
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereodivergent organocatalysis for the asymmetric synthesis of chiral annulated furans†
Charlie
Verrier
b and
Paolo
Melchiorre
 *ab
*ab
aICREA – Catalan Institution for Research and Advanced Studies, Pg. Lluís Companys 23, 08010 Barcelona, Spain. E-mail: pmelchiorre@iciq.es; Tel: +34 977920208
bICIQ – Institute of Chemical Research of Catalonia, Av. Països Catalans 16, 43007 Tarragona, Spain
First published on 27th April 2015
Abstract
Disclosed herein is a stereoselective method for the synthesis of 2,3-furan fused carbocycles bearing adjacent quaternary and tertiary carbon stereocenters. The chemistry is based on an asymmetric addition of β-ketoesters to 2-(1-alkynyl)-2-alkene-1-ones catalysed by natural cinchona alkaloids followed by a silver-catalysed intramolecular cycloisomerisation. By exploiting the distinct catalysis modes of quinine, which can act either as a general base or, upon opportune modifications, as a phase transfer catalyst, a complete switch of the enforced sense of diastereoinduction is achieved. The stereodivergent systems enable access to the full matrix of all possible stereoisomeric products.
Introduction
This research project was motivated by our interest in devising a direct and versatile strategy for stereoselectively assembling chiral annulated furans.1 As shown in Fig. 1, many biologically active compounds and natural products possess a furan system fused to rings of various sizes and adorned with multiple stereocenters.2 Despite significant advances in preparing racemic 2,3-furan-fused carbocycles,3 there are only a few catalytic strategies for their direct stereoselective synthesis.4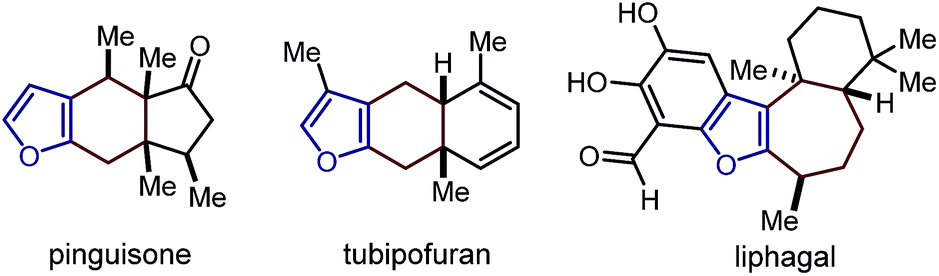 | ||
| Fig. 1 Naturally occurring chiral annulated furans. | ||
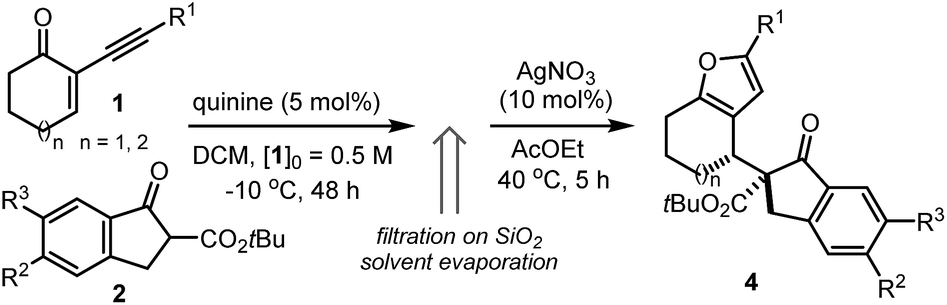
Herein, we describe a straightforward synthetic strategy for accessing six- and seven-membered-ring furan derivatives bearing adjacent quaternary and tertiary carbon stereocentres in very high yields and stereoselectivities. The chemistry, which uses readily available substrates and catalysts, is based on a two-step sequential process whereby an organocatalytic asymmetric addition of β-ketoesters 2 to cyclic 2-(1-alkynyl)-2-alkene-1-ones 1 (ref. 5 and 6) is followed by a silver-catalysed intramolecular cycloisomerisation of the transient allenyl ketone7 intermediate 3 (Scheme 1). Significantly, we have identified two distinct catalytic systems which infer complementary diastereoselectivities, thereby enabling access to the full complement of stereoisomers of the annulated products 4 and 5 at will.8
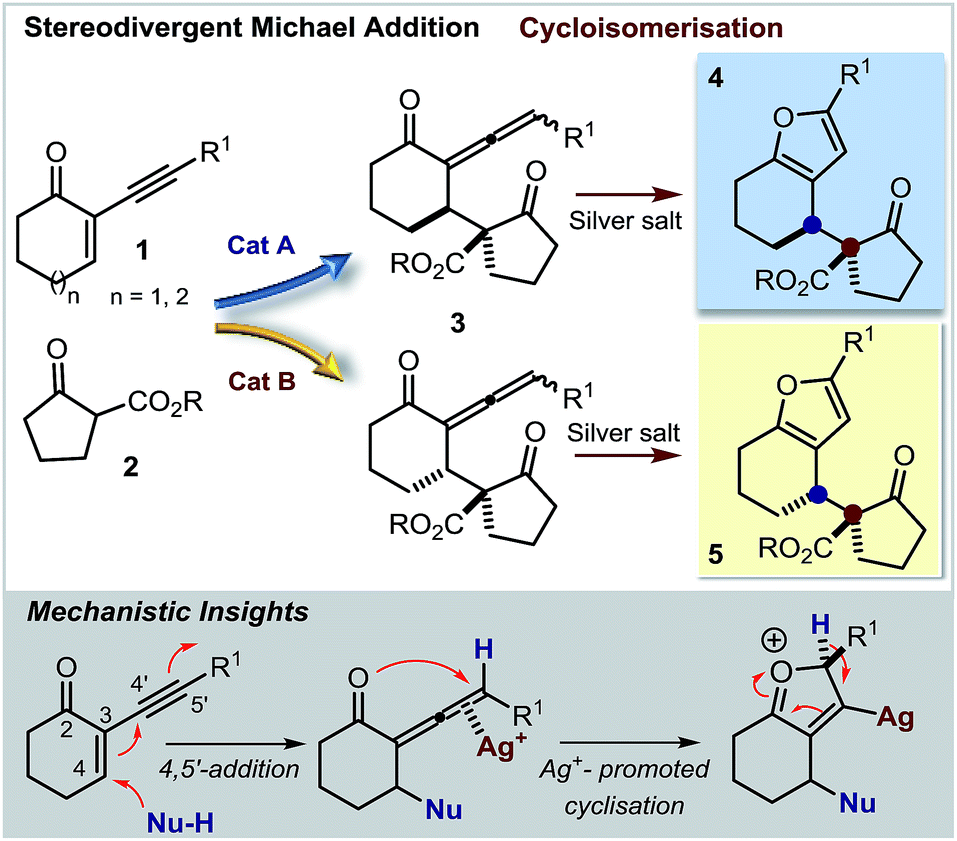 | ||
| Scheme 1 Diastereodivergent and enantioselective synthesis of chiral 2,3-furan fused carbocycles via an organocatalytic 4,5′-addition/cycloisomerisation sequence promoted by distinct organocatalysts (Cat A & Cat B) and a silver catalyst, respectively. | ||
Results and discussion
Our initial explorations focused on the reaction between 2-phenylethynyl-2-cyclohexen-1-one 1a (ref. 9) and the cyclic ketoester 2a in DCM and in the presence of 5 mol% of a chiral tertiary amine, which could act as a general base catalyst (deprotonative activation of 2a). The process was conducted in two sequential steps, with the initial organocatalytic path exclusively providing the corresponding allenyl ketone of type 3 (ref. 10) (Scheme 1) by means of a selective 4,5′-addition.6 Upon filtering out the chiral amine through SiO2 and evaporation of the solvent, the cycloisomerisation of crude 3a was achieved by applying a slightly modified Marshall procedure7,11 (using 10 mol% AgNO3, and AcOEt at 40 °C) to afford the diastereomeric adducts 4 and 5.We focused on identifying chiral organocatalysts that could infer high stereocontrol in the initial 4,5′-addition reaction. Representative results of our extensive studies are listed in Table 1, with more details reported in the ESI.† Intriguingly, natural cinchona alkaloid derivatives,12 acting as general base catalysts,13 afforded impressive levels of stereoselectivity (4a formed with a dr of up to 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and an ee of up to 99%, entries 1–4), largely outperforming any other synthetic catalyst tested (see ESI†).
:1, and an ee of up to 99%, entries 1–4), largely outperforming any other synthetic catalyst tested (see ESI†).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Yieldb (%) |
4a:5a ratioc |
ee (%) major isomerd |
|
a Reactions were performed in DCM at −10 °C on a 0.2 mmol scale using 1.2 equiv. of 2a, with [1a]0 = 0.1 M. After 48 hours, the 4,5′-addition was quenched by filtration through a pad of silica. Upon evaporation of the solvent, the cycloisomerisation of the intermediate 3a was conducted by dissolving the crude residue in 2 mL of AcOEt and adding 10 mol% of AgNO3.
b Yield of the isolated products 4a and 5a (diastereomeric mixture).
c Diastereomeric ratio determined by 1H NMR analysis of the crude mixture upon cycloisomerisation.
d Enantiomeric excess, as determined by HPLC analysis on chiral stationary phases, refers to the major diastereoisomer; the absolute configuration is specified between brackets.
e [1a]0 = 0.5 M.
f Performed at −20 °C in a 5:1 mixture of DCM/33% K2CO3 aq., with [1a]0 = 0.2 M.
g Performed using 10 mol% of catalyst.
|
||||
| 1 | QN | 66 | 19:1 |
99 (1R,2R) |
| 2 | CD | 62 | 12:1 |
99 (1R,2R) |
| 3 | QD | 82 | 18:1 |
98 (1S,2S) |
| 4 | CN | 84 | 17:1 |
98 (1S,2S) |
| 5e | QN | 83 | 19:1 |
99 (1R,2R) |
| 6 | OMe-QN | 15 | 1:1.5 |
<5 |
| 7 | QN-OH | <5 | — | — |
| 8f,g | PTC-QN | 73 | 1:13 |
86 (1R,2S) |
| 9f | PTC-QD | 89 | 1:19 |
87 (1S,2R) |
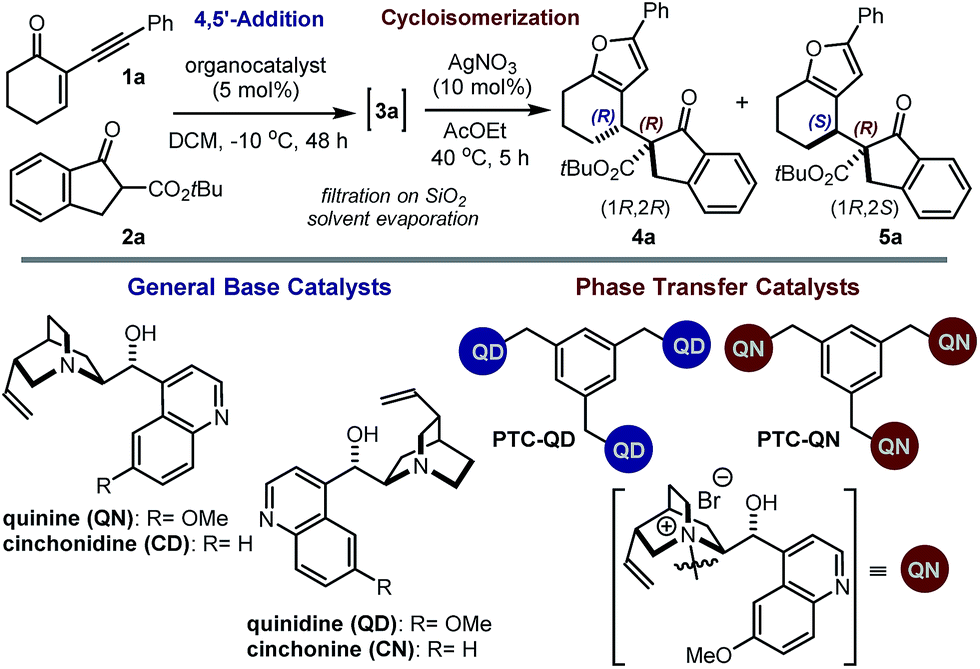
Importantly, the use of the “pseudoenantiomeric” catalysts quinine (QN) and quinidine (QD) secured access to both antipodes of the adduct 4 with excellent selectivity (entries 5 & 3, respectively). Protection of the quinine hydroxy moiety resulted in a greatly reduced reactivity along with a complete loss of stereocontrol (entry 6). Mechanistically, this suggests that the cinchona catalysts might operate through a bifunctional activation mode, simultaneously binding and activating the two reacting partners.13a,14 Interestingly, the cupreidine derivative QN-OH, a catalyst with a proven ability to promote the highly stereoselective addition of β-ketoesters to cyclic enones, remained completely inactive in our system (entry 7).15
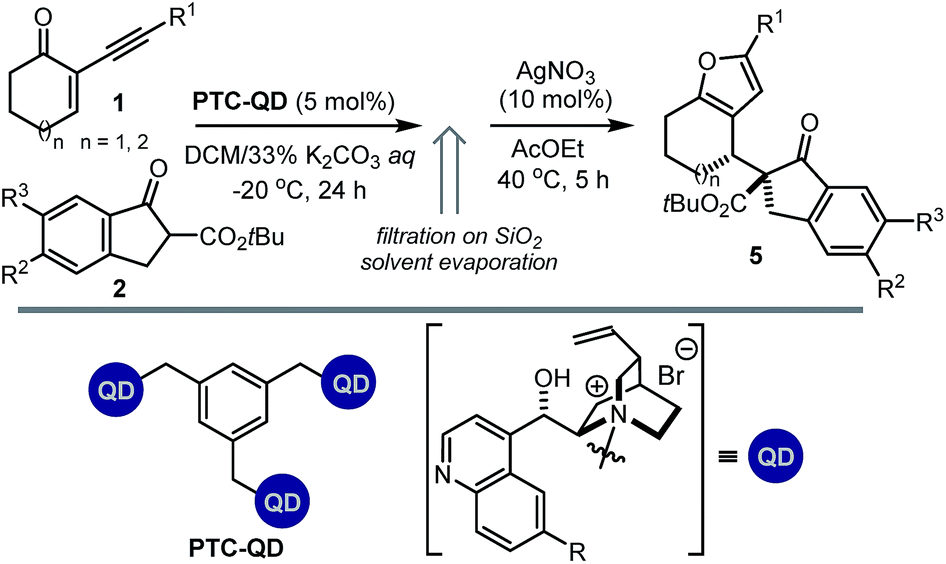
We then modified the cinchona alkaloid scaffold by alkylating the basic bridgehead nitrogen of the quinuclidine core, the classical approach for achieving catalysts suitable for use as phase transfer catalysts (PTCs).16 Among the many PTCs tested in the model reaction (see ESI† for details), the cinchona-derived trimeric species PTC-QN and PTC-QD,17 easily obtained by the poly-alkylation of quinine and quinidine with 1,3,5-tris(bromomethyl)benzene, provided the most interesting results. When performing the reaction in DCM and in the presence of 33% K2CO3 aq., a complete switch of the enforced sense of diastereoinduction was achieved, so that the adduct 5 was almost exclusively formed in high optical purity (entries 8 & 9).
These findings allowed us to fully control the stereochemical outcome of the process, enabling the generation of any stereoisomer of the annulated furans 4 and 5 at will. This is considered a challenging goal because when asymmetric catalysis is applied to processes that generate two stereogenic centres in one product, there is generally no obvious means of modifying a catalyst to modulate the relative sense of those two centres.8 In this case, the two diastereodivergent systems are based on different organocatalysts, but are derived from the common chiral core of natural cinchona alkaloids. The divergent stereocontrol arises from the ability of the cinchona catalysts to execute distinct modes of catalysis for activating the reagents (base catalysis vs. phase transfer catalysis, Scheme 2).
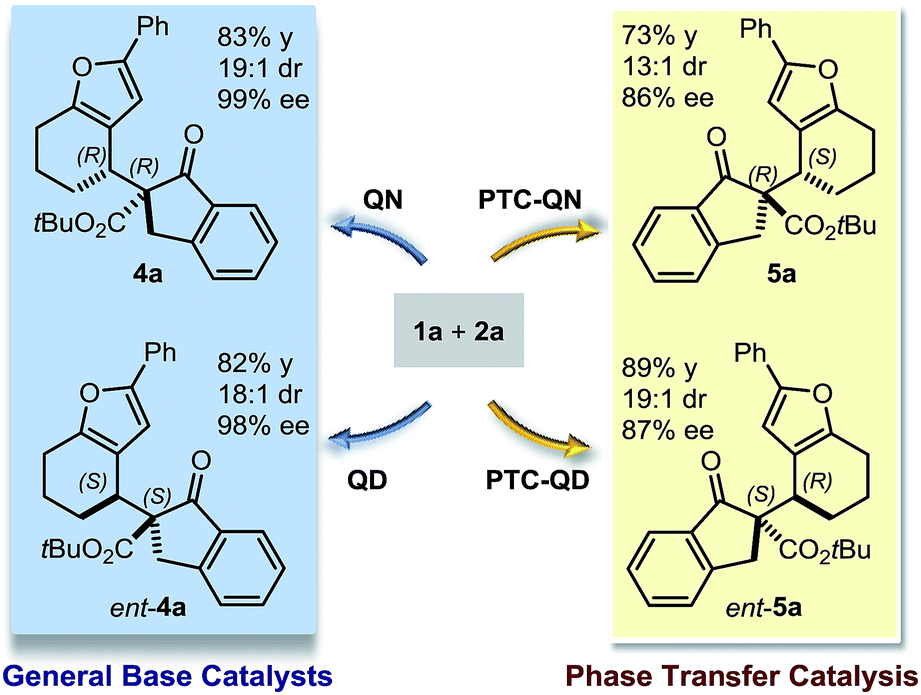 | ||
| Scheme 2 Accessing the full matrix of the stereoisomers of the annulated furans 4a and 5a. When QN and QD function as general base catalysts, both enantiomers of the diastereoisomer 4a could be accessed. Modifying the catalyst structure to induce a different activation pattern, namely phase transfer catalysis, enables direct access to both antipodes of 5a using PTC-QN and PTC-QD. | ||
Having identified two distinct catalytic systems that can selectively channel the reaction manifolds toward complementary diastereochemical outcomes, we examined the scope of the two-step process using QN (5 mol%) as a general base catalyst. As revealed in Table 2, the method shows a wide substrate generality and an excellent level of stereoselectivity, providing access to a variety of complex annulated furans 4 adorned with two vicinal quaternary and tertiary stereocentres. We first tested the possibility of modifying the cyclic scaffold of the 2-(1-alkynyl)-2-alken-1-one component 1. The cycloheptenone derivative reacted smoothly to provide the seven-membered-ring furan 4b in high optical purity (entry 2). In contrast, the cyclopentenone derivative was not a suitable substrate, since a complete lack of reactivity in the silver-catalysed intramolecular cycloisomerisation step was observed. Different substitution patterns on the aromatic moiety of 1 were well-tolerated, regardless of their electronic properties (entries 3–5). In addition, an alkyne bearing a vinylic substituent (entry 6) provided the corresponding furan 4f with high stereocontrol, albeit with a moderate chemical yield. A limitation of the system is that we have thus far failed to react alkynes bearing alkyl or TMS groups, and linear substrates.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R 1, n | R 2 | R 3 | 4 | Yieldb (%) | drc | eed (%) |
| a Reactions were performed at −10 °C on a 0.2 mmol scale and using 1.2 equiv. of 2. After 48 hours, the organocatalytic 4,5′-addition was quenched by filtration through a pad of silica. Upon evaporation of the solvent, the crude residue was dissolved in 2 mL of AcOEt and 10 mol% of AgNO3 was added. b Yield of the isolated products 4 (diastereomeric mixture). c Diastereomeric ratio determined by 1H NMR analysis of the crude mixture upon cycloisomerization. d Enantiomeric excess determined by HPLC analysis on chiral stationary phases. e The absolute configuration of 4h was unambiguously inferred by X-ray analysis, see ref. 18. | |||||||
| 1 | Ph, 1 | H | H | a | 83 | 19:1 |
99 |
| 2 | Ph, 2 | H | H | b | 75 | 12:1 |
97 |
| 3 | 4-MeOC6H4, 1 | H | H | c | 83 | 19:1 |
99 |
| 4 | 4-CF3C6H4, 1 | H | H | d | 90 | 19:1 |
99 |
| 5 | 4-MeC6H4, 1 | H | H | e | 83 | 19:1 |
99 |
| 6 |
|
H | H | f | 25 | 19:1 |
95 |
| 7 | Ph, 1 | MeO | H | g | 62 | 19:1 |
99 |
| 8 | Ph, 1 | H | Br | h | 82 | 19:1 |
98 |
| 9 | Ph, 1 | MeO | MeO | i | 67 | 19:1 |
99 |
| 10 | Ph, 1 | H | Me | j | 87 | 19:1 |
97 |
| 11 | Ph, 1 | F | H | k | 88 | 19:1 |
98 |
As for the nucleophilic partners 2, electronic variations in the indanone ring were possible, as both electron donating and withdrawing substituents gave the desired products in high yields and excellent diastereo- and enantioselectivities (entries 7–11). Efforts to react six-membered cyclic and linear β-ketoesters need further optimization, since only traces of the corresponding products could be obtained.
We then evaluated the synthetic potential of the PTC-mediated system. As depicted in Table 3, the reactions catalysed by 5 mol% of PTC-QD showed a comparable versatility to the system under general base catalysis, but secured a complementary diastereoselectivity, since the opposite diastereoisomers 5 of the annulated furans were almost exclusively formed with high enantiocontrol.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R 1, n | R 2 | R 3 | 5 | Yieldb (%) | drc | eed (%) |
|
a Reactions were performed at −20 °C in a 5:1 mixture of DCM/33% K2CO3 aq., on a 0.2 mmol scale and using 1.2 equiv. of 2; with [1a]0 = 0.2 M. After 24 hours, the 4,5′-addition was quenched by filtration through a pad of silica. Upon evaporation of the solvent, the crude residue was dissolved in 2 mL of AcOEt and 10 mol% of AgNO3 was added.
b Yield of the isolated products 5 (diastereomeric mixture).
c Diastereomeric ratio determined by 1H NMR analysis of the crude mixture upon cycloisomerisation.
d Enantiomeric excess determined by HPLC analysis on chiral stationary phases.
e The absolute configuration of 5a was unambiguously inferred by X-ray analysis, see ref. 18.
f Reactions were performed at −10 °C using 1.5 mol% of catalyst.
|
|||||||
| 1e | Ph, 1 | H | H | a | 89 | 19:1 |
87 |
| 2 | Ph, 2 | H | H | b | 81 | 9.5:1 |
93 |
| 3 | 4-MeOC6H4, 1 | H | H | c | 87 | 19:1 |
89 |
| 4 | 4-CF3C6H4, 1 | H | H | d | 84 | 8:1 |
81 |
| 5 | 4-MeC6H4, 1 | H | H | e | 82 | 19:1 |
91 |
| 6f |
|
H | H | f | 53 | 19:1 |
92 |
| 7f | Ph, 1 | MeO | MeO | i | 62 | 16:1 |
72 |
| 8f | Ph, 1 | H | Br | h | 78 | 11:1 |
77 |
| 9 | Ph, 1 | H | Me | j | 87 | 19:1 |
87 |
| 10 | Ph, 1 | F | H | k | 87 | 15:1 |
85 |
| 11 | Ph, 1 | Br | H | l | 70 | 19:1 |
83 |
The relative and absolute configuration of products 4h and 5a were unambiguously inferred by anomalous dispersion X-ray crystallographic analysis.18
Conclusions
In conclusion, we have developed an operationally simple, two-step process to access a variety of stereochemically dense 2,3-furan fused carbocycles bearing adjacent quaternary19 and tertiary stereocentres. The salient feature of our study is that complementary organocatalytic systems, both using natural cinchona-derived catalysts, have been identified, which ensure highly selective access to the full matrix of all possible stereoisomeric products at will.Acknowledgements
This work was supported by the ICIQ Foundation, and MINECO (CTQ2013-45938-P). C.V. thanks CMIRA 2013-EXPLO'RA PRO, grant number: 006402-01 (region Rhone-Alpes, France) for postdoctoral support. We also thank MINECO for support through the Severo Ochoa Excellence Accreditation 2014–2018 (SEV-2013-0319) and the CELLEX Foundation through the CELLEX-ICIQ high throughput experimentation platform.Notes and references
- For recent reviews on furan synthesis, see: (a) W. J. Moran and A. Rodríguez, Org. Prep. Proced. Int., 2012, 44, 103 CrossRef CAS PubMed; (b) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed; (c) H. N. C. Wong, K.-S. Yeung and Z. Yang, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katrizky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, ch. 3.06, p. 407 Search PubMed.
- (a) B. A. Keay, J. M. Hopkins and P. W. Dibblein, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katrizky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, ch. 3.08, p. 571 Search PubMed; (b) H. H. Issa, J. Tanaka and T. Higa, J. Nat. Prod., 2003, 66, 251 CrossRef CAS PubMed. Pinguisone: (c) S. Bernasconi, P. Gariboldi, G. Jommi, S. Montanari and M. Sisti, J. Chem. Soc., Perkin Trans. 1, 1981, 2394 RSC. Tubipofuran: (d) K. Iguchi, K. Mori, M. Suzuki, H. Takahashi and Y. Yamada, Chem. Lett., 1986, 1789 CrossRef CAS; (e) E. P. Kündig, R. Cannas, M. Laxmisha, L. Ronggang and S. Tchertchian, J. Am. Chem. Soc., 2003, 125, 5642 CrossRef PubMed. Liphagal: (f) F. Marion, D. E. Williams, B. O. Patrick, I. Hollander, R. Mallon, S. C. Kim, D. M. Roll, L. Feldberg, R. van Soest and R. J. Andersen, Org. Lett., 2006, 8, 321 CrossRef CAS PubMed; (g) J. J. Day, R. M. McFadden, S. C. Virgil, H. Kolding, J. L. Alleva and B. M. Stoltz, Angew. Chem., Int. Ed., 2011, 50, 6814 CrossRef CAS PubMed.
- For selected examples, see: (a) A. S. K. Hashmi, P. Haufe, C. Schmid, A. Rivas Nass and W. Frey, Chem.–Eur. J., 2006, 12, 5376 CrossRef CAS PubMed; (b) Z. Dong, C.-H. Liu, Y. Wang, M. Lin and Z.-X. Yu, Angew. Chem., Int. Ed., 2013, 52, 14157 CrossRef CAS PubMed; (c) L. Zhou, M. Zhang, W. Li and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6542 CrossRef CAS PubMed.
- (a) F. Liu, D. Qian, L. Li, X. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6669 CrossRef CAS PubMed; (b) V. Rauniyar, Z. J. Wang, H. E. Burks and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 8486 CrossRef CAS PubMed; (c) Z.-M. Zhang, P. Chen, W. Li, Y. Niu, X.-L. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4350 CrossRef CAS PubMed.
- It is of note that, to date, only a single example of enantioselective catalytic Michael addition to activated enynes to afford enantio-enriched 2,3-allenoates has been reported, see: H. Qian, X. Yu, J. Zhang and J. Sun, J. Am. Chem. Soc., 2013, 135, 18020 CrossRef CAS PubMed.
- For examples of non-stereoselective 4,5′-additions of nucleophiles to 1 leading to racemic 1,2-allenyl ketones of type 3, see: (a) X. Yu, H. Ren, Y. Xiao and J. Zhang, Chem.–Eur. J., 2008, 14, 8481 CrossRef CAS PubMed; (b) X. Yu and J. Zhang, Adv. Synth. Catal., 2011, 353, 1265 CrossRef CAS PubMed.
- (a) J. A. Marshall and X. J. Wang, J. Org. Chem., 1991, 56, 960 CrossRef CAS; (b) J. A. Marshall and G. S. Bartley, J. Org. Chem., 1994, 59, 7169 CrossRef CAS . For a review, see; (c) P. Belmont and E. Parker, Eur. J. Org. Chem., 2009, 6075 CrossRef CAS PubMed.
- For highlight articles on diastereo-divergent strategies that give access to the full matrix of product stereoisomers from the same set of starting materials, see: (a) C. S. Schindler and E. N. Jacobsen, Science, 2013, 340, 1052 CrossRef CAS PubMed; (b) M. T. Oliveira, M. Luparia, D. Audisio and N. Maulide, Angew. Chem., Int. Ed., 2013, 52, 13149 CrossRef CAS PubMed. For selected examples, see: (c) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed; (d) S. Krautwald, M. A. Schafroth, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 3020 CrossRef CAS PubMed; (e) X. Tian, C. Cassani, Y. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 17934 CrossRef CAS PubMed; (f) M. Luparia, M. T. Oliveira, D. Audisio, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631 CrossRef CAS PubMed; (g) A. Nojiri, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 3779 CrossRef CAS PubMed; (h) B. Simmons, A. M. Walji and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2009, 48, 4349 CrossRef CAS PubMed; (i) Y. Chi, S. T. Scroggins and J. M. Fréchet, J. Am. Chem. Soc., 2008, 130, 6322 CrossRef CAS PubMed; (j) B. Wang, F. Wu, Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2007, 129, 768 CrossRef CAS PubMed.
- Metal-catalysed cycloisomerisation of 2-(1-alkynyl)-2-alkene-1-ones 1 has been widely used in the non-stereoselective preparation of furans, see: (a) T. Yao, X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 11164 CrossRef CAS PubMed; (b) T. Yao, X. Zhang and R. C. Larock, J. Org. Chem., 2005, 70, 7679 CrossRef CAS PubMed; (c) N. T. Patil, H. Wu and Y. Yamamoto, J. Org. Chem., 2005, 70, 4531 CrossRef CAS PubMed . For stereoselective variants, see ref. 4a and b.
- Under the reaction conditions detailed in Table 1, the allenyl ketone intermediate 3a is produced with very poor diastereomeric purity (dr ranging from 1:1 to 1.5:1) as a consequence of the lack of stereocontrol over the axial chirality. This stereochemical information, however, is eventually lost during the cycloisomerisation to give the annulated furans.
- All attempts to perform the organocatalytic 4,5′-addition/cycloisomerisation sequence in one-pot have met with failure. For recently reported examples showing the compatibility of silver with organocatalysis, see: (a) D. Hack, P. Chauhan, K. Deckers, G. N. Hermann, L. Mertens, G. Raabe and D. Enders, Org. Lett., 2014, 16, 5188 CrossRef CAS PubMed and references therein; (b) I. Ortín and D. J. Dixon, Angew. Chem., Int. Ed., 2014, 53, 3462 CrossRef PubMed.
- C. E. Song, Cinchona Alkaloids in Synthesis and Catalysis; Wiley-VCH: Weinheim, Germany, 2009 Search PubMed.
- For a pioneering work, see: (a) H. Hiemstra and H. Wynberg, J. Am. Chem. Soc., 1981, 103, 417 CrossRef CAS. For a review: (b) S. Ingemann and H. Hiemstra, in Comprehensive Enantioselective Organocatalysis, ed. P. Dalko, Wiley-VCH, Weinheim, Germany, 2013, vol. 1, ch. 6, p. 119 Search PubMedFor selected examples of highly enantioselective reactions catalyzed by natural cinchona alkaloids, see: (c) S. Lou, B. M. Taoka, A. Ting and S. E. Schaus, J. Am. Chem. Soc., 2005, 127, 11256 CrossRef CAS PubMed; (d) G. Bartoli, M. Bosco, A. Carlone, A. Cavalli, M. Locatelli, A. Mazzanti, P. Ricci, L. Sambri and P. Melchiorre, Angew. Chem., Int. Ed., 2006, 45, 4966 CrossRef CAS PubMed.
- For a computational study, see: C. S. Cucinotta, M. Kosa, P. Melchiorre, A. Cavalli and F. L. Gervasio, Chem.–Eur. J., 2009, 15, 7913 CrossRef CAS PubMed.
- F. Wu, H. Li, R. Hong and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 947 CrossRef CAS PubMed.
- Asymmetric Phase Transfer Catalysis, ed. K. Maruoka, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- (a) W. E. Siew, C. Ates, A. Merschaert and A. G. Livingston, Green Chem., 2013, 15, 663 RSC; (b) R. Sallio, S. Lebrun, N. Schifano-Faux, J.-F. Goossens, F. Agbossou-Niedercorn, E. Deniau and C. Michon, Synlett, 2013, 24, 1785 CrossRef CAS PubMed.
- ESI† The absolute configuration of the light-atom molecule 5a was assigned using the X-ray diffraction methodology described in: E. C. Escudero-Adán, J. Benet-Buchholz and P. Ballester, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 660 Search PubMed.
- For a review highlighting modern catalytic enantioselective methods to forge quaternary carbon stereocentres, see: K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and full compound characterisation, including HPLC traces and NMR spectra (PDF). CCDC 1046068 and 1046069. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01052g |
| This journal is © The Royal Society of Chemistry 2015 |