
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed cross-coupling of α-bromocarbonyls and allylic alcohols for the synthesis of α-aryl dicarbonyl compounds†
Yang
Yu
and
Uttam K.
Tambar
*
The University of Texas Southwestern Medical Center at Dallas, Department of Biochemistry, 5323 Harry Hines Boulevard, Dallas, Texas 75390-9038, USA. E-mail: Uttam.Tambar@utsouthwestern.edu; Fax: +1-214-648-8856; Tel: +1-214-648-0580
First published on 17th March 2015
Abstract
The palladium-catalyzed coupling of olefins and organohalides is a versatile approach for synthesizing complex molecules from simple starting materials. We have developed a palladium-catalyzed coupling of α-bromocarbonyl compounds with allylic alcohols for the generation of acyclic aryl-substituted dicarbonyl compounds. The reaction proceeds via a tandem olefin insertion of an α-acyl radical followed by a 1,2-aryl migration. In addition to providing preliminary evidence for a free radical mediated mechanism, we demonstrate unprecedented levels of 1,3-stereoinduction for the 1,2-migration step.
Introduction
Since the initial reports of the Mizoroki–Heck reaction over 40 years ago,1 the palladium-catalyzed coupling of olefins and organohalides has become one of the most effective strategies for the generation of new carbon–carbon bonds.2,3 The widespread use of this reaction manifold is often attributed to the functional group tolerance of palladium catalysts, along with the affordability, accessibility, and stability of olefins and organohalides.We were interested in expanding the synthetic utility of the Mizoroki–Heck reaction by exploiting the unique reactivity of allylic alcohols. This specific class of olefin substrates reacts with organohalides under palladium catalysis to yield traditional Mizoroki–Heck products after β-hydride elimination (Scheme 1a).4 We hypothesized that under modified conditions, the traditional β-hydride elimination pathway could be suppressed and allylic alcohols could be coupled with organohalides to undergo a 1,2-migration (Scheme 1b). Moreover, the use of bromocarbonyl compounds5 as the organohalide would furnish acyclic substituted 1,5-dicarbonyl compounds with multiple stereocenters, which are valuable synthons for many biologically active natural products and pharmaceutical drug candidates.6
 | ||
| Scheme 1 Palladium-catalyzed coupling of allylic alcohols and organohalides. | ||
In this communication, we describe a new strategy for synthesizing acyclic aryl dicarbonyl compounds. Bromocarbonyls and allylic alcohols are coupled in the presence of a palladium catalyst and a silver salt to furnish products with broad substrate scope. We provide evidence for the intermediacy of free radicals in this process and present preliminary data for the control of stereochemistry, with unprecedented levels of 1,3-stereoinduction in the 1,2-aryl migration.7
Results and discussion
Initial exploration of this new approach to aryl dicarbonyls was performed with allylic alcohol 1 and bromoester 2 in the presence of various palladium sources, phosphine ligands, and metal salts as additives (Table 1).8 With [PdCl2(PhCN)2] as the precatalyst and dppe as the ligand, product 3 was not formed in the presence of NaOAc or Cu(OAc)2 (entries 1–2). To our delight, AgOAc furnished the desired product, albeit in low yield (entry 3), and Ag2CO3 proved to be a more effective additive (entry 4). The identity of the ligand also affected the efficiency of the reaction (entries 4–9), and the bidendate ligand dppe was optimal. After a survey of common organic solvents, trifluorotoluene (TFT) was identified as the reaction medium of choice (entries 10–12).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Pd source (5 mol%) | Ligand (10 mol%) | Additive (2 equiv.) | Solvent | Temp. (°C) | Yielda (%) |
| a HNMR yield with 1,4-dimethoxybenzene as an internal standard. b Isolated yield. c Ethyl chloroacetate (0.2 mmol) used as substrate. d Ethyl iodoacetate (0.2 mmol) used as substrate. e Reaction conditions: 1 (0.1 mmol), 2 (0.2 mmol), palladium source (5 mol%), ligand (10 mol%), additive (0.2 mmol), and solvent (0.1 M), 12 h. | ||||||
| 1 | [PdCl2(PhCN)2] | dppe | NaOAc | PhMe | 110 | 0 |
| 2 | [PdCl2(PhCN)2] | dppe | Cu(OAc)2 | PhMe | 110 | 0 |
| 3 | [PdCl2(PhCN)2] | dppe | AgOAc | PhMe | 110 | 24 |
| 4 | [PdCl2(PhCN)2] | dppe | Ag2CO3 | PhMe | 110 | 66 |
| 5 | [PdCl2(PhCN)2] | — | Ag2CO3 | PhMe | 110 | 22 |
| 6 | [PdCl2(PhCN)2] | PPh3 | Ag2CO3 | PhMe | 110 | 63 |
| 7 | [PdCl2(PhCN)2] | P(p-Tol)3 | Ag2CO3 | PhMe | 110 | 55 |
| 8 | [PdCl2(PhCN)2] | P(o-Tol)3 | Ag2CO3 | PhMe | 110 | 43 |
| 9 | [PdCl2(PhCN)2] | PCy3 | Ag2CO3 | PhMe | 110 | 47 |
| 10 | [PdCl2(PhCN)2] | dppe | Ag2CO3 | DMF | 120 | 0 |
| 11 | [PdCl2(PhCN)2] | dppe | Ag2CO3 | Dioxane | 100 | 63 |
| 12 | [PdCl2(PhCN)2] | dppe | Ag2CO3 | PhCF3 | 120 | 93(81)b |
| 13 | — | dppe | Ag2CO3 | PhMe | 110 | 0 |
| 14 | [PdCl 2 (PhCN) 2 ] | dppe | Ag 2 O | PhCF 3 | 120 | 94(83) |
| 15c | [PdCl2(PhCN)2] | dppe | Ag2O | PhCF3 | 120 | 0 |
| 16d | [PdCl2(PhCN)2] | dppe | Ag2O | PhCF3 | 120 | 50 |

The unique reactivity of silver salts may be due to their ability to act as single electron oxidants in radical transformations.9 In the absence of palladium, product 3 was not formed, which suggests that silver is not mediating this process by itself (entry 13). Another less basic silver salt, Ag2O, proved to be an effective additive for the process (entry 14). This observation supports the role of silver salts as single electron oxidants rather than inorganic bases.
Interestingly, the corresponding chloroester did not yield the desired product (entry 15), presumably because of the greater bond dissociation energy of C–Cl than C–Br.10 In addition, the analogous iodoester reacted with allylic alcohol 1 to furnish 1,5-dicarbonyl 3 in considerably lower yield (entry 16), which may be a result of competing reaction pathways for this more reactive α-haloester.
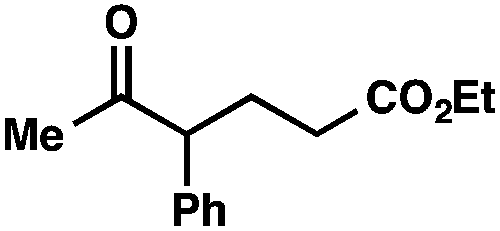
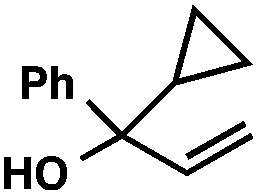
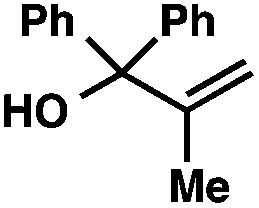
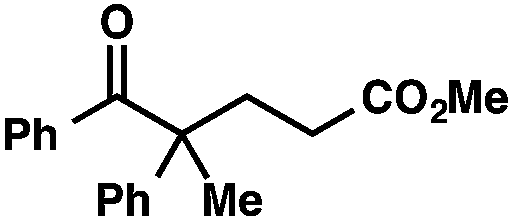
With optimal reaction conditions in hand (Table 1, entry 14), we examined the scope of allylic alcohols that can be coupled with bromoester 5 (Table 2). Symmetrical allylic alcohols (R = Ar) with electron-rich and electron-deficient aromatic rings yielded the aryl ketone products in synthetically useful yields (entries 1–6). The reaction was tolerant of ortho-, meta-, and para-substitution on the aromatic ring. Interestingly, unsymmetrical allylic alcohols coupled with bromoester 5 usually with preferential migration of the electron-deficient aromatic ring (entries 7–8), which sheds light on the mechanism of the 1,2-migration (vide infra). A simple aliphatic group (R = Me) resisted migration, which resulted in the formation of an acyclic unsymmetrical aryl ketone (entry 10). Accessing this ketone by many other methods would be plagued by low regioselectivity in functionalization. Alternatively, a cyclopropyl group (R = c-Pr) underwent 1,2-migration to yield a cyclopropyl phenylketone, albeit as the minor product (entry 11).11 Finally, a 2-substituted allylic alcohol was a competent substrate for the coupling reaction, which furnished a quaternary carbon (entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Allylic alcohol | Product | Yielda (%) | |
a Isolated yield.
b Structural isomer ratio in parentheses (A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B). Structural isomers were separable by column chromatography.
c Ethyl bromoacetate used as substrate (0.4 mmol).
d Reaction conditions: 4 (0.2 mmol), 5 (0.4 mmol), [PdCl2(PhCN)2] (5 mol%), dppe (10 mol%), Ag2O (0.4 mmol), α,α,α-trifluorotoluene (0.1 M), at 120 °C, 12 h. :B). Structural isomers were separable by column chromatography.
c Ethyl bromoacetate used as substrate (0.4 mmol).
d Reaction conditions: 4 (0.2 mmol), 5 (0.4 mmol), [PdCl2(PhCN)2] (5 mol%), dppe (10 mol%), Ag2O (0.4 mmol), α,α,α-trifluorotoluene (0.1 M), at 120 °C, 12 h.
|
||||
| 1 |
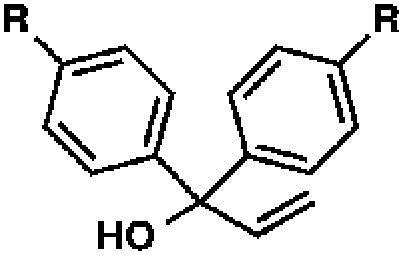
|
R = H |
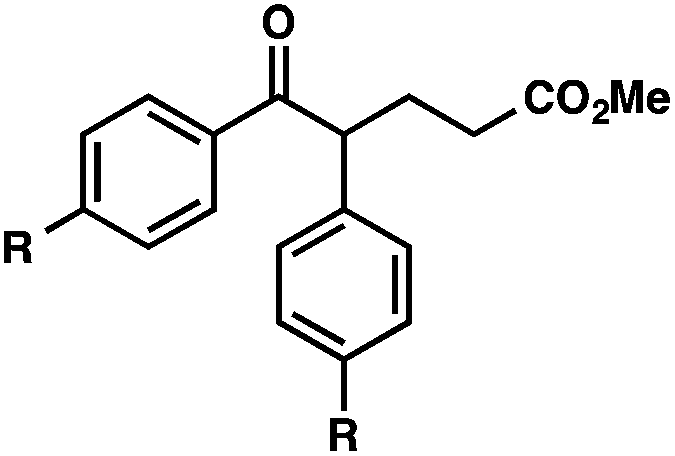
|
84 |
| 2 | R = Me | 56 | ||
| 3 | R = OMe | 85 | ||
| 4 | R = Cl | 66 | ||
| 5 |
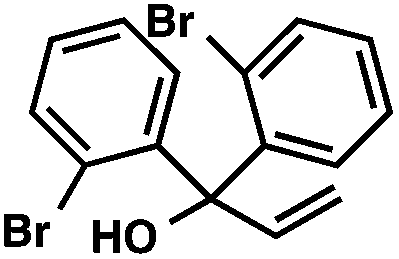
|
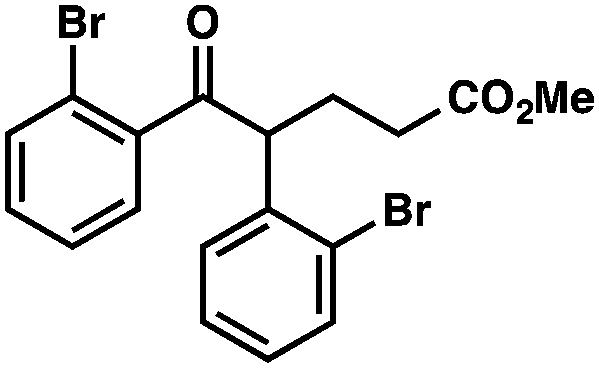
|
42 | |
| 6 |
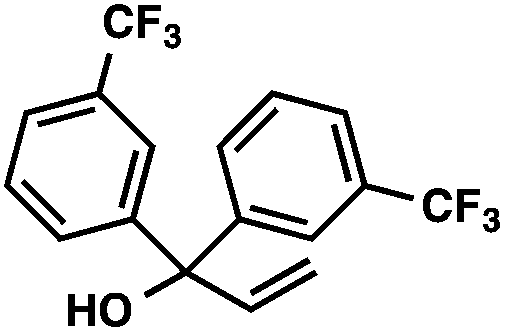
|
|
88 | |
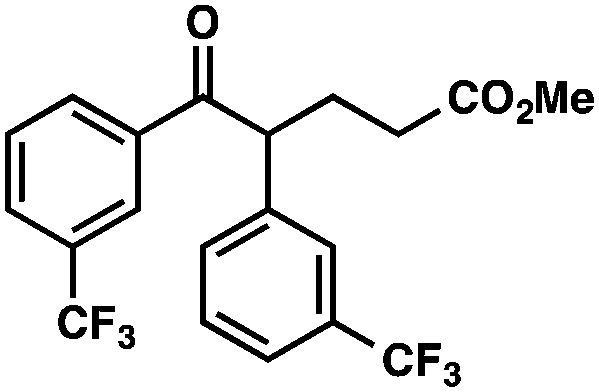
| 7 |
|
Ar = p-OMePh |

|
66 (1.7:1)b |
| 8 | Ar = p-CF3Ph | 81 (1:8)b |
||
| 9 | Ar = 2-Thiophene | 46 (0:1)b |
||
| 10c |
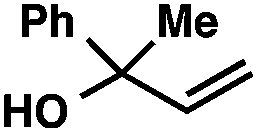
|
|
53 | |
| 11 |
|

|
43 (2.5:1)b |
|
| 12 |
|
|
73 | |
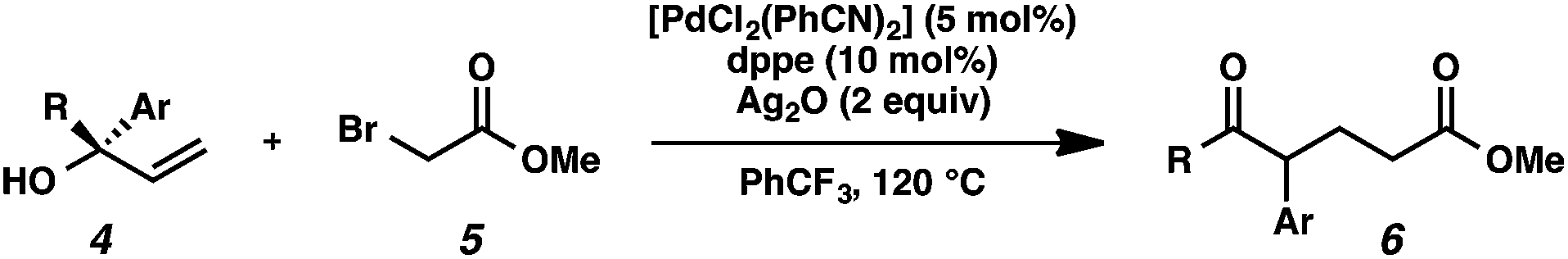
We recognized the potential of this novel coupling reaction to generate aryl dicarbonyl compounds with multiple stereocenters (Table 3). First, we determined that substitution did not affect the efficiency of the overall process (entry 1). Next, the diastereoselectivity of product formation with methyl bromocarbonyl compounds was examined (entries 2–7). An examination of the literature reveals that the 1,4-addition of the enolate of an aryl ketone to a methacrylate derivative leads to low 1,3-stereoinduction in the formation of 1,5-dicarbonyl compounds such as 8.12 We therefore focused on the diastereoselective generation of product 8. Under our palladium-catalyzed coupling conditions, methyl bromoesters generated coupling products with low diastereoselectivity regardless of the steric bulk of the ester (entries 2–4). Weinreb amides,13 on the other hand, exhibited modest diastereoselectivity in product formation (entries 5–6). To our delight, a piperidine-substituted methyl bromoamide was converted to the aryl ketone product with a synthetically useful diastereomeric ratio of 5:1 (entry 7), which is the highest level of 1,3-stereoinduction in 1,2-aryl migrations reported to date.14 The relative stereochemistry of the major diastereomer of the product was confirmed by X-ray crystallography.15
|
|
||||
|---|---|---|---|---|
| Entry | Bromocarbonyl | Product | Yielda (%) | |
|
a Isolated yield.
b Diastereomeric ratio in parentheses (syn:anti).
c Reaction conditions: 1 (0.2 mmol), 7 (0.4 mmol), [PdCl2(PhCN)2] (5 mol%), dppe (10 mol%), Ag2O (0.4 mmol), α,α,α-trifluorotoluene (0.1 M), at 120 °C, 12 h.
|
||||
| 1 |

|
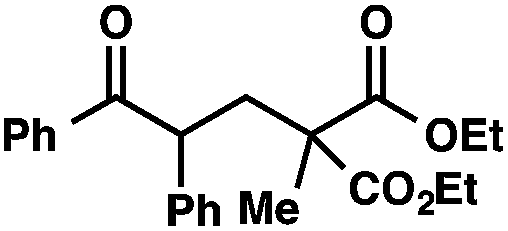
|
72 | |
| 2 |
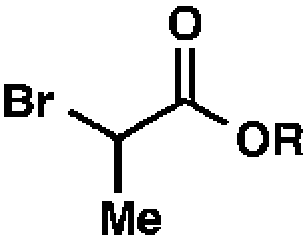
|
R = Me |

|
98 (1.3:1)b |
| 3 | R = Et | 99 (1.3:1)b |
||
| 4 | R = t-Bu | 69 (1.7:1)b |
||
| 5 |
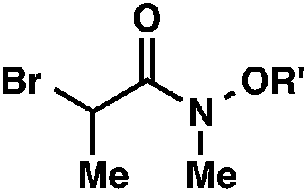
|
R′ = Me |
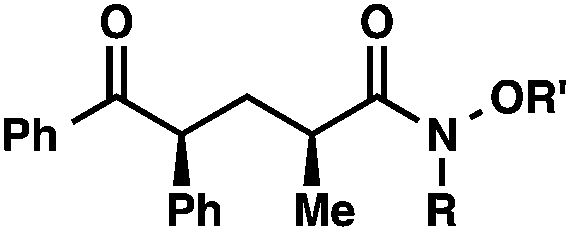
|
92 (3:1)b |
| 6 | R′ = t-Bu | 75 (3.6:1)b |
||
| 7 |
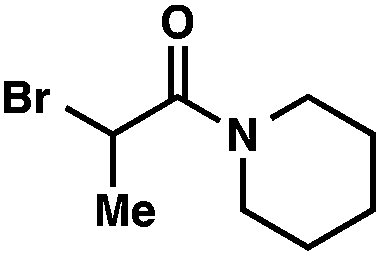
|
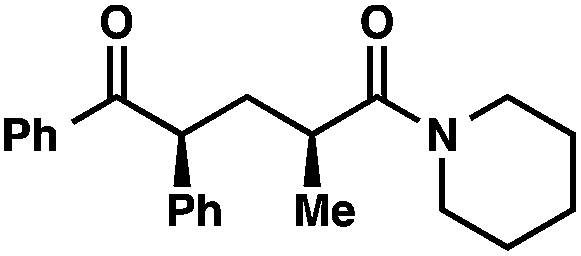
|
95 (5:1)b |
|
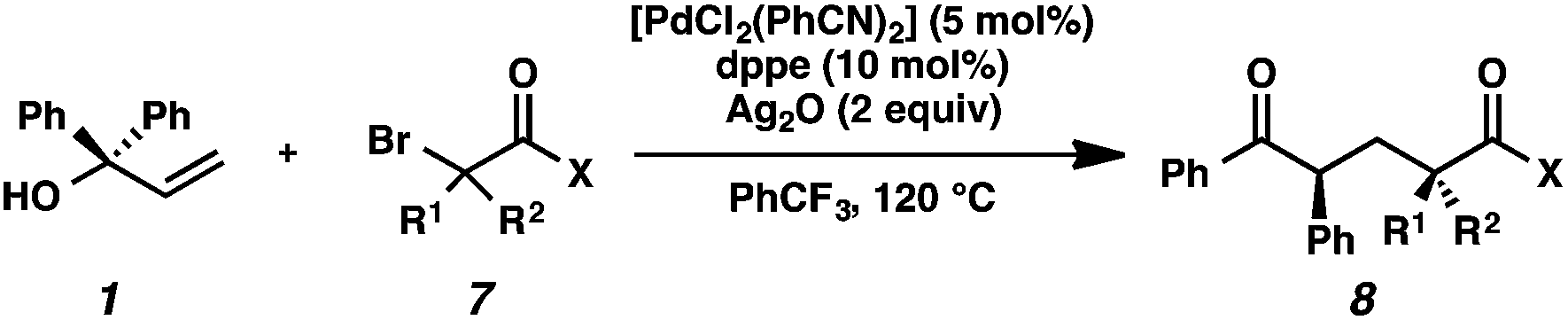
Initial mechanistic studies favor a free radical pathway (Scheme 2, path b) rather than organopalladium intermediates (path a). Inclusion of TEMPO in the reaction medium resulted in the formation of hydroxylamine 14.16 The TEMPO adduct of free radical intermediate 15 was not observed, presumably because 1,2-aryl migrations are kinetically competitive with free radical coupling with TEMPO.17,18 As shown previously, electron-deficient aromatic rings exhibited a greater proclivity for migration than electron-rich aromatic rings (Table 2, entries 7–8), which suggests a neophyl-type rearrangement via spiro[2,5]octadienyl radical 16.19 The stereochemical outcome of subjecting highly enantioenriched allylic alcohols 19a and 19b to the reaction conditions is also consistent with a mechanism that involves acyclic free radical 15. The loss of enantiomeric excess in the formation of aryl ketones 20a and 20b may be the result of 1,2-aryl migration to either stereotopic face of the radical center in intermediate 15.20
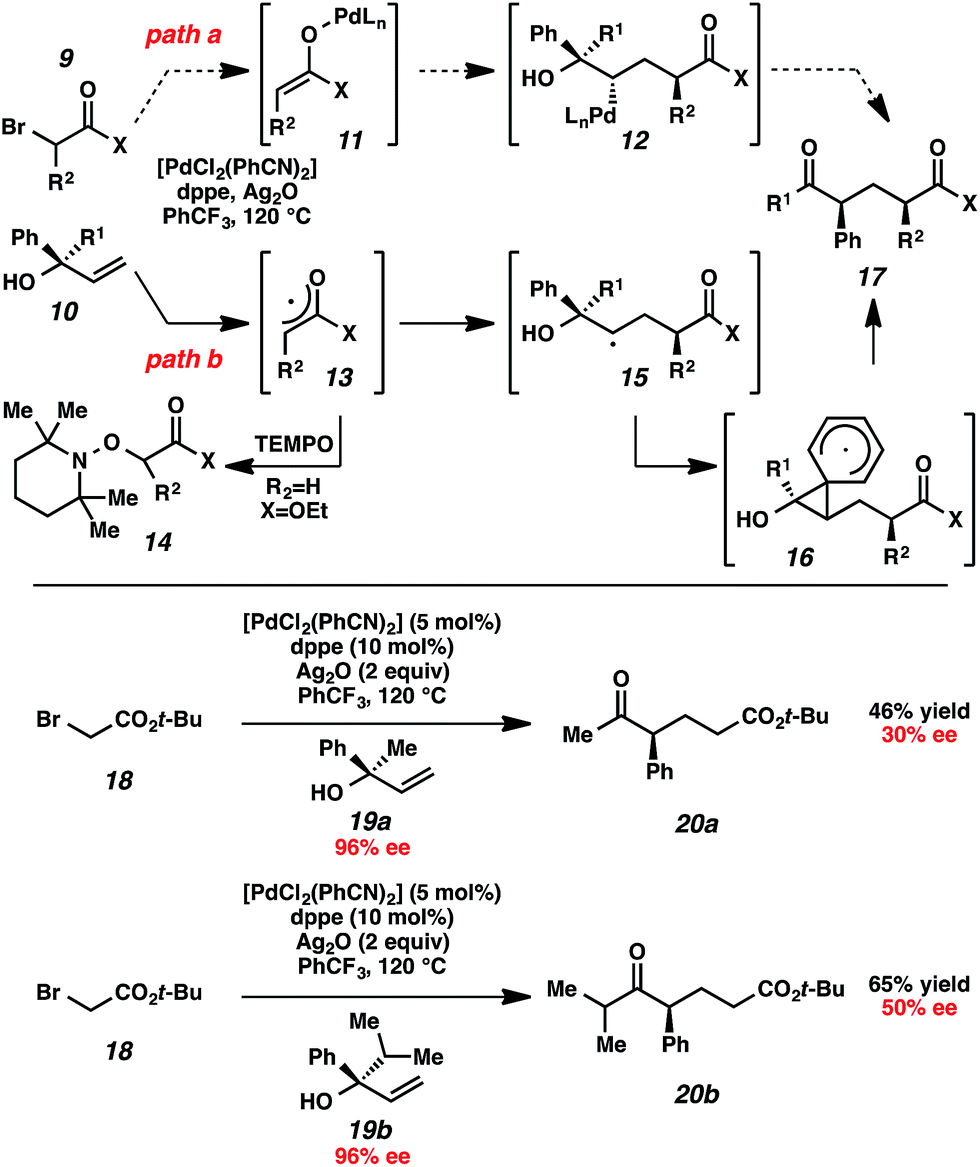 | ||
| Scheme 2 Mechanistic studies. | ||
Conclusions
In conclusion, we have discovered a new mode of reactivity for the palladium-catalyzed coupling of allylic alcohols with bromo carbonyl compounds to generate a broad range of aryl 1,5-dicarbonyls. Despite the intermediacy of acyclic free radicals in the proposed mechanism, we have preliminary data for the formation of acyclic 1,5-dicarbonyl products with synthetically useful levels of 1,3-stereoinduction. We are currently exploring a catalytic enantioselective version of this process and its application to the synthesis of complex natural products.Experimental
In an 8 mL reaction vial, a solution of 1,1-diphenylprop-2-en-1-ol 1 (0.200 mmol, 42.0 mg, 1.0 equiv.), Pd(PhCN)2Cl2 (3.80 mg, 5 mol%), dppe (8.0 mg, 10 mol%), and Ag2O (0.400 mmol, 92.7 mg, 2.0 equiv.), in α,α,α-trifluorotoluene (1.0 mL, 0.2 M) was treated with ethyl bromoacetate 2 (0.400 mmol, 66.9 mg, 2.0 equiv.). The reaction vial was charged with nitrogen for 5 minutes and then sealed. The mixture was stirred at 120 °C for 12 h. After the reaction was finished, the reaction mixture was cooled to room temperature and concentrated under reduced pressure. The resulting residue was purified by silica gel flash chromatography (gradient eluent pentane/diethyl ether) to afford the desired product 3 (49.2 mg, 83% yield) as a clear oil.Acknowledgements
Financial support was provided by the W. W. Caruth, Jr Endowed Scholarship, the Robert A. Welch Foundation (Grant I-1748), the National Institutes of Health (1R01GM102604-01), and the Sloan Research Fellowship. We thank Dr Vincent Lynch for X-ray structural analysis.Notes and references
- (a) R. F. Heck, J. Am. Chem. Soc., 1968, 90, 5518–5526 CrossRef CAS; (b) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581–581 CrossRef CAS; (c) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- For reviews of the Mizoroki–Heck reaction, see: (a) A. de Meijere and F. E. Meyer, Angew. Chem., Int. Ed., 1995, 33, 2379–2411 CrossRef; (b) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed; (c) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2964 CrossRef CAS PubMed; (d) R. F. Heck, Org. React., 1982, 27, 345–390 CAS; (e) M. Oestreich and Editor, The Mizoroki–Heck Reaction, John Wiley & Sons Ltd., 2009 Search PubMed; (f) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS; (g) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- For recent advances in the Mizoroki–Heck reaction, see: (a) Y. Ikeda, T. Nakamura, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2002, 124, 6514–6515 CrossRef CAS PubMed; (b) W. Affo, H. Ohmiya, T. Fujioka, Y. Ikeda, T. Nakamura, H. Yorimitsu, K. Oshima, Y. Imamura, T. Mizuta and K. Miyoshi, J. Am. Chem. Soc., 2006, 128, 8068–8077 CrossRef CAS PubMed; (c) L. Firmansjah and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 11340–11341 CrossRef CAS PubMed; (d) K. S. Bloome and E. J. Alexanian, J. Am. Chem. Soc., 2010, 132, 12823–12825 CrossRef CAS PubMed; (e) K. S. Bloome, R. L. McMahen and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 20146–20148 CrossRef CAS PubMed; (f) M. E. Weiss, L. M. Kreis, A. Lauber and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 11125–11128 CrossRef CAS PubMed; (g) E. W. Werner and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 9692–9695 CrossRef CAS PubMed; (h) E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455–1458 CrossRef CAS PubMed; (i) Z. Yang and J. Zhou, J. Am. Chem. Soc., 2012, 134, 11833–11835 CrossRef CAS PubMed; (j) T. W. Liwosz and S. R. Chemler, Org. Lett., 2013, 15, 3034–3037 CrossRef CAS PubMed; (k) T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830–6833 CrossRef CAS PubMed; (l) E. A. Standley and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 1585–1592 CrossRef CAS PubMed; (m) M. R. Harris, M. O. Konev and E. R. Jarvo, J. Am. Chem. Soc., 2014, 136, 7825–7828 CrossRef CAS PubMed; (n) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340–344 CrossRef CAS PubMed; (o) M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 2282–2285 CrossRef CAS PubMed; (p) L. Xu, M. J. Hilton, X. Zhang, P.-O. Norrby, Y.-D. Wu, M. S. Sigman and O. Wiest, J. Am. Chem. Soc., 2014, 136, 1960–1967 CrossRef CAS PubMed.
- For examples of Mizoroki–Heck reactions with allylic alcohols, see: (a) A. J. Chalk and S. A. Magennis, J. Org. Chem., 1976, 41, 273–278 CrossRef CAS; (b) A. J. Chalk and S. A. Magennis, J. Org. Chem., 1976, 41, 1206–1209 CrossRef CAS; (c) J. B. Melpolder and R. F. Heck, J. Org. Chem., 1976, 41, 265–272 CrossRef CAS; (d) Y. Tamaru, Y. Yamada and Z.-i. Yoshida, Tetrahedron Lett., 1977, 18, 3365–3368 CrossRef; (e) Y. Tamaru, Y. Yamada and Z.-i. Yoshida, Tetrahedron Lett., 1978, 19, 919–922 CrossRef; (f) J.-M. Gaudin, Tetrahedron Lett., 1991, 32, 6113–6116 CrossRef CAS; (g) D. Basavaiah and K. Muthukumaran, Tetrahedron, 1998, 54, 4943–4948 CrossRef CAS; (h) B. M. Trost, J. R. Corte and M. S. Gudiksen, Angew. Chem., Int. Ed., 1999, 38, 3662–3664 CrossRef CAS; (i) M.-S. Schiedel, C. A. Briehn and P. Bäuerle, Angew. Chem., Int. Ed., 2001, 40, 4677–4680 CrossRef CAS; (j) F. Berthiol, H. Doucet and M. Santelli, Tetrahedron Lett., 2004, 45, 5633–5636 CrossRef CAS PubMed; (k) F. Berthiol, H. Doucet and M. Santelli, Appl. Organomet. Chem., 2006, 20, 855–868 CrossRef CAS; (l) F. Berthiol, H. Doucet and M. Santelli, Tetrahedron, 2006, 62, 4372–4383 CrossRef CAS PubMed; (m) V. Calò, A. Nacci, A. Monopoli and V. Ferola, J. Org. Chem., 2007, 72, 2596–2601 CrossRef PubMed; (n) G. Satyanarayana and M. E. Maier, Org. Lett., 2008, 10, 2361–2364 CrossRef CAS PubMed; (o) E. A. Voight, H. Yin, S. V. Downing, S. A. Calad, H. Matsuhashi, I. Giordano, A. J. Hennessy, R. M. Goodman and J. L. Wood, Org. Lett., 2010, 12, 3422–3425 CrossRef CAS PubMed; (p) A. Sauza, J. A. Morales-Serna, M. García-Molina, R. Gaviño and J. Cárdenas, Synthesis, 2012, 272–282 CAS.
- For examples of Mizoroki–Heck reactions with halocarbonyl compounds, see: (a) Q. Liu, H. Yi, J. Liu, Y. Yang, X. Zhang, Z. Zeng and A. Lei, Chem.–Eur. J., 2013, 19, 5120–5126 CrossRef CAS PubMed; (b) T. Nishikata, Y. Noda, R. Fujimoto and T. Sakashita, J. Am. Chem. Soc., 2013, 135, 16372–16375 CrossRef CAS PubMed.
- For recent discussions of the value of aryl dicarbonyl compounds as synthons, see: (a) J. Christoffers, J. Chem. Soc., Perkin Trans. 1, 1997, 3141–3150 RSC; (b) M. J. Chapdelaine and M. Hulce, Org. React., 2004, 38, 225–653 Search PubMed; (c) F. Guo, M. D. Clift and R. J. Thomson, Eur. J. Org. Chem., 2012, 4881–4896 CrossRef CAS PubMed; (d) A.-M. R. Dechert-Schmitt, D. C. Schmitt, X. Gao, T. Itoh and M. J. Krische, Nat. Prod. Rep., 2014, 31, 504–513 RSC; (e) Y. Zhao, A. Aguilar, D. Bernard and S. Wang, J. Med. Chem., 2015, 58, 1038–1052 CrossRef CAS PubMed.
- For recent examples of 1,2-migration of allylic alcohols, see: (a) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962–6966 CrossRef CAS PubMed; (b) X.-Q. Chu, H. Meng, Y. Zi, X.-P. Xu and S.-J. Ji, Chem. Commun., 2014, 50, 9718–9721 RSC; (c) X.-Q. Chu, Y. Zi, H. Meng, X.-P. Xu and S.-J. Ji, Chem. Commun., 2014, 50, 7642–7645 RSC; (d) A. Bunescu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2015, 54, 3132–3135 CrossRef CAS PubMed.
- See ESI† for more complete optimization tables.
- (a) M. Naodovic and H. Yamamoto, Chem. Rev., 2008, 108, 3132–3148 CrossRef CAS PubMed; (b) J.-M. Weibel, A. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149–3173 CrossRef CAS PubMed; (c) J.-H. Fan, W.-T. Wei, M.-B. Zhou, R.-J. Song and J.-H. Li, Angew. Chem., Int. Ed., 2014, 53, 6650–6654 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- The observation of small amounts of cyclopropyl-migration and no methyl-migration (entries 11 vs. 10) may be rationalized by a ring-expanded cyclobutyl intermediate during the 1,2-migration step in Table 2, entry 11.
- For a recent example, see: B. S. Lucas, B. Fisher, L. R. McGee, S. H. Olson, J. C. Medina and E. Cheung, J. Am. Chem. Soc., 2012, 134, 12855–12860 CrossRef CAS PubMed.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS.
- A. Studer and M. Bossart, Tetrahedron, 2001, 57, 9649–9667 CrossRef CAS.
- See ESI† for detailed X-ray crystallographic data.
- M. Newcomb, Tetrahedron, 1993, 49, 1151–1176 CrossRef CAS.
- D. A. Lindsay, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1984, 106, 7087–7093 CrossRef CAS.
- Although it is possible that the TEMPO adduct of free radical 15 is not observed because acyl-radical 13 is completely consumed by TEMPO before 15 is generated, our observation of trace amounts of product 17 even in the presence of TEMPO is not consistent with this alternate explanation.
- (a) W. H. Urry and M. S. Kharasch, J. Am. Chem. Soc., 1944, 66, 1438–1440 CrossRef CAS; (b) A. Effio, D. Griller, K. U. Ingold, J. C. Scaiano and S. J. Sheng, J. Am. Chem. Soc., 1980, 102, 6063–6068 CrossRef CAS; (c) A. N. Abeywickrema, A. L. J. Beckwith and S. Gerba, J. Org. Chem., 1987, 52, 4072–4078 CrossRef CAS; (d) R. Leardini, D. Nanni, G. F. Pedulli, A. Tundo, G. Zanardi, E. Foresti and P. Palmieri, J. Am. Chem. Soc., 1989, 111, 7723–7732 CrossRef CAS.
- For a discussion of the challenges in acyclic stereocontrol with free radical intermediates, see: (a) B. Giese, Angew. Chem., Int. Ed., 1989, 28, 969–980 CrossRef; (b) N. A. Porter, B. Giese and D. P. Curran, Acc. Chem. Res., 1991, 24, 296–304 CrossRef CAS; (c) A. Gansäuer and H. Bluhm, Chem. Rev., 2000, 100, 2771–2788 CrossRef PubMed; (d) M. P. Sibi, S. Manyem and J. Zimmerman, Chem. Rev., 2003, 103, 3263–3296 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1042318. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00505a |
| This journal is © The Royal Society of Chemistry 2015 |