
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHyperpolarization of amino acid derivatives in water for biological applications†
S.
Glöggler
a,
S.
Wagner
b and
L.-S.
Bouchard
*acd
aDepartment of Chemistry and Biochemistry, University of California at Los Angeles, Los Angeles, California 90095-1569, USA. E-mail: bouchard@chem.ucla.edu
bBiomedical Imaging Research Institute, Cedars Sinai Medical Center, 8700 Beverly Blvd, Davis Building G149E, Los Angeles, California 90048-1804, USA
cCalifornia NanoSystems Institute, 570 Westwood Plaza, Building 114, Los Angeles, California 90095-1569, USA
dDepartment of Bioengineering, University of California at Los Angeles, 420 Westwood Plaza, RM 5121 Engineering V, P.O. Box 951600, Los Angeles, California 90095-1569, USA
First published on 26th May 2015
Abstract
We report on the successful synthesis and hyperpolarization of N-unprotected α-amino acid ethyl propionate esters and extensively, on an alanine derivative hyperpolarized by PHIP (4.4 ± 1.0% 13C-polarization), meeting required levels for in vivo detection. Using water as solvent increases biocompatibility and the absence of N-protection is expected to maintain biological activity.
Introduction
NMR is an established analytical technique broadly used in chemistry and biomedicine, recognized as a highly effective, yet insensitive, diagnostic tool. This lack of sensitivity prohibits the detection of low concentration molecules, such as metabolites or pharmaceuticals, within manageable scanning times. Various hyperpolarization techniques have been developed to overcome the sensitivity problem. These techniques rely on the creation of large non-equilibrium population differences of nuclear spin polarization in the magnetic energy sublevels of the nuclear spins. This population difference is substantially larger than the thermal (Boltzmann) population difference, potentially resulting in signal enhancements over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fold. This tremendous increase in signal offers the potential to design new contrast agents for improved disease marker detection with MRI.
000 fold. This tremendous increase in signal offers the potential to design new contrast agents for improved disease marker detection with MRI.
Different hyperpolarization methods have been applied to pursue this goal.1–11 For example, hyperpolarized xenon by means of spin exchange optical pumping has been successfully implemented in lung imaging and has also been caged in functionalized cryptophane structures to act as a biosensor.1–5 Dynamic Nuclear Polarization (DNP), in which the polarization from an unpaired electron in a free radical is transferred to the nuclei, has been employed to hyperpolarize biologically active molecules and pursue their metabolism in vivo by monitoring the subsequent transformation of hyperpolarized products.6–11 DNP has been the most utilized hyperpolarization to date for metabolic image; it is biocompatible and has recently been adopted in patient studies.11
The PHIP technique12,13 is another very promising and more cost-effective hyperpolarization method, however, it requires the hydrogenation of a substrate molecule with an unsaturated bond using para-hydrogen (p-H2), followed by a subsequent conversion of the nuclear spin singlet to a triplet state to induce polarizations.12,13 The production of p-H2 depends on cooling hydrogen gas to low temperatures in the presence of a catalyst.12,13 The cooling of hydrogen gas to 25 K can yield nearly 100% enrichment and thus, an almost pure quantum singlet spin order.12–15 The application of PHIP to biomolecular imaging builds on efficient pairwise addition of p-H2 to the substrate in aqueous solution. Based on the scarcity of discovered biological relevant substrates suited to be hyperpolarized in biologically compatible solvents, such as water, the application to living systems has lagged behind other methods. Molecular tracers must be designed and synthesized for each application of interest to incorporate viable water solubility and a hyperpolarizable PHIP moiety. The design of these molecules must provide a binding site for the p-H2 adjacent to a heteronuclear species, such as 13C, to transfer the polarization. The heteronuclear recipient generally exhibits longer relaxation times allowing for extended in vivo traceability.16
The substrates that have been hyperpolarized to significant extents are mostly metabolites (or derivatives thereof) involved in glycolysis or the Kreb's cycle. While such applications to metabolic imaging are extremely promising, it is desirable to expand the range of molecular probes available to experimentalists and eventually for diagnostic purposes, so that additional physiological phenomenons can be explored, such as cell signaling, enzyme metabolism, and binding events to a target.
Recently, PHIP was applied to hyperpolarize select metabolites for contrast-enhanced angiography in animals.16–20 Biologically active and biocompatible molecules that can be traced inside an organism provide the essential groundwork for molecular imaging.21–24 A synthetic compound has also been used to detect the presence of fatty plaque in mice through chemical shift alteration induced by binding.25 Among candidate tracer molecules, amino acids and their derivatives are of particular interest because they can potentially serve as tracers for metabolism or as indicators of binding events. The amino acids can then act as building blocks to produce peptides. One recent achievement towards the investigation of binding events includes the functionalization of a peptide, sunflower trypsin inhibitor 1, which was hyperpolarized while the enzyme remained intact.26 Molecular interactions occur in the range of seconds to a few minutes within the decay time of the hyperpolarized signal. For example, DNP polarized glutamine is taken up and metabolized to glutamate within 20 seconds after injection in vivo by cancer cells and binding of cancer targeting peptides were reported in vivo within one to a few minutes.27–29
Most PHIP investigations with amino acids and their derivatives have been performed in non-aqueous solvents, such as chloroform, methanol or acetone, predominantly with protected amine functional groups. These studies30–34 not only yielded lower polarization than reported here (in particular, a 13C polarization of 1.3% in ref. 33), but more importantly, the reaction exhibited little to no polarization in aqueous solution. We note that most of the previous studies were performed with 50% p-H2 enrichment. Taking this into account, an increase in polarization by a factor of 3 can be estimated.35 The lack of sufficient polarization however will prevent any successful clinical application. Also, to maintain biological activity and biocompatibility respectively, the design of biomarkers with active amine sites is desirable and making the use of water as solvent preferable (see discussion on amino acid polarization in ESI†).
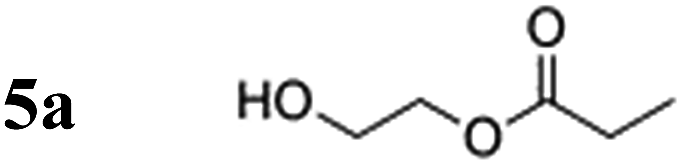
In this article, we show the synthesis of N-unprotected α-amino acid ethyl acrylate esters and in particular, an alanine derivative, for which the highest 13C polarization in D2O to date was achieved when the ester was treated with para-hydrogen in the presence of a water-soluble rhodium complex, i.e. bis(norbornadiene)rhodium(I) tetrafluoroborate (Sigma-Aldrich). Utilizing a comparable compound, a 10% 13C-polarization with 2-hydroxyethylacrylate (HEA), and a polarization of 4.4 ± 1.0% was obtained with our amino acid derivative based on alanine (nearly 100% p-H2 enrichment). The measured polarization level can be further improved by reducing the transport time to the magnet (currently 20 s), as explained below.
HEA was chosen as the polarization-carrying moiety to obtain hyperpolarization in amino acid derivatives based on prior successes in PHIP experiments and because the hyperpolarized product, hydroxyethyl propionate (HEP) has an in vivo LD50 value corresponding to that of fumarate, a metabolite naturally occurring in the citric acid cycle36 (for further discussion of toxic and metabolic behaviour please refer to the ESI†). This research represents the first significant step toward practical in vivo applications of PHIP techniques for peptides. The crucial contributions made by this work are three-fold: (1) generation of unprecedented 13C nuclear spin polarization of amino acid derivatives to levels sufficient for in vivo applications, (2) use of (deuterated) water as the solvent to increase biocompatibility; and (3) design of N-unprotected amino acid derivatives with hyperpolarizable PHIP moieties that can function as peptide building blocks to produce biologically active peptides.
Results and discussion
When designing a potential hyperpolarized biomarker, the critical factors are fast delivery to the target location inside the organism, as well as, sufficiently long retention of the polarization. For 13C polarization specifically, the time to the target needs to be within one to two minutes. The timescale over which nuclear spins depolarize and return to their thermal equilibrium is the longitudinal relaxation time (T1). As reported here, spin polarization is defined as:| P = (Nβ − Nα)(Nβ + Nα)−1, |
HEA has been frequently used in PHIP hyperpolarization studies, including at times as a MRI contrast agent for angiography in animals.16,20 It is commercially available in both, deuterated- and 13C-labelled forms. When partly deuterated, one of the 13C nuclei of the acrylate functionality possesses a T1 close to two minutes in deuterated solvents in low magnetic fields, and a polarization of P > 10% is routinely detected in the magnet through the use of a custom-built polarizer apparatus.16,37 The α-amino acids derivatized in this study were chosen to be glycine, alanine and glutamine for their potential relevance as tracers in molecular imaging experiments. Glycine is the simplest proteinogenic amino acid and has recently been identified as a metabolite associated with cancer proliferation, it is used for cellular purine synthesis, a building block for nucleic acids and thus, DNA.38 The metabolic pathways of alanine are closely connected to pyruvate, a metabolite often overexpressed in cancer cells.9 Glutamine is currently being discussed as a potential tracer in MRI studies as it is overexpressed in some cancer cells.39 Multiple studies with positron emission tomography tracers or amino acid modified gadolinium complexes indicated that significantly altered amino acids, even with considerably larger modifications than presented here, are still biologically active and internalized by cells according to the same pathways as amino acids on a minute timescale. This has been evaluated for alanine and glutamine derivatives.39–42
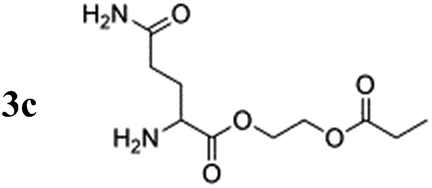
The amino acid derivatives used in our experiments (Fig. 1) were synthesized, as detailed in the Methods section. The hyperpolarized products yielded amino acid ethyl propionate esters (1c–3c; Fig. 1c) that show a characteristic triplet and quartet at 1.0 and 2.4 ppm, respectively, in the 1H NMR spectrum for the investigated substrates (Fig. 2). Typical yields for the proton experiments ranged from 5 to 10% for unoptimized mixing conditions. The reported polarization values have been normalized to the yield. The observed phase pattern corresponds to Iz–Sz magnetization as expected from an ALTADENA experiment but is shown in magnitude mode for comparison with the thermal spectrum.43 Hydrogenation experiments were conducted at two different temperatures: 20 °C and 80 °C.
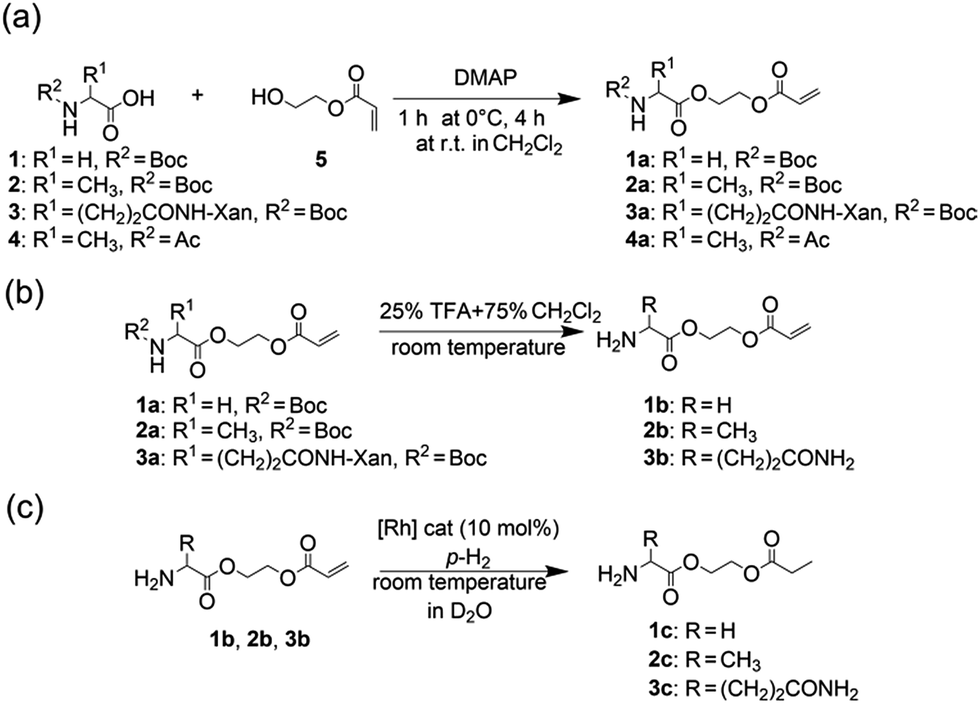 | ||
| Fig. 1 (a) Synthesis of the protected amino acid ethyl acrylate esters from N-protected amino acids and hydroxyethylacrylate (b) deprotection of the amine function with TFA yields N-unprotected α-amino acid derivatives. (c) Hydrogenation with para-hydrogen in deuterated water catalyzed by a water-soluble rhodium complex. | ||
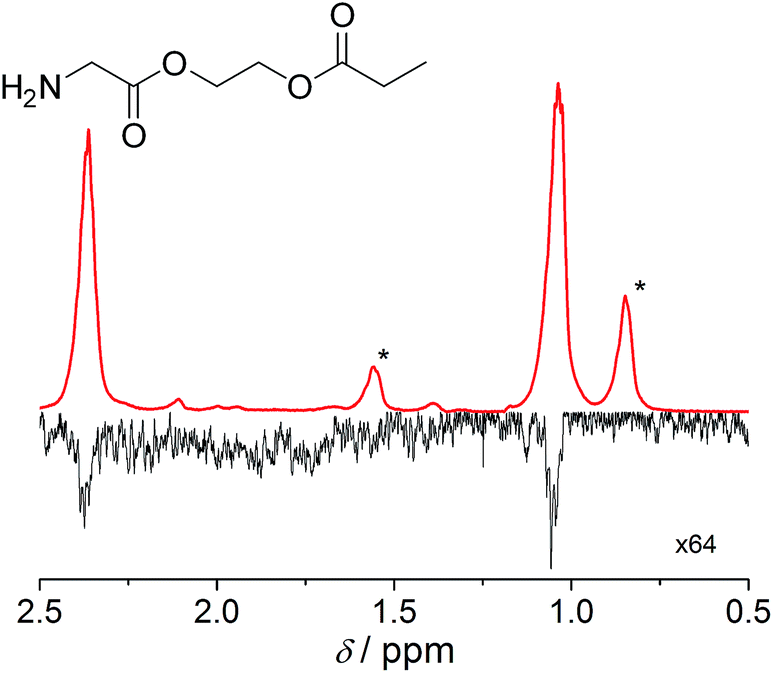 | ||
| Fig. 2 Hydrogenation of glycine derivative in D2O using water-soluble rhodium complex. Top trace: hyperpolarized section of the proton spectrum following hydrogenation performed with p-H2 at 80 °C. After the esters were treated with p-H2, physical transport of the sample to the NMR magnet took 10 s. Bottom trace: thermal polarization of propionate protons following equilibration at 14.1 T and 20 °C. The bottom trace has been magnified by a factor of 64 and both spectra are shown in magnitude (absolute value) mode. Peaks attributed to the decomposition products of the catalyst are marked with an asterisk. The level of polarization in the hyperpolarized product corresponds to 0.73% after transport. | ||
Notably, the proton polarization of all amino acid derivatives was at least a factor of 2 higher at the elevated temperature (Table 1). Hyperpolarization of the glycine and alanine derivatives, both containing a single amine function, resulted in a measured proton polarization of P(gly) = 0.73% and P(ala) = 0.70%. All experiments were performed at a pH = 6.5 ± 0.5 as indicated by pH test paper. A large difference from a neutral pH may result in a better polarization but has a disadvantage: the polarization product requires dilution in buffer prior to injection to regulate the pH value to a biocompatible level. Dilution of the compounds can result in concentrations of the potential tracers that are too low. Table 1 summarizes the determined proton polarization values of all hyperpolarized molecules investigated. The T1 relaxation times of the protons range between 2–6 s (see ESI†) at the detection field. However, due to the transportation time between polarization and detection (10 s), significant polarization is lost before acquisition. Since T1 is not constant during the transportation process from earth's magnetic field into high magnetic field, the loss in polarization cannot be precisely quantified here but is considered to be substantial on account of validation through 13C transfer experiments. An example 1H NMR spectrum of the hyperpolarized region of the glycine derivative is shown in Fig. 2. Here, a signal enhancement of ≈200 relative to the thermal polarized signal at B0 = 14.1 T (600 MHz proton frequency) can be clearly discerned. In high temperature experiments, additional hyperpolarized peaks can be identified, which are attributed to the decomposition of the catalyst. These peaks are discussed in more detail in the ESI.†
| Hyperpolarized species | Polarization at 20 °C [%] | Polarization at 80 °C [%] |
|---|---|---|
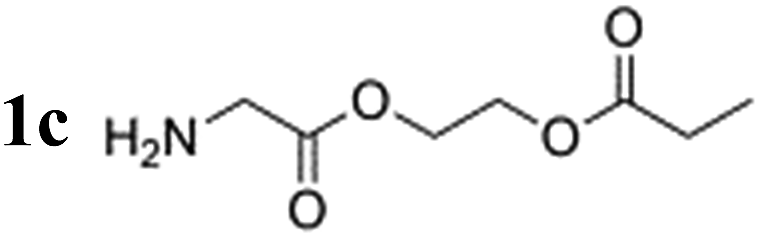
|
0.33 | 0.73 |
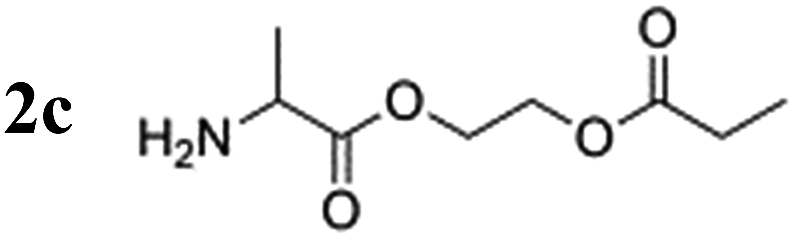
|
0.13 | 0.70 |
|
0.02 | 0.52 |
|
0.48 | 1.02 |
|
0.74 | 1.26 |
For the hyperpolarized glutamine derivative, which contains two unprotected amine groups, proton polarization of only 0.02% was observed at 20 °C, but 0.52% was observed at 80 °C. Previous studies on rhodium transition metal complexes with phosphine ligands indicate that catalytic activity may be reduced if unprotected amine groups are present in the reaction mixture during hydrogenation; the observed trend in polarization can be deduced as follows: the reaction proceeds faster with increasing temperature and the quantum correlation of para-hydrogen used to create hyperpolarization can be transferred more effectively. This effect is contrary to the progressive deactivation of the catalyst by free amine groups, which leads to catalyst decomposition slowing down the addition of para-hydrogen and leading to a decrease in observable proton polarization.44 In this case, the catalyst is sacrificed in the reaction to produce more product carrying a higher polarization.
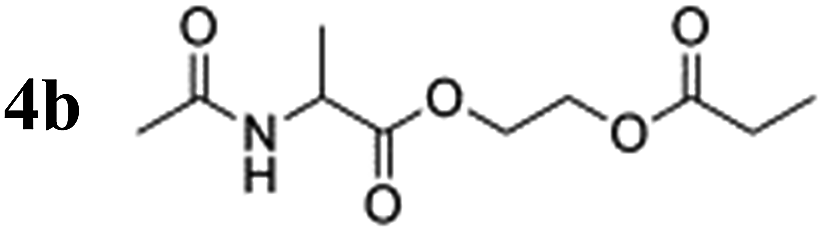
To verify the effect of the free amine on the achieved polarization, a protected alanine-derived amine was synthesized. Because the commonly used tert-butoxycarbonyl (Boc)-protected amino acid ethyl acrylate ester is sparingly soluble in water, an acetyl protecting group was selected to protect the amine (4a). Hyperpolarization was performed accordingly to the procedure described with 10 mol% catalyst and 5 bar para-hydrogen pressure. In the experiments, the level of proton polarization achieved of the acetyl protected alanine derivative was approximately a factor of four (P = 0.48%) higher at 20 °C and ≈1.5 (P = 1.02%) at 80 °C relative to that obtained with the non-acetylated form, which suggests that the free amine group deactivates the rhodium complex, probably as described above. With amine protection, the polarization is within 80% of the hydroxyethyl propionate standard (HEP, 5a).
Subsequently, transfer of the proton polarization to a 13C nucleus was investigated using the alanine derivative. When HEP is polarized with a device that features optimal reaction conditions and mixing techniques to react para-hydrogen with the substrate and pulse sequences for polarization transfer, a 13C polarization of more than 10% can be reached.16,45 By following the same approach, 13C and deuterium-labelled HEA were utilized for the synthesis of a labelled alanine derivative and hyperpolarized, followed by subsequent detection in a 9.4 T NMR magnet (Fig. 3). The polarizer used in this experiment has been recently described.46 The T1 values of 13C nuclei in carbonyl-HEP moieties in non-deuterated molecules and in deuterated solvent are 14–17 s (with air present at 11.7 T, see the ESI† for detailed T1 data) and 21.5 s in the deuterated compound at 14.1 T. These values not only affect the polarization transfer but are typical values necessary for in vivo studies; however, they are expected to be smaller in non-deuterated solvents and in vivo47 (as mentioned previously, a minimum of tens of seconds to minutes are required for transport of tracer molecules to targets of interest). However, it has been pointed out that the relaxation times of 13C in carbonyl groups are strongly influenced by the chemical shift anisotropy, which is proportional to the magnetic field strength.48 The determined T1 = 21.5 s was measured at 14.1 T. Close to the earth's magnetic field, where polarization transfer and transport take place, T1 is expected to be much longer and may be close to 2 minutes as for HEA. The yielded polarization was P = 4.4 ± 1.0% for the alanine derivative and P = 10% for HEA with 100% conversion to the desired product. These values were obtained in a 9.4 T field after an unoptimized transportation process that took 20 s. The 20 s time was due to the physical transport of the sample tube from the polarizer to the center of the magnet. Improvements should be possible, shortening the distance from the polarizer to the magnet. Extrapolating back to time t = 0, which would be prior to the transportation process, (to compare the polarization e.g. with ref. 49) the 13C polarization of the alanine derivative immediately following chemical reaction is estimated to be P = 12%, if a T1 value of 21.5 s is assumed. This gives an indication of the maximally achievable 13C polarization value but may in effect be smaller due to the T1 being greater in low magnetic fields during the transport. Hence, the 13C polarization level for this amino acid derivative may be comparative to polarization levels achieved with metabolites such as pyruvate.11 This result highlights that, despite the presence of free amine groups, the polarization required for in vivo applications is feasible. A 13C polarization of P = 12% would correspond to a signal enhancement of ≈86000 compared to thermal polarizations in a standard clinical MRI scanner (B0 = 1.5 T) or ≈17000000 at 5 mT (see the field strength of interest in ref. 49). Key to achieving such high levels of polarization for amino acid derivatives is the use of the sequence introduced by Goldman et al.16 This sequence works perfectly for the demonstrated derivatives but may not always be applicable for some amino acid derivatives, as discussed in the ESI.†
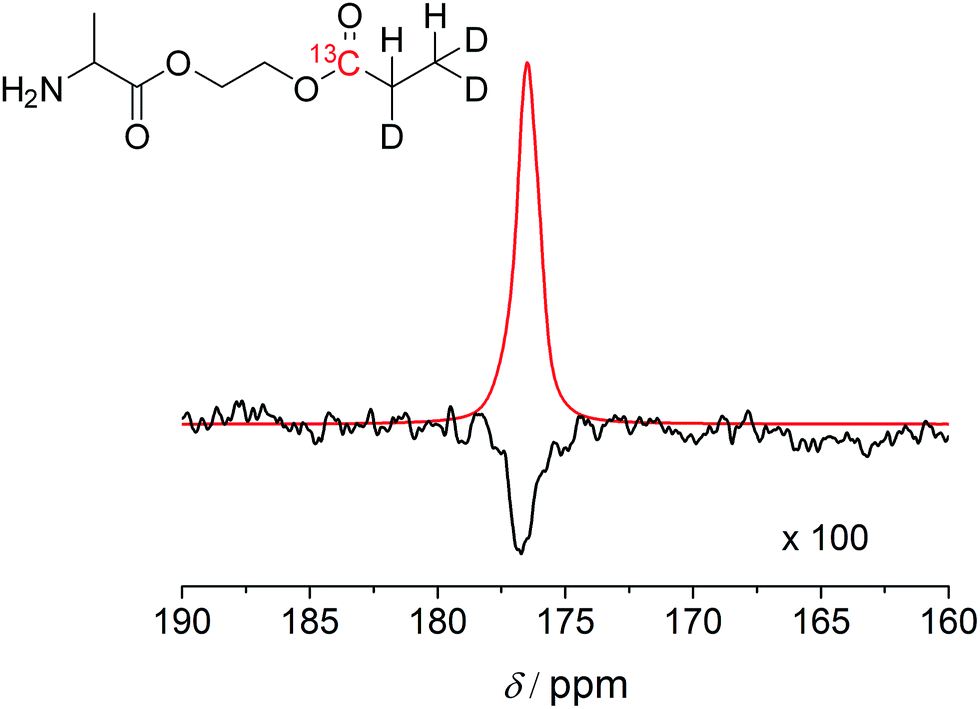 | ||
| Fig. 3 The substrate, deuterium-labeled 2c, was hyperpolarized using a PHIP polarizer device followed by a polarization transfer sequence to 13C nuclei and physical transport of the polarized substrate to the NMR magnet for measurement. Top trace: hyperpolarized signal from 13C NMR spectroscopy and deuterium-labeled (real spectrum) 2c. The physical transport to the NMR magnet took 20 s. Bottom trace: thermally polarized signal after 16 scans following equilibration at 9.4 T and 20 °C. This spectrum was magnified 100 times. The level of polarization in the hyperpolarized product corresponds to 4.4 ± 1.0% after transport. | ||
Conclusions
The N-unprotected α-amino acid ethylacrylate esters based on glycine, alanine and glutamine with hyperpolarizable PHIP moieties were successfully synthesized and applied. The alanine derivative yielded 13C polarization levels in sufficient concentrations (10 mM) in deuterated water and sufficient signal for in vivo applications.9,19 The high polarization, which correlates with the decrease of free amine moieties in the synthesized molecules, indicates a gain of catalyst activity during the hydrogenation reaction. Our findings suggest that amino acid based acrylates can potentially serve as new contrast agents either by themselves or as labelled “building blocks” within peptides that retain partial bioactivity with a limited number of unprotected amine groups. Although, amino acid derivatives are likely to be taken up by cells, it is unclear whether the derivatives take part in metabolic processes that can be observed. Work is currently underway in our laboratory to investigate the metabolic behavior and to extend this method to small cancer-binding peptides that may be of particular interest in combination with recently developed nanoparticles showing significant level of polarization utilizing heterogeneous PHIP in water, thereby mitigating catalyst toxicity effects.50Acknowledgements
The authors gratefully acknowledge financial support from NSF, grant CHE-1153159, equipment grant CHE-1048804, the Jonsson Comprehensive Cancer Center (JCCC) at UCLA and the Arnold and Mabel Beckman Foundation through a Young Investigator Award. Manuscript editing by Schlicht Scientific Editing. The authors thank Philipp Schleker for constructive discussions.Notes and references
- T. G. Walker and W. Happer, Rev. Mod. Phys., 1997, 69, 629 CrossRef CAS.
- S. Appelt, B.-A. Baranga, C. J. Erickson, M. V. Romalis, A. R. Young and W. Happer, Phys. Rev. A, 1998, 58, 1412 CrossRef CAS.
- M. S. Albert, G. D. Cates, B. Driehuys, W. Happer, B. Saam, C. S. Springer Jr and A. Wishnia, Nature, 1994, 370, 199 CrossRef CAS PubMed.
- M. M. Spence, S. M. Rubin, I. E. Dimitrov, E. J. Ruiz, D. E. Wemmer, A. Pines, S. Q. Yao, F. Tian and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10654 CrossRef CAS PubMed.
- L. Schröder, T. J. Lowery, C. Hilty, D. E. Wemmer and A. Pines, Science, 2006, 314, 446 CrossRef PubMed.
- K. Golman, R. in 't Zandt and M. Thaning, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11270 CrossRef CAS PubMed.
- K. Golman, J. H. Ardenkjær-Larsen, J. S. Petersson, S. Mansson and I. Leunbach, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10435 CrossRef CAS PubMed.
- J. H. Ardenkjær-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158 CrossRef PubMed.
- S. J. Kohler, Y. Yen, J. Wolber, A. P. Chen, M. J. Albers, R. Bok, V. Zhang, J. Tropp, S. Nelson, D. B. Vigneron, J. Kurhanewicz and R. E. Hurd, Magn. Reson. Med., 2007, 58, 65 CrossRef CAS PubMed.
- F. A. Gallagher, M. I. Kettunen, S. E. Day, M. Lerche and K. M. Brindle, Magn. Reson. Med., 2008, 60, 253 CrossRef CAS PubMed.
- S. J. Nelson, J. Kurhanewicz, D. B. Vigneron, P. E. Z. Larson, A. L. Harzstark, M. Ferrone, M. van Criekinge, J. W. Chang, R. Bok, I. Park, G. Reed, L. Carvajal, E. J. Small, P. Munster, V. K. Weinberg, J. H. Ardenkjaer-Larsen, A. P. Chen, R. E. Hurd, L.-I.- Odegardstuen, F. J. Robb, J. Tropp and J. A. Murray, Sci. Transl. Med., 2013, 14, 98ra108 Search PubMed.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645 CrossRef CAS.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541 CrossRef CAS.
- T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 8089 CrossRef CAS.
- R. A. Green, R. W. Adams, S. B. Duckett, R. E. Mewis, D. C. Williamson and G. G. R. Green, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 67, 1 CrossRef CAS PubMed.
- M. Goldman, H. Johannesson, O. Axelsson and M. Karlsson, C. R. Chim., 2006, 9, 357 CrossRef CAS PubMed.
- K. Golman, O. Axelsson, H. Johannesson, S. Mansson, C. Olofsson and J. S. Petersson, Magn. Reson. Med., 2001, 46, 1 CrossRef CAS PubMed.
- P. Bhattacharya, K. Harris, A. P. Lin, M. Mansson, V. A. Norton, W. H. Perman, D. P. Weitekamp and B. D. Ross, Magn. Reson. Mater. Phys., Biol. Med., 2005, 18, 245 CrossRef CAS PubMed.
- P. Bhattacharya, E. Y. Chekmenev, W. H. Perman, K. C. Harris, A. P. Lin, V. A. Norton, C. T. Tan, B. D. Ross and D. P. Weitekamp, J. Magn. Reson., 2007, 186, 150 CrossRef CAS PubMed.
- M. Goldman, H. Johannesson, O. Axelsson and M. Karlsson, Magn. Reson. Imaging, 2005, 23, 153 CrossRef CAS PubMed.
- F. Reineri, A. Viale, S. Ellena, D. Alberti, T. Boi, G. B. Giovenzana, R. Gobetto, S. S. D. Premkumar and S. Aime, J. Am. Chem. Soc., 2012, 134, 11146 CrossRef CAS PubMed.
- M. Roth, J. Bargon, H. W. Spiess and A. Koch, Magn. Reson. Chem., 2008, 46, 713 CrossRef CAS PubMed.
- F. Reineri, A. Viale, S. Ellena, D. Alberti, T. Boi, G. B. Giovenzana, R. Gobetto, S. S. D. Premkumar and S. Aime, J. Am. Chem. Soc., 2008, 130, 15047 CrossRef CAS PubMed.
- M. Plaumann, U. Bommerich, T. Trantzschel, D. Lego, S. Dillenberger, G. Sauer, J. Bargon, G. Buntkowsky and J. Bernarding, Chem.–Eur. J., 2013, 19, 6334 CrossRef CAS PubMed.
- P. Bhattachary, E. Y. Chekmenev, W. F. Reynolds, S. Wagner, N. Zacharias, H. R. Chan, R. Bünger and B. D. Ross, NMR Biomed., 2011, 24, 1023 CrossRef PubMed.
- G. Sauer, D. Nasu, D. Tietze, T. Gutmann, S. Englert, O. Avrutina, H. Kolmar and G. Buntkowsky, Angew. Chem., Int. Ed., 2014, 53, 12941 CrossRef CAS PubMed.
- C. Cabella, M. Karlsson, C. Canape, G. Catanzaro, S. Colombo Serra, L. Miragoli, L. Poggi, F. Uggeri, L. Venturi, P. R. Jensen, M. H. Lerche and F. Tedoldi, J. Magn. Reson., 2013, 232, 45 CrossRef CAS PubMed.
- J. Dreischalück, C. Schwöppe, T. Spieker, T. Kessler, K. Tiemann, R. Liersch, C. Schliemann, M. Kreuter, A. Kolkmeyer, H. Hintelmann, R. M. Mesters and W. E. Berdel, Int. J. Oncol., 2010, 37, 1389 Search PubMed.
- M. Oostendorp, K. Douma, T. M. Heckeng, M. A. M. J. van Zandvoort, M. J. Post and W. H. Backes, Contrast Media Mol. Imaging, 2010, 5, 9 CAS.
- F. Gruppi, X. Xu, B. Zhang, J. A. Tnag, A. Jerschow and J. W. Canary, Angew. Chem., Int. Ed., 2012, 51, 11787 CrossRef CAS PubMed.
- S. Glöggler, R. Müller, M. Emondts, M. Dabroski, B. Blümich and S. Appelt, Phys. Chem. Chem. Phys., 2011, 13, 13759 RSC.
- M. Körner, G. Sauer, A. Heil, D. Nasu, M. Empting, D. Tietze, S. Voigt, H. Weidler, T. Gutmann, O. Avrutina, H. Kolmar, T. Ratajczyk and G. Buntkowsky, Chem. Commun., 2013, 49, 7839 RSC.
- P. C. Soon, X. Xu, B. Zhang, F. Gruppi, J. Canary and A. Jerschow, Chem. Commun., 2013, 49, 5304 RSC.
- T. Trantzschel, M. Plaumann, J. Bernarding, D. Lego, T. Ratajczyk, S. Dillenberger, G. Buntkowksy, J. Bargon and U. Bommerich, Appl. Magn. Reson., 2013, 44, 267 CrossRef CAS.
- D. Canet, C. Aroulanda, P. Mutzenhardt, S. Aime, R. Gobetto and F. Reineri, Concepts Magn. Reson., Part A, 2006, 28, 321 CrossRef PubMed.
- H. R. Chan, P. Bhattacharya, A. Imam, A. Freundlich, T. Tran, W. H. Perman, A. P. Lin, K. Harris, E. Y. Chekmenev, M. Ingram and B. D. Ross, Proc. Int. Soc. Mag. Reson. Med., 2009, 17, 2448 Search PubMed.
- R. V. Shchepin, A. M. Coffey, K. W. Wadell and E. Y. Chekemenev, J. Phys. Chem. Lett., 2012, 3, 3281 CrossRef CAS PubMed.
- M. Jain, R. Nilsson, S. Sharma, N. Madhusudhan, T. Kitami, A. L. Souza, R. Kafri, M. W. Kirschner, C. B. Clish and V. K. Mootha, Science., 2012, 336, 1040 CrossRef CAS PubMed.
- R. Stefania, L. Tei, A. Arge, S. G. Crich, I. Szabo, C. Cabella, G. Cravotto and S. Aime, Chem.–Eur. J., 2009, 15, 76 CrossRef CAS PubMed.
- P. A. Pelligrini, N. R. Howell, R. K. Shepherd, N. A. Lengkeek, E. Oehlke, A. G. Katsifis and I. Greguric, Molecules, 2013, 18, 7160 CrossRef PubMed.
- J. M. Webster, C. A. Morton, B. F. Johnson, H. Yuang, M. J. Rishel, B. D. Lee, Q. Miao, C. Pabba, D. T. Yapp and P. Schaffer, J. Nucl. Med., 2014, 55, 657 CrossRef CAS PubMed.
- L. Wang, Z. Zha, W. Qu, H. Qiao, B. P. Lieberman, K. Plössl and H. F. Kung, Nucl. Med. Biol., 2012, 39, 983 Search PubMed.
- M. G. Pravica and D. P. Weitekamp, Chem. Phys. Lett., 1988, 145, 255 CrossRef CAS.
- K. B. Hansen, T. Rosner, M. Kubryk, P. G. Dormer and J. D. Armstrong III, Org. Lett., 2005, 22, 4935 CrossRef PubMed.
- J. B. Hövener, E. Y. Chekmenev, K. C. Harris, W. H. Perman, L. W. Robertson, B. D. Ross and P. Bhattacharya, Magn. Reson. Mater. Phys., Biol. Med., 2009, 22, 111 CrossRef PubMed.
- J. Agraz, A. Grunfeld, K. Cunningham, D. Li and S. Wagner, J. Magn. Reson., 2013, 235, 77 CrossRef CAS PubMed.
- K. R. Keshari and D. M. Wilson, Chem. Soc. Rev., 2014, 43, 1627 RSC.
- F. Reineri, A. Viale, W. Dastrù, R. Gobetto and S. Aime, Contrast Media Mol. Imaging, 2011, 6, 77 CrossRef CAS PubMed.
- R. V. Shchepin, A. M. Coffey, K. W. Waddell and E. Y. Chekmenev, Anal. Chem., 2014, 86, 5601 CrossRef CAS PubMed.
- S. Glöggler, A. M. Grunfeld, Y. N. Ertas, J. McCormick, S. Wagner, P. P. M. Schleker and L.-S. Bouchard, Angew. Chem., Int. Ed., 2015, 54, 2452 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, para-hydrogen experiments, relaxation times of amino acid derivatives, catalyst decomposition and remarks on amino acid PHIP. See DOI: 10.1039/c5sc00503e |
| This journal is © The Royal Society of Chemistry 2015 |