7,8 whereas IVb was obtained when Ph3P–gold chloride was used.8 In 2012, the same group isolated a novel planar tetra-metalated boron complex Va from the reaction of V with (NHC)AuCl, and proposed unique σ and π metal–boron bonding interactions in Va.9
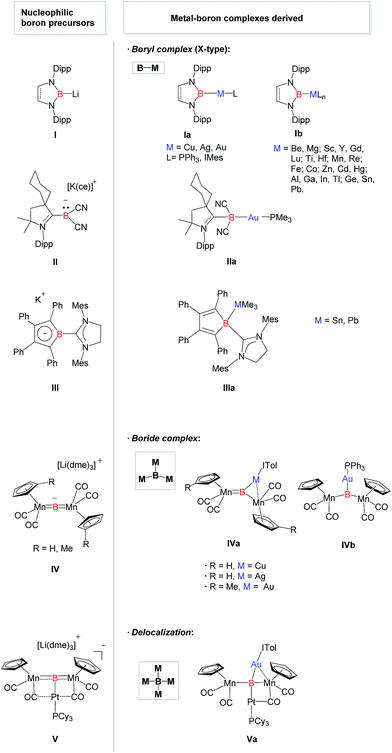 |
| | Fig. 1 Structurally characterized metal complexes Ia–Va featuring a boron-centered ligand derived from the corresponding nucleophilic boron species I–V. [Cy = cyclohexyl, Dipp = 2,6-diisopropylphenyl, IMes = 1,3-bis(mesityl)imidazol-2-ylidene, ITol = 1,3-bis(tolyl)imidazol-2-ylidene, Mes = 2,4,6-trimethylphenyl, dme = 1,2-dimethoxyethane, ce = dibenzo-18-crown-6]. | |
Although a variety of M–B bonding modes have been achieved through reactions between metal precursors and nucleophilic boron species as mentioned above, such an approach is limited to the construction of M–B single bonds mainly in their covalent fashion, and cannot be applied in the formation of the B:→M bonding mode despite the significance from the fundamental and application points of view.1 In addition, conventional synthetic methodology for the complexes bearing B:→M bonds cannot be applicable to all metals due to the lack of the reducing strength of some metals towards boron–halide bonds, such as for coinage metals. Undoubtedly, it will be straightforward if direct coupling between metal precursors and ligands possessing a lone pair on the boron is available. In 2011, Bertrand and co-workers reported the first isolation of tricoordinate nucleophilic organoboron VI that is isoelectronic with amines (Fig. 2).10,11 They also developed an efficient synthetic route to relevant derivatives VII and VIII.12 Despite the development of unique compounds VI–VIII, studies dealing with them are limited to their protonation reaction with Brønsted acids that afforded boronium species, and oxidation to generate radical cations. Recently, we reported the synthesis of L2PhB: (1) (L = oxazol-2-ylidene) and elucidated that 1 can readily react with (thf)Cr(CO)5 to afford the complex (L2PhB)Cr(CO)5, underlining its nucleophilic character (Fig. 2).13 This result prompted us to investigate the further reactivity of 1 towards metal precursors. Herein, we report the reactivity of L2PhB: (1) towards alkali and late transition metal derivatives, and discuss the outcome of these reactions based on spectroscopic analysis, single-crystal X-ray diffraction and computational studies.
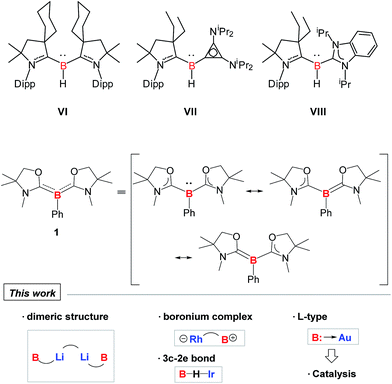 |
| | Fig. 2 Isolated examples of tricoordinate organoborons VI–VIII and 1 that are isoelectronic with amines. | |
Results
Reactivity of 1 towards alkali metal salts
First, we attempted the reaction between 1 and lithium trifluoromethanesulfonate (LiOTf). Treatment of 1 with an equivalent of LiOTf in toluene for 48 h at ambient temperature afforded a yellow precipitate. After removal of the solvent under vacuum, the residue was washed with n-hexane to afford a yellow powder of 2 in 90% yield (Scheme 1). The 11B NMR spectrum of 2 in C6D6 displays a singlet at 1.74 ppm, which is shifted only 0.07 ppm downfield compared with that of 1, implying that the boron center is inert towards lithium salt.
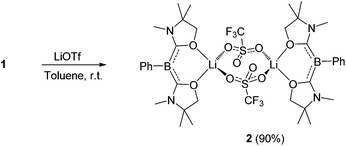 |
| | Scheme 1 Reaction of 1 with LiOTf. | |
Single crystals were obtained from a saturated toluene solution of 2 and the solid state structure was confirmed by an X-ray diffraction study (Fig. 3).14 Compound 2 contains two L2PhB: units and two LiOTf molecules. Two oxygen atoms from each oxazol-2-ylidene coordinate to a lithium atom forming a BC2O2Li six-membered ring. The two lithium atoms are linked by two OTf units in which two oxygen atoms from each OTf unit coordinate to different lithium atoms to form a LiO2S2O2Li eight-membered ring, with the CF3 groups on the S atoms in a trans orientation. The geometry around the boron centers remains trigonal planar (sum of the angles = 360.0°) and structural parameters in each of the L2PhB: moieties are nearly identical to those in compound 1. To gain insight into the electronic properties, we performed quantum chemical calculations for 2. Electron delocalization over the two C–B–C units was confirmed in the HOMO (−5.355 eV) and the HOMO−1 (−5.357 eV) that are nearly degenerate (Fig. 3). Interestingly, these MOs are lower in energy than the HOMO (−4.80 eV) of compound 1, presumably caused by the coordination of oxygen atoms to the Lewis acidic Li atoms, which reduces the electron density in the L2PhB: units. Thus, the nucleophilicity of 1 might be tunable depending on the property of the units coordinating to these O-atoms. The formation of 2 rather than B:→Li coordination can be rationalized by the hard acidic and oxophilic properties of lithium in contrast to the nature of the boron center as a soft base. Although compound 2 is thermally stable, it decomposes upon exposure to air. Attempts to react 1 with NaOTf and KOTf, containing metals slightly softer than Li, afforded neither B:→M (M = Na, K) coordination nor the corresponding analogues to 2.
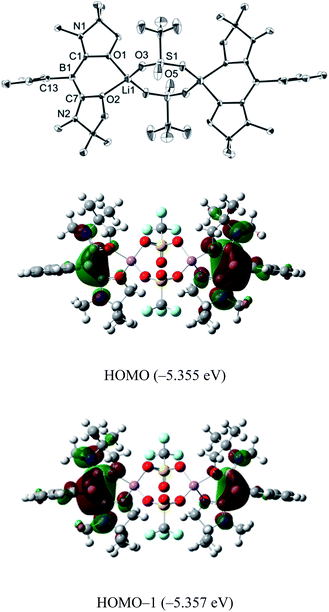 |
| | Fig. 3 Solid-state structure of 2 (top) (thermal ellipsoids are set at the 50% probability level. Hydrogen atoms are omitted for clarity). Plots of the HOMO (middle) and the HOMO−1 (bottom) of 2 (calculated at the M05-2X/6-311G(d,p) level of theory). | |
Reactivity of 1 towards group 9 metals
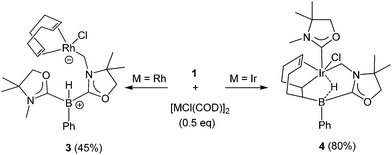
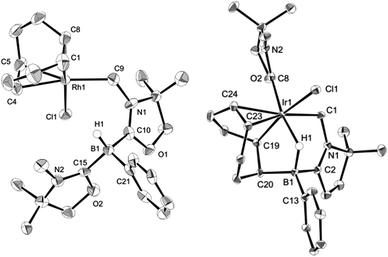
Reactivity of 1 towards late transition metals was investigated employing rhodium and iridium complexes. To a THF solution of 1, 0.5 equivalent of [RhCl(COD)]2 was added at ambient temperature. After removal of the solvent in vacuo, the residue was washed with cold n-hexane to afford a yellow solid of 3 in 45% yield (Scheme 2). The 11B NMR spectrum of 3 displays a doublet at −21.0 ppm which is due to boron–hydrogen coupling (1JBH = 34.0 Hz). In the 13C NMR spectrum, a new peak for CH2 appeared at 39.7 ppm as a doublet (1JRhC = 31.8 Hz), indicating the formation of a rhodium–carbon bond. The solid structure of 3 was determined by a single crystal X-ray diffraction study, which revealed the unique zwitterionic property of 3 involving a boronium cation and an anionic rhodium(I) center (Fig. 4, left).14 Note that only a few anionic Rh(I) complexes have been described and structurally characterized thus far, and to the best of our knowledge, no rhodium complexes including a boronium fragment have been reported before.15 The anionic Rh center coordinated by COD is covalently bonded to a chlorine atom and the CH2 carbon [Rh–Cl: 2.408(3) Å. Rh–CH2: 2.100(10) Å]. The distance between the Rh and the B atoms is greater than 3.6791(5) Å, indicating no interaction between them. The geometric parameters around the boron center are almost identical to those previously reported in the boronium [L2PhBH]OTf.13
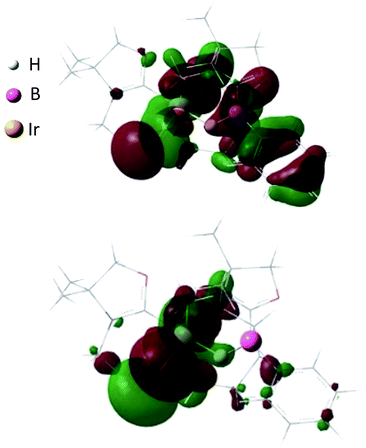
Reaction of 1 with 0.5 equivalent of [IrCl(COD)]2 led to the formation of a new complex 4 in 80% yield (Scheme 2). In the 1H NMR spectrum of 4, a characteristic broad peak was observed at −7.54 ppm, indicating a strong interaction of the H atom with the iridium center. The 11B NMR spectrum of 4 showed a broad peak at −22.7 ppm, corresponding to the tetracoordinate boron. Single crystals of 4 were obtained from a saturated toluene solution at room temperature. In the solid structure of 4, one of the oxazol-2-ylidenes coordinates to the Ir center in an η1-fashion (Fig. 4, right).14 In contrast to the bonding situation in 3 where two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C moieties in COD coordinate to the Rh center, both the Ir and the B atoms in 4 form covalent bonds with carbons in the COD, and only one CC part interacts with the Ir atom in an η2-fashion. Taking NMR data and the structural parameters of 4, the presence of the B–H–Ir bonding interaction can be postulated. Indeed, the experimental geometry is well reproduced by a DFT calculation on 4, where the Ir–H and B–H distances are 1.78 Å and 1.31 Å; these are 0.06 Å and 0.16 Å longer than the sum of the covalent radii of Ir–H and B–H respectively.16 Thus, it can be illustrated as a 3c-2e Ir–H–B bond.17 The B–H and Ir–H bonding interactions are involved in the HOMO and HOMO−1, respectively (Fig. 5). Natural Bond Orbital (NBO) analysis gave Wiberg bond index (WBI) values for the B–H bond (0.57) and Ir–H bond (0.30). The solid-state IR spectrum of 4 displays an Ir–H–B bending vibration at 1618 cm−1, which is in line with the computational result (mode 167: 1589 cm−1, see the ESI†).
C moieties in COD coordinate to the Rh center, both the Ir and the B atoms in 4 form covalent bonds with carbons in the COD, and only one CC part interacts with the Ir atom in an η2-fashion. Taking NMR data and the structural parameters of 4, the presence of the B–H–Ir bonding interaction can be postulated. Indeed, the experimental geometry is well reproduced by a DFT calculation on 4, where the Ir–H and B–H distances are 1.78 Å and 1.31 Å; these are 0.06 Å and 0.16 Å longer than the sum of the covalent radii of Ir–H and B–H respectively.16 Thus, it can be illustrated as a 3c-2e Ir–H–B bond.17 The B–H and Ir–H bonding interactions are involved in the HOMO and HOMO−1, respectively (Fig. 5). Natural Bond Orbital (NBO) analysis gave Wiberg bond index (WBI) values for the B–H bond (0.57) and Ir–H bond (0.30). The solid-state IR spectrum of 4 displays an Ir–H–B bending vibration at 1618 cm−1, which is in line with the computational result (mode 167: 1589 cm−1, see the ESI†).
 |
| | Scheme 2 Reactions of 1 with [MCl(COD)2] (M = Rh, Ir). | |
 |
| | Fig. 4 Solid-state structures of 3 (left) and 4 (right) (hydrogen atoms except for those on the B atom are omitted for clarity). Thermal ellipsoids are set at the 50% probability level. | |
 |
| | Fig. 5 Plots of the HOMO (−7.043 eV) (top) and HOMO−1 (−7.161 eV) (bottom) of 4 (calculated at the M05-2X/6-311G(d,p) level of theory with the LANL2TZ(f) pseudopotential applied for the iridium atom). | |
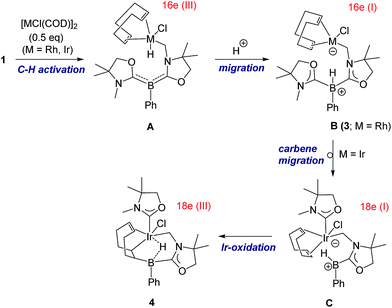
A proposed reaction pathway for the formation of 3 and 4 is drawn in Scheme 3. As no metal complexes containing B:→M bonding were observed in these reactions even when reactions were conducted at low temperature, the reactions may be initiated by C–H activation at one of the methyl groups on nitrogen atoms to generate 16 e metal hydride A. Subsequent proton migration from the metal center to the boron atom would lead to zwitterionic species B in the +I oxidation state. Note that it can be regarded as the first example of deprotonation from a metal hydride by the basic boron center. For the case of Ir, further migration of oxazol-2-ylidene from the boron to the iridium center would afford intermediate C which involves an anionic 18 e Ir(I) atom and a borenium cation. Addition of the Ir and the B atoms across one of the CC bonds in COD results in the formation of the iridium borane complex 4 bearing a 3c-2e B–H–Ir bond.
 |
| | Scheme 3 Proposed reaction pathway for the formation of 3 and 4 from reactions of 1 with [MCl(COD)]2 (M = Rh, Ir). | |
Reactivity of 1 towards coinage metal compounds
Numerous coinage metals featuring boron-based ligand have been isolated and structurally characterized.18 Although various types of bonding interaction between coinage metals and boron atoms have been described,19 to the best of our knowledge, the B:→M (M = Cu, Ag, Au) bonding mode is still unknown to date. We were interested in introducing 1 as the ligand onto coinage metals, not only for the fundamental curiosity in developing the novel B:→M bonding, but also considering their further application since a variety of coinage metal complexes exhibit catalytic activity in organic transformation.20 First, we investigated the reaction between 1 and coinage metal chlorides MCl (M = Cu, Ag, Au). Addition of two equivalents of MCl to a THF solution of 1 at room temperature immediately afforded a black precipitate, presumably metal (0), indicating the reduction of coinage metal chlorides by 1. In fact, quantitative formation of the resulting boronium 5 was observed (Scheme 4, top), and 5 was fully characterized by HRMS and NMR spectroscopy which are identical to previous data for 5 with trifluoromethanesulfonate (−OTf) as the counter anion.13 We reasoned that the presence of a strong σ-donating ligand on the M(I) would increase the electron density at the metal center, which will prevent the electron transfer from 1 to the metals. To examine this hypothesis, we chose N-heterocyclic carbene–gold chloride (IPr)AuCl as a precursor. A THF solution of 1 and (IPr)AuCl was stirred at ambient temperature for 10 min. As expected, no precipitation of black metal was detected, and a clear yellow solution was generated instead. After evaporation of the solvent in vacuo and washing of the residue with toluene, 6 was obtained as a white solid in 95% yield (Scheme 4, bottom). In the 11B NMR spectrum of 6, an upfield shift from the precursor was observed at −15.1 ppm, which is in line with the formation of a new gold complex bearing the L2PhB: ligand. Recrystallization from a saturated THF solution of 6 at −25 °C under argon afforded single crystals, and the solid-state structure of cationic gold(I) complex 6 was confirmed by a single-crystal X-ray diffraction study (Fig. 6).14 Both the L2PhB: and IPr ligands coordinate to the cationic gold center heteroleptically, and the B–Au–C(carbene) angle (179.1(5)°) deviates only slightly from linearity. The Au–B distance of 2.170(12) Å is significantly shorter than those (2.31–2.66 Å) reported for gold–borane complexes;21 however, it correlates well with those of gold–boryl and boride complexes Ia, IIa, IVa, IVb, and Va (2.07–2.21 Å). It is also in good agreement with the calculated result (2.176 Å). Significantly, a slight elongation of the Au–C(carbene) bond (2.068(10) Å) with respect to that in [(IPr)Au(PPh3)]BF4 (2.039(5) Å)22 was observed which suggests a trans influence of L2PhB: comparable with triphenylphosphine. To gain further insight into the bonding nature of the Au–B bond, geometry optimization, NBO analysis, and NPA calculation for 6 were performed. Fig. 7 shows the HOMO of 6 which mainly corresponds to the Au–B bond. The Au–B bond of 6 is formed by coordination of electrons in a high-p-character hybrid orbital (s: 16.08%, p: 83.89%) of the boron atom to an unoccupied orbital of the cationic gold atom (s: 84.51%, p: 0.18%, d: 15.29%). Remarkably, the WBI value of the Au–B bond (0.48) is even larger than that of the Au–C(carbene) bond (0.46), supporting the strong σ-donating property of the L2PhB: ligand. Indeed, NPA revealed that a charge transfer of 0.52e occurs from the L2PhB: fragment to the cationic gold(I) whereas a lesser charge (0.36e) is transferred from the IPr fragment to the gold(I) center. The calculated bond dissociation energy (BDE) for the B:→Au and Au←:C(carbene) bonds in 6 are 92.6 and 72.0 kcal mol−1, respectively. The solid-state IR spectrum of 6 displays Au–B stretching vibrations at 570 cm−1, that are consistent with the computational result (mode 89: 582 cm−1, see the ESI†).
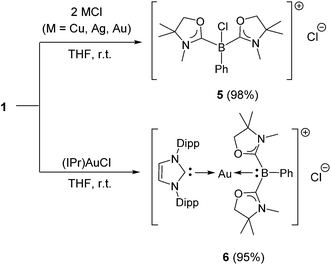 |
| | Scheme 4 Reactions of 1 with MCl (M = Cu, Ag, Au) and (IPr)AuCl (Dipp = 2,6-diisopropylphenyl). | |
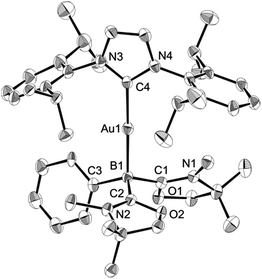 |
| | Fig. 6 Solid-state structure of 6 (hydrogen and chlorine atoms and solvent molecules are omitted for clarity). Thermal ellipsoids are set at the 50% probability level. | |
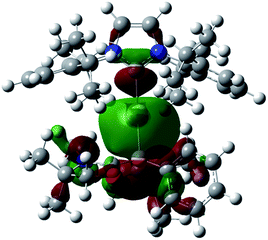 |
| | Fig. 7 Plot of the HOMO (−9.138 eV) of 6 (calculated at the M05-2X/6-311G(d,p) level of theory, with the LANL2TZ(f) pseudopotential applied for the gold atom). | |
Reactions with (IPr)CuCl and (IPr)AgCl were also examined. A CD3CN solution of 1 and (IPr)CuCl was prepared in a J. Young NMR tube. 1H and 11B NMR spectra were measured immediately, and showed unidentified complex mixtures. Meanwhile, a reaction mixture of 1 with an equivalent of (IPr)AgCl in CD3CN displayed a characteristic signal at −15.6 ppm in the 11B NMR spectrum, presumably indicating the generation of [(L2PhB)Ag(IPr)]Cl. However, this intermediate was not thermally stable: thus, it decomposed completely after 4 h at ambient temperature, and the formation of [(IPr)2Ag]Cl was confirmed concomitant with the reproduction of 1 (see the ESI†). The reactivity of 1 towards triphenylphosphine-ligated group 11 metal chlorides (Ph3P)MCl (M = Cu, Ag, Au) was also investigated. A mixture of 1 and an equivalent of (Ph3P)CuCl was dissolved in CD3CN and the reaction was monitored by NMR spectroscopy (see the ESI†). In the 11B NMR spectrum, a singlet appeared at −16.8 ppm which is in line with the formation of (L2PhB)CuCl 7 (Scheme 5, top). In fact, a quantitative generation of free Ph3P was confirmed by 31P NMR. However, compound 7 gradually decomposed to 5 within 24 h at room temperature. Reaction of 1 with (Ph3P)AgCl afforded 5 directly, and no corresponding Ag complex intermediate was detected even when the reaction was conducted at low temperature (Scheme 5, middle). Interestingly, a contrasting result was obtained when (Ph3P)AuCl was employed. An acetonitrile solution of 1 and (Ph3P)AuCl was stirred at ambient temperature for 10 min. After removing the solvent in vacuo and washing the residue with n-hexane, the neutral gold chloride complex 8 was obtained as a white solid in 80% yield. Formation of 8via the smooth replacement of triphenylphosphine by 1 on the gold atom demonstrates the strong nucleophilicity of the L2PhB: ligand 1. Note that a variety of gold complexes featuring strong σ-donating neutral ligands are widely used as efficient catalysts in organic synthesis.23 The 11B NMR of 8 displays a sharp singlet at −18.6 ppm which is shifted upfield compared to that of 1 (−1.1 ppm). The complex 8 is not only moisture sensitive but light sensitive, and quickly decomposes if exposed to air or light.
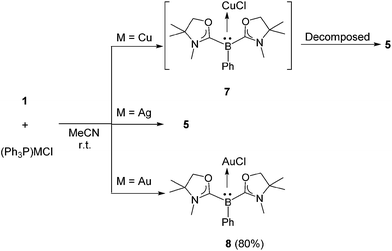 |
| | Scheme 5 Reactions of 1 with (Ph3P)MCl (M = Cu, Ag, Au). | |
Single crystals of 8 were obtained from an acetonitrile/toluene mixed solvent at room temperature, and X-ray diffraction analysis revealed that 8 features an almost linear geometry (B1–Au1–Cl1: 175.2(4)°), and the tetracoordinate boron is bound to the gold(I) center in an η1-fashion (Fig. 8).14 The Au–B distance of 2.166(14) Å is nearly identical to that of 6. These parameters are in good accordance with the calculated results for the model compounds (Ph3P)2(BH)AuCl and (NHCMe)2(BH)AuCl.24 We performed quantum chemical calculations to investigate the property of the Au–B bond in 8. The HOMO of 8 displays the coordination of electrons of the boron to the gold atom (Fig. 9). NBO analysis gave a Wiberg bond index (WBI) value of the Au–B bond (0.50), which is formed from the high-p-character hybrid orbital (s: 15.49%, p: 84.47%) of the boron atom and mainly the s-orbital of the gold atom (s: 84.66%, p: 1.72%, d: 13.60%). Natural Population Analysis (NPA) indicates an overall charge transfer of 0.43e from the L2PhB: fragment to the gold chloride. The calculated BDE of the donor–acceptor bond between 1 and the gold chloride in 8 is 78.4 kcal mol−1, which is comparable to that (77.3 kcal mol−1) of (Ph3P)2(BH)AuCl.24 Comparatively, much lower BDEs were estimated for the corresponding bonds in 1-CuCl (64.3 kcal mol−1) and 1-AgCl (55.8 kcal mol−1), which may account for the instability of compound 7. An analogous trend of the bond energies for the metal fragments (Au > Cu > Ag) was also found in their carbene, silylene and germylene complexes.25 In the solid state IR spectrum of 8, a characteristic peak was observed at 586 cm−1, which is assigned to a Au–B stretch based on computational results (mode 41: 577 cm−1, see the ESI†).
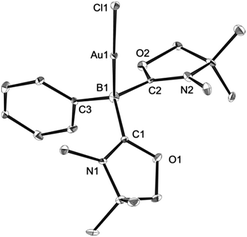 |
| | Fig. 8 Solid-state structure of 8 (hydrogen atoms are omitted for clarity). Thermal ellipsoids are set at the 50% probability level. | |
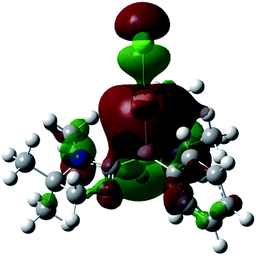 |
| | Fig. 9 Plot of the HOMO (−6.779 eV) of 8 (calculated at the M05-2X/6-311G(d,p) level of theory, with the LANL2TZ(f) pseudopotential applied for the gold atom). | |
Preliminary investigation of catalysis with complex 8
Next, we turned our attention to the potential ability of complex 8 as a catalyst. We chose the hydroamination reaction between aniline 9a and phenylacetylene 10a as a model reaction. A brief screening of solvents (Table 1, entries 1–5) revealed that benzene is suitable for the reaction; an imine derivative 11aa was obtained in 94% yield after 12 h at room temperature (entry 5). During this reaction we observed a main signal in the 11B NMR spectrum at −16.1 ppm, in addition to the peak at −16.6 for B(C6F5)4 and a small peak at −19.3 ppm. Control reactions suggest that the main signal may correspond to either (L2PhB)Au(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh) or σ,π-acetylide complex [(L2PhB)Au]2(CCPh)·B(C6F5)4, proposing the resting state of the catalyst (see the ESI†). Hence, this result supports the innocence of colloidal gold, demonstrating the essential role of 8 in the catalytic reaction. With AgOTf as an additive, little formation of 11aa (<5%) was observed (entry 6).
CPh) or σ,π-acetylide complex [(L2PhB)Au]2(CCPh)·B(C6F5)4, proposing the resting state of the catalyst (see the ESI†). Hence, this result supports the innocence of colloidal gold, demonstrating the essential role of 8 in the catalytic reaction. With AgOTf as an additive, little formation of 11aa (<5%) was observed (entry 6).
Table 1 Optimization of the reaction conditionsa
|
|
| Entry |
Additive (5 mol%) |
Solvent |
Yieldb (%) |
|
Reaction conditions: 9a (0.5 mmol), 10a (0.5 mmol), solvent (0.5 mL).
Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxy benzene as an internal standard.
|
| 1 |
KBArF4 |
CD3CN |
9 |
| 2 |
KBArF4 |
THF-D8 |
12 |
| 3 |
KBArF4 |
CDCl3 |
73 |
| 4 |
KBArF4 |
CD2Cl2 |
74 |
| 5 |
KBArF4 |
C6D6 |
94 |
| 6 |
AgOTf |
C6D6 |
<5 |
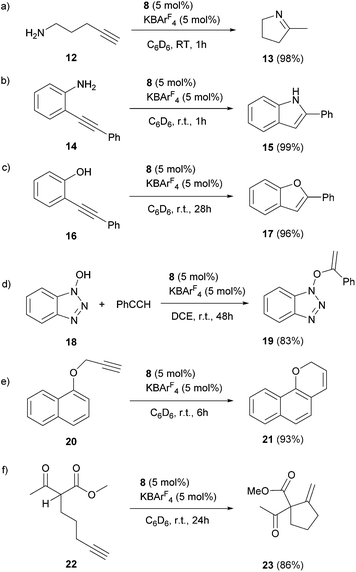
The scope of the catalytic reaction was briefly examined with various primary amine and terminal alkyne derivatives at ambient temperature (Table 2). The addition reactions of aniline 9a, p-anisidine 9b or 3,5-dimethylaniline 9c to phenylacetylene 10a or 4-ethynylanisole 10b were effectively catalyzed to afford the corresponding imines in excellent yields: 11aa (98%), 11ab (93%), 11ba (90%), 11bb (94%) and 11ca (91%) (entries 1, 2, 4, 5, 7). Lower activity was observed when the sterically hindered amine substrates 9d, e were used (entries 8–10); i.e. from the reaction of 9e and 10b, only moderate formation of the corresponding imine 11eb (44%) was observed after 72 h (entry 10). Although a longer reaction time was required, the catalyst 8 was also effective for the hydroamination with more challenging electron-poor alkynes, such as 1-bromo-4-ethynylbenzene 10c. Thus, imine derivatives 11ac, 11bc and 11ec were slowly formed from the reaction of 10c and the corresponding amines (9a, 9b and 9e) in low to good yields (entries 3, 6 and 11, respectively). Not surprisingly, complex 8 also catalyzed intramolecular hydroamination (Scheme 6a and b), in which N-heterocyclic products 13 and 15 were obtained quantitatively. We also tested O–H additions to alkynes with a catalytic amount of 8 (5 mol%). At room temperature, compound 16 was smoothly converted to 2-phenylbenzofuran 17 (96%) (Scheme 6c). When N-hydroxy benzotriazole 18 was treated with phenylacetylene, 19 was formed in good yield (83%) via an intermolecular O–H addition (Scheme 6d). Because selective C–H activation as well as the formation of C–C bonds is significant in organic synthesis, we also investigated the catalytic transformation of compounds 20 and 22. In the reaction with 1-naphthalenyl propargyl ether 20, addition of a C(sp2)–H bond across the C–C triple bond took place which afforded a benzo[h]chromene derivative 21 in 93% yield after 6 h (Scheme 6e). The cyclisation reaction of compound 22 also occurred effectively via activation of a C(sp3)–H bond, and β-ketoester 23 was obtained in good yield (86%) (Scheme 6f). Since it has been already demonstrated that with colloidal gold, higher temperature, irradiation of light, or longer reaction times are required to conduct the relevant reactions,26 its involvement may be ruled out here. Note that some of the results shown here were comparable to or even better than those with (Ph3P)AuCl as a precatalyst under similar conditions.27 It is also noteworthy that these results demonstrated the first application of metal complexes featuring a neutral boron-based ligand in catalysis.
Table 2 Catalytic hydroamination of alkynes with primary aminesa
|
|
| Entry |
9
|
10
|
Time (h) |
11, Yieldb (%) |
|
Reaction conditions: 9 (2 mmol), 10 (2 mmol) benzene (3 mL).
Isolated yields.
|
| 1 |
9a (R = Ph) |
10a (R′ = Ph) |
24 |
11aa, 98 |
| 2 |
9a
|
10b (R′ = 4-MeOC6H4) |
24 |
11ab, 93 |
| 3 |
9a
|
10c (R′ = 4-BrC6H4) |
72 |
11ac, 86 |
| 4 |
9b (R = 4-MeOC6H4) |
10a
|
36 |
11ba, 90 |
| 5 |
9a
|
10b
|
36 |
11bb, 94 |
| 6 |
9a
|
10c
|
72 |
11bc, 54 |
| 7 |
9c (R = 3,5-Me2C6H3) |
10a
|
36 |
11ca, 91 |
| 8 |
9d (R = 2,4,6-Me3C6H2) |
10b
|
72 |
11db, 73 |
| 9 |
9e (R = 2,6-iPr2C6H3) |
10a
|
72 |
11ea, 46 |
| 10 |
9e
|
10b
|
72 |
11eb, 44 |
| 11 |
9e
|
10c
|
72 |
11ec, 30 |
 |
| | Scheme 6 Catalytic addition of X–H (X = N, O, C) bonds to alkynes. | |
Conclusions
In conclusion, we have demonstrated the diverse reactivity of L2PhB: (1) towards metal precursors. Addition of hard acidic LiOTf to 1 induced coordination of the oxygen atoms in the oxazol-2-ylidenes to the Li atoms, and the unique compound 2 containing two L2PhB: moieties bridged via a cyclic Li(OTf)2Li unit was obtained. Reaction of 1 with [RhCl(COD)]2 afforded the hitherto unknown zwitterionic complex 3 involving an anionic rhodium and a boronium cation whereas iridium borane complex 4 featuring a 3c-2e Ir–H–B bond was isolated from reaction with [IrCl(COD)]2. In the formation of complexes 3 and 4, deprotonation of the metal hydrides by the boron atom is proposed, which illustrates the strongly basic property of the boron center in 1. Direct complexation between 1 and gold chloride derivatives bearing carbene or phosphine ligands produced the first isolable gold complexes 6 and 8 featuring B:→Au bonding. Depending on the ligand on the gold chloride precursors, either a cationic or a neutral gold(I) complex supported by the L2PhB: ligand can be obtained selectively via Au–Cl bond dissociation or ligand exchange, respectively. X-ray diffraction analysis and computational studies revealed the unique Au–B bonding formed by the coordination of lone-pair electrons on the boron atom to the gold center as well as the strong σ-donating property of the L2PhB: ligand. Complex 8 showed good catalytic activity when employed as a precatalyst for both intra- and inter-molecular X–H (X = N, O, C) addition to alkynes. Further developments of metal complexes bearing 1 and its application in catalysis are currently under investigation in our laboratory.
Acknowledgements
The authors gratefully acknowledge financial support from Nanyang Technological University and A*STAR (PSF/SERC 1321202066) of Singapore.
Notes and references
- For recent reviews on transition metal boron complexes, see:
(a) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197 RSC;
(b) D. Vidovic and S. Aldridge, Chem. Sci., 2011, 2, 601 RSC;
(c) H. Braunschweig, R. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924 CrossRef CAS PubMed;
(d) D. Vidovic, G. A. Pierce and S. Aldridge, Chem. Commun., 2009, 1157 RSC;
(e) C. E. Anderson, H. Braunschweig and R. D. Dewhurst, Organometallics, 2008, 27, 6381 CrossRef CAS;
(f) H. Braunschweig, C. Kollann and D. Rais, Angew. Chem., Int. Ed., 2006, 45, 5254 CrossRef CAS PubMed;
(g) H. Braunschweig and G. Whittell, Chem.–Eur. J., 2005, 11, 6128 CrossRef CAS PubMed;
(h) H. Braunschweig and D. Rais, Heteroat. Chem., 2005, 16, 566 CrossRef CAS;
(i) S. Alridge and D. L. Coombs, Coord. Chem. Rev., 2004, 248, 535 CrossRef PubMed;
(j) H. Braunschweig, Adv. Organomet. Chem., 2004, 51, 163 CrossRef CAS.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113 CrossRef CAS PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710 CrossRef CAS PubMed.
-
(a) T. Arnold, H. Braunschweig, W. C. Ewing, T. Kramer, J. Mies and J. K. Schuster, Chem. Commun., 2015, 51, 737 RSC;
(b) R. Frank, J. Howell, R. Tirfoin, D. Dange, C. Jones, D. M. P. Mingos and S. Aldridge, J. Am. Chem. Soc., 2014, 136, 15730 CrossRef CAS PubMed;
(c) A. V. Protchenko, D. Dange, A. D. Schwarz, C. Y. Tang, N. Phillips, P. Mountford, C. Jones and S. Aldridge, Chem. Commun., 2014, 50, 3841 RSC;
(d) A. V. Protchenko, D. Dange, J. R. Harmer, C. Y. Tang, A. D. Schwarz, M. J. Kelly, N. Phillips, R. Tirfoin, K. H. Birjkumar, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, Nat. Chem., 2014, 6, 315 CrossRef CAS PubMed;
(e) N. Dettenrieder, C. Schädle, C. Maichle-Möcssmer, P. Sirsch and R. Anwander, J. Am. Chem. Soc., 2014, 136, 886 CrossRef CAS PubMed;
(f) N. Dettenrieder, H. M. Dietrich, C. Schaedle, C. Maichle-Möessmer, K. W. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2012, 51, 4461 CrossRef CAS PubMed;
(g) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500 CrossRef CAS PubMed;
(h) Y. Okuno, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2011, 50, 920 CrossRef CAS PubMed;
(i) S. Li, J. Cheng, Y. Chen, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 6360 CrossRef CAS PubMed;
(j) L. M. A. Saleh, K. H. Birjkumar, A. V. Protchenko, A. D. Schwarz, S. Aldridge, C. Jones, N. Kaltsoyannis and P. Mountford, J. Am. Chem. Soc., 2011, 133, 3836 CrossRef CAS PubMed;
(k) T. Terabayashi, T. Kajiwara, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14162 CrossRef CAS PubMed;
(l) T. Kajiwara, T. Terabayashi, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2008, 47, 6606 CrossRef CAS PubMed;
(m) M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570 CrossRef CAS PubMed;
(n) Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710 CrossRef CAS PubMed.
- D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590 CrossRef CAS PubMed.
- R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453 CrossRef CAS PubMed.
- H. Braunschweig, P. Brenner, R. D. Dewhurst, M. Kaupp, R. Müller and S. Östreicher, Angew. Chem., Int. Ed., 2009, 48, 9735 CrossRef CAS PubMed.
- H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kramer, S. Östreicher, K. Radacki and A. Vargas, J. Am. Chem. Soc., 2013, 135, 2313 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Kraft, S. Östreicher and K. Radacki, Angew. Chem., Int. Ed., 2012, 51, 2183 CrossRef CAS PubMed.
-
(a) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610 CrossRef CAS PubMed;
(b) For an amino borylene supported by only one CAAC ligand, see: F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159 CrossRef CAS PubMed.
- For different viewpoints on the use of dative arrows, see:
(a) D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 370 CrossRef CAS PubMed;
(b) G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040 CrossRef CAS PubMed;
(c) D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 6047 CrossRef CAS PubMed;
(d)
G. Frenking and S. Shaik, The Chemical Bond 2, Wiley-VCH, Weinheim, 2014 Search PubMed.
- D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837 RSC.
- L. Kong, Y. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280 CrossRef CAS PubMed.
- ESI†.
- Only a few transition metals involving boronium have been reported. For selected recent examples, see:
(a) B. Bentivegna, C. I. Mariani, J. R. Smith, S. Ma, A. L. Rheingold and T. J. Brunker, Organometallics, 2014, 33, 2820 CrossRef CAS;
(b) S. De, G. A. Pierce, D. Vidovic, D. L. Kays, N. D. Coombs, E. D. Jemmis and S. Aldridge, Organometallics, 2009, 28, 2961 CrossRef CAS;
(c) G. A. Pierce, D. Vidovic, D. L. Kays, N. D. Coombs, A. L. Thompson, E. D. Jemmis, S. De and S. Aldridge, Organometallics, 2009, 28, 2947 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- Some representative papers describing the Ir–H–B 3c-2e bond:
(a) H. Kameo and H. Nakazawa, Organometallics, 2012, 31, 7476 CrossRef CAS;
(b) M. D. Spicer and J. Reglinski, Eur. J. Inorg. Chem., 2009, 1553 CrossRef CAS;
(c) N. Tsoureas, T. Bevis, C. P. Butts, A. Hamilton and G. R. Owen, Organometallics, 2009, 28, 5222 CrossRef CAS;
(d) N. Tsoureas, G. R. Owen, A. Hamilton and A. G. Orpen, Dalton Trans., 2008, 6039 RSC.
-
(a) D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036 CrossRef CAS PubMed;
(b) D. S. Laitar, P. Mueller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196 CrossRef CAS PubMed; Reviews on transformations catalyzed by coinage metal boryl complexes generated in situ:
(c) R. Barbeyron, E. Benedetti, J. Cossy, J. J. Vasseur, S. Arseniyadis and M. Smietana, Tetrahedron, 2014, 70, 8431 CrossRef CAS PubMed;
(d) L. Dang, Z. Lin and T. B. Marder, Chem. Commun., 2009, 3987 RSC.
- σ-Coordination:
(a) H. Braunschweig, K. Radacki and R. Shang, Chem. Commun., 2013, 49, 9905 RSC. Reviews on “Z-type” boron ligands:
(b) A. Amgouneab and D. Bourissou, Chem. Commun., 2011, 47, 859 RSC;
(c) H. Braunschweig and R. D. Dewhurst, Dalton Trans., 2011, 40, 549 RSC . See also ref. 3, 4h, 4l, 4n, 7–9, 18a and 18b.
-
(a) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed;
(b) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379 CrossRef PubMed.
-
(a) S. Bontemps, G. Bouhadir, W. Gu, M. Mercy, C.-H. Chen, B. M. Foxman, L. Maron, O. V. Ozerov and D. Bourissou, Angew. Chem., Int. Ed., 2008, 47, 1481 CrossRef CAS PubMed;
(b) M. Sircoglou, S. Bontemps, G. Bouhadir, N. Saffron, K. Miqueu, W. Gu, M. Mercy, C.-H. Chen, B. M. Foxman, L. Maron, O. V. Ozerov and D. Bourissou, J. Am. Chem. Soc., 2008, 130, 16729 CrossRef CAS;
(c) M. Sircoglou, S. Bontemps, M. Mercy, N. Saffron, M. Takahashi, G. Bouhadir, L. Maron and D. Bourissou, Angew. Chem., Int. Ed., 2007, 46, 8583 CrossRef CAS PubMed;
(d) S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056 CrossRef CAS PubMed.
- S. Gaillard, P. Nun, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 5402 CrossRef CAS.
- For recent reviews on homogeneous gold catalysis, see:
(a) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC;
(b) P. Garcia, M. Malacria, C. Aubert, V. Gandon and L. Fensterbank, ChemCatChem, 2010, 2, 493 CrossRef CAS;
(c) A. Arcadi, Chem. Rev., 2008, 108, 3266 CrossRef CAS PubMed;
(d) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed;
(e) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed;
(f) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed.
- M. A. Celik, R. Sure, S. Klein, R. Kinjo, G. Bertrand and G. Frenking, Chem.–Eur. J., 2012, 18, 5676 CrossRef CAS PubMed.
- P. Jerabek, H. Roesky, G. Bertrand and G. Frenking, J. Am. Chem. Soc., 2014, 136, 17123 CrossRef CAS PubMed; C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801 CrossRef.
-
(a) J. Zhao, Z. Zheng, S. Bottle, A. Chou, S. Sarina and H. Zhu, Chem. Commun., 2013, 49, 2676 RSC;
(b) L.-A. Schaper, X. Wei, S. J. Hock, A. Pöthig, K. Öfele, M. Cokoja, W. A. Herrmann and F. E. Kühn, Organometallics, 2013, 32, 3376 CrossRef CAS;
(c) V. Belting and N. Krause, Org. Lett., 2006, 8, 4489 CrossRef CAS PubMed;
(d) C. Efe, I. N. Lykakis and M. Stratakis, Chem. Commun., 2011, 47, 803 RSC;
(e) J. J. Kennedy-Smith, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 4526 CrossRef CAS PubMed;
(f) J. Oliver-Meseguer, A. Leyva-Pérez and A. Corma, ChemCatChem, 2013, 5, 3509 CrossRef CAS.
-
(a) D. Malhotra, M. K. Mashuta, G. B. Hammond and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 4456 CrossRef CAS PubMed;
(b) M. Kumar, M. Scobie, M. S. Mashuta, G. B. Hammond and B. Xu, Org. Lett., 2013, 15, 724 CrossRef CAS PubMed;
(c) I. N. Lykakis, C. Efe, C. Gryparis and M. Stratakis, Eur. J. Org. Chem., 2011, 2334 CrossRef CAS;
(d) H. Duan, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2009, 131, 12100 CrossRef CAS PubMed;
(e) A. Corma, Adv. Synth. Catal., 2009, 351, 2876 CrossRef;
(f) Y. Zhang, Z. Xin, J. Xu and Y. Li, Chin. J. Chem., 2008, 26, 1461 CrossRef CAS;
(g) A. Leyva, E. Mizushima, T. Hayashi and M. Tanaka, Org. Lett., 2003, 5, 3349 CrossRef PubMed . See also ref. 26e.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and calculation details, and crystallographic information for 2, 3, 4, 6, 8. CCDC 1038665, 1038666, 1038667, 1011534, and 1011533. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00404g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 















