
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrodialysis SPR: diffusion-gated sensing in blood†
Julien
Breault-Turcot
a and
Jean-Francois
Masson
*ab
aDepartement de chimie, Université de Montréal, CP 6128 Succ. Centre-Ville, Montreal, QC H3C 3J7, Canada. E-mail: jf.masson@umontreal.ca; Tel: +1-514-343-7342
bCentre for Self-Assembled Chemical Structures (CSACS), Canada
First published on 8th May 2015
Abstract
Chemical measurements are rarely performed in crude blood due to the poor performance of sensors and devices exposed to biofluids. In particular, biosensors have been severely limited for detection in whole blood due to surface fouling from proteins, the interaction of cells with the sensor surface and potential optical interference when considering optical methods of analysis. To solve this problem, a dialysis chamber was introduced to a surface plasmon resonance (SPR) biosensor to create a diffusion gate for large molecules. This dialysis chamber relies on the faster migration of small molecules through a microporous membrane towards a sensor, located at a specified distance from the membrane. Size filtering and diffusion through a microporous membrane restricted the access of blood cells and larger biomolecules to a sensing chamber, while smaller, faster diffusing biomolecules migrated preferentially to the sensor with limited interference from blood and serum. The affinity of a small peptide (DBG178) with anti-atherosclerotic activity and targeting type B scavenger receptor CD36 was successfully monitored at micromolar concentrations in human serum and blood without any pre-treatment of the sample. This concept could be generally applied to a variety of targets for biomolecular interaction monitoring and quantification directly in whole blood, and could find potential applications in biochemical assays, pharmacokinetic drug studies, disease treatment monitoring, implantable plasmonic sensors, and point-of-care diagnostics.
Introduction
Blood remains one of the most important biofluids for gathering information about the health of an individual. The concentrations of proteins, blood cells, metabolites and therapeutic molecules circulating in blood can provide important information about the health of patients, the status of many biological processes and functions, and the progress of therapy. A common example involves the analysis of blood to determine the level of certain biomarkers for use in disease screening.2,3 Blood is also a vehicle for disease treatment, and it is important to be able to detect the concentration of circulating therapeutic drugs, as well as monitoring the fate of these drugs.4 However, most contemporary techniques fail to analyze molecules directly in whole blood and thus require time consuming and labour intensive sample preparation to avoid interference from the cells and proteins contained in blood. This factor has severely limited the development of several technologies in biomedical sciences such as point-of-care tests and pharmacokinetic studies which utilise biosensors.The ability to carry out an analysis in blood has many advantages, including the reduced sample preparation needed, which is applicable to point-of-care diagnostics. Additionally, it would provide a more realistic environment for biomolecular interaction assays that would account for the potential molecular reactions (degradation, complexation or metabolism) that occur in a native biological environment. For example, the high concentration of proteins in blood will impact the pharmacokinetics of certain drugs, affecting their efficiency,4–6 and this has been reported for insulin, among other therapeutic drugs.7 Testing in blood would therefore provide a more realistic model to understand biochemical events or interactions occurring within a living organism. Among several other applications, a sensor that could measure biomolecular affinities in whole blood could also provide useful in vitro data on the fate of therapeutic targets.
A number of strategies have been proposed to eliminate interference from blood cells and proteins on sensors. The isolation of blood cells from serum (if clotting occurs) or plasma (without clotting) leads to a solution free of large particles. Cells are traditionally separated from blood with sedimentation or diffusion-based techniques. Centrifugation is commonly used and exploits the faster sedimentation rate of cells, however it can be time consuming and costly.8 Fluidic based systems have also been proposed to analyse blood samples9 and they have been employed in several applications such as clinical diagnosis, environmental analysis, and ligand screening.10,11 Fluidic devices can isolate, capture or filter blood cells based on size dependent particle separation.12,13 In addition to interference from cells, biosensors have also been hampered by the nonspecific adsorption of proteins in serum or plasma.14 Nonspecific adsorption of proteins can be limited by using depletion columns,15 protein precipitation16 or coating the sensor with an ultralow fouling surface17 composed of polyethylene glycol (PEG),18 zwitterionic molecules19 or peptide monolayers.20 The combination of fluidic devices sufficient in removing cells with the appropriate surface chemistry could thus enable whole blood sensing.
Additionally, current sensing techniques generally rely on direct contact of the sample with the surface of the sensor. Hence, all molecules interact with the surface at essentially the same time, effectively reducing the ability of the sensor to discriminate between molecules. However, diffusion can be drastically different between small molecules, proteins and cells and this can be exploited by creating a diffusion gate, which can be used to specify the distance the molecules must travel from the sample to the sensor. Fluid contact must be maintained between the sensor and the sample, however, this can be done with a transfer fluid suitable for the biosensor. Meeting these conditions, faster moving molecules would preferentially reach the sensor surface, leaving slower moving molecules in the bulk solution. Implantable electrochemical glucose and gas sensors often rely on the concept of diffusion membranes.21,22 This concept could prove highly efficient for the analysis of small molecules, such as metabolites, contaminants, or therapeutic drugs in blood, in the presence of proteins and cells, using a wide variety of surface-based optical sensors.
Fluidic devices relying on dialysis could efficiently integrate sample preparation in biosensing systems. In particular, surface plasmon resonance (SPR) sensing has gained broad acceptance in biomolecular interaction analysis.17 SPR sensing currently suffer from high background signals from biofluids and therefore, have been limited to the analysis of relatively pure solutions.17 In dialysis, the concentration gradient between blood (high concentration) and the dialysate (low concentration) forces molecules through a semi-permeable membrane. Dialysis is more efficient for fast diffusing molecules, and thus performs well for small molecules. Strategically placing a microporous membrane at the interface between two fluid chambers in a SPR instrument, one of which is allocated to the blood sample and the other to the sensor would enable sample pre-treatment in situ. The microporous membrane would serve to filter cells and create a diffusion gate to the SPR sensor. Thereby, a concentration gradient would rapidly transfer small molecules from blood to a chamber containing a SPR sensor specific for a target of interest, while retarding slower diffusing molecules. The interference from slow moving, abundant and large proteins in blood could be avoided as they would reach the sensor surface later, and the time delay would be a function of diffusion coefficient, molecular size and distance travelled.
The concept of diffusion membranes has not been successfully implemented to SPR sensors for bioanalysis in blood. In the rare literature examples citing the use of diffusion membranes in SPR, the membrane was either directly deposited on the surface of the SPR sensor for methanol–raffinose analysis23 or positioned off-line for sample preparation for the analysis of cortisol in saliva.24 Here we report on such a dialysis-based sensor for use in combination with SPR. As a proof-of-principle experiment, the detection of a small peptide which exhibits anti-atherosclerotic activity is performed directly in whole blood using a CD36 based biosensor, the affinity of which has previously been reported.20
Experimental
Microdialysis SPR sensor
Materials are provided in the ESI.† Custom made PDMS fluidics and spacers (Fig. 1) were prepared by mixing 184 silicone elastomer base and 184 silicone elastomer curing agent in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Spacers of different thicknesses were fabricated between 150 μm and 1.0 mm. Curing of the PDMS components was carried out at 80 °C for 1 hour and cooled to room temperature before de-moulding. Dove shaped glass prisms of 12 × 20 × 3 mm were cleaned with piranha solution (sulphuric acid + hydrogen peroxide – 3:1 at 80 °C) for 90 minutes. Caution: piranha solution is highly corrosive! The prisms were then vigorously washed with deionised water to remove any traces of acid. Sputtering of 0.5 nm Cr and 45 nm Au was performed (Cressington 308R sputter coater, Ted Pella Inc. Redding, CA) on the long face of the prisms to create the SPR surface. The small, portable 4-channel SPR device used for analysis has previously been reported.25 The fluidic was modified according to Fig. 1. Three channels were used as the sensing channels while the single channel served as a reference for instrumental fluctuation. The dialysis fluidic chamber was created by placing the spacer directly on the SPR prism, completely filling the chamber of the spacer with PBS buffer to avoid the presence of air bubbles. Then, the microporous membrane was placed on top of the spacer before latching the fluidic cell in place. The SPR instrument was calibrated with or without the microporous membrane using successive injections of 1 mL of sucrose solutions with different refractive indices ranging from 1.33772 to 1.35589.
:1 ratio. Spacers of different thicknesses were fabricated between 150 μm and 1.0 mm. Curing of the PDMS components was carried out at 80 °C for 1 hour and cooled to room temperature before de-moulding. Dove shaped glass prisms of 12 × 20 × 3 mm were cleaned with piranha solution (sulphuric acid + hydrogen peroxide – 3:1 at 80 °C) for 90 minutes. Caution: piranha solution is highly corrosive! The prisms were then vigorously washed with deionised water to remove any traces of acid. Sputtering of 0.5 nm Cr and 45 nm Au was performed (Cressington 308R sputter coater, Ted Pella Inc. Redding, CA) on the long face of the prisms to create the SPR surface. The small, portable 4-channel SPR device used for analysis has previously been reported.25 The fluidic was modified according to Fig. 1. Three channels were used as the sensing channels while the single channel served as a reference for instrumental fluctuation. The dialysis fluidic chamber was created by placing the spacer directly on the SPR prism, completely filling the chamber of the spacer with PBS buffer to avoid the presence of air bubbles. Then, the microporous membrane was placed on top of the spacer before latching the fluidic cell in place. The SPR instrument was calibrated with or without the microporous membrane using successive injections of 1 mL of sucrose solutions with different refractive indices ranging from 1.33772 to 1.35589.
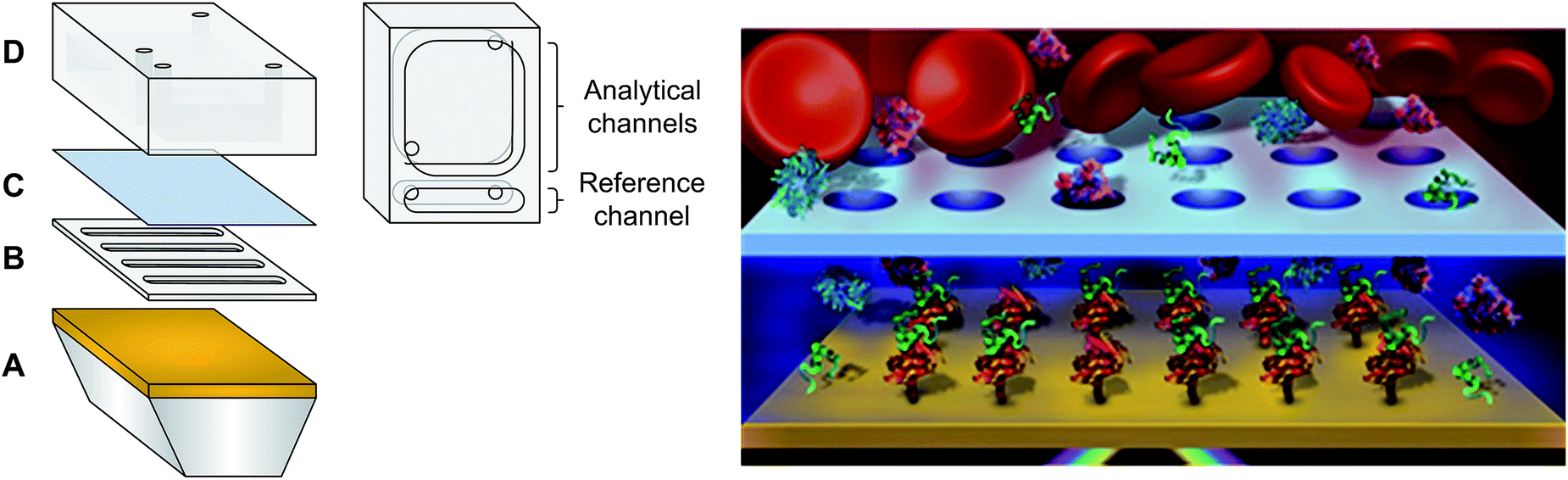 | ||
| Fig. 1 (Left) Microfluidic system with a diffusion barrier: (A) gold coated prism, (B) PDMS spacer (sensing chamber), (C) microporous membrane and (D) PDMS fluidic reservoir (reservoir chamber). (Right) Schematic of the dialysis chamber with blood in the reservoir chamber (not to scale). | ||
Diffusion measurements
The impact of molecular weight on diffusion was assessed with the dialysis chamber and a SPR sensor modified with a 3-MPA-LHDLHD-OH monolayer to minimize nonspecific adsorption. Diffusion of sucrose, a small molecule, was compared with larger molecules such as poly(acrylic acid) (MW = 2000 Da), bovine serum albumin (BSA) (MW = 66.4 kDa), hemoglobin (MW = 64.0 kDa), and IgG (MW = 150 kDa). Sucrose and polymer solutions were prepared at 10 mg mL−1, while protein solutions were prepared at 1 mg mL−1. The solutions were also injected without the microporous membrane in the fluidic. Analysis times of 10 and 90 min were used, respectively, without and with the microporous membrane.Peptide biosensing
The CD36 biosensor was constructed as previously described.20 In brief, 3-MPA-LHDLHD-OH was self-assembled on the SPR sensor in an overnight procedure, then reacted with Nα,Nα-bis(carboxymethyl)-L-lysine (NTA) using EDC/NHS coupling chemistry and subsequently chelated to Cu2+ to create the sensor competent for binding hexahistidine-tagged CD36. All these surface chemistry modification were also performed offline the SPR instrument. The functionalized SPR sensors are stable for at least 6 months and can form a complex with any protein containing a His-tag. The SPR sensors were functionalized with CD36 in the sensing and reference channels using the fluidic cell presented in Fig. 1, while the SPR parameters were previously described by Zhao et al.25 Following the functionalization steps, the dialysis chamber was mounted on the SPR sensor. CD36 was expressed as previously reported20 and immobilized to the SPR surface by chelation with the monolayer. Small peptides (DBG178 and CP-2B(i)) were detected with the CD36 SPR sensor at concentrations in the micromolar range in PBS to evaluate the performance of the dialysis chamber. DBG178 was used as a positive control and CP-2B(i) as a negative control. All peptides were injected for 1.5 hours in the reservoir chamber to ensure equilibrium measurements in the sensing chamber. The experiments were repeated in human serum and whole human blood for DBG178 in the same concentration range.IgG biosensing
A human gamma immunoglobulin (IgG) biosensor was constructed on a 16-mercaptohexadecanoic acid (16-MHA) monolayer as previously described.26 The sensor was created by activating the 16-MHA monolayer with EDC/NHS and reacted with anti-IgG. The remaining NHS esters on the SPR sensor were deactivated with ethanolamine. IgG was detected at the nanomolar level in PBS with the anti-IgG biosensor and the dialysis chamber. Binding equilibrium was reached after 8 hours.Results and discussion
Characteristics and optimization of the dialysis fluidic chamber
The microfluidic possesses three main components: a spacer, a microporous membrane and a reservoir fluidic chamber (Fig. 1). The microporous membrane created a partition and a diffusion barrier separating the reservoir chamber containing the biofluid from the sensing chamber. The reservoir was significantly larger than the sensing chamber to ensure minimal dilution of the samples between the reservoir and the sensing chamber. Due to their localization within the SPR instrument and their small size, the reservoir and the sensing chambers could not be stirred and mixing relied exclusively upon molecular diffusion inside the pores and in the chambers.To validate the performance of the SPR instrument with the microdialysis chamber, changes in refractive index were measured with the injection of sucrose solutions into the reservoir chamber while monitoring the SPR response from the sensing chamber. SPR responses were successfully monitored in accordance with the changes in the refractive index of the reservoir using the 1 mm PDMS spacer. In classical experiments, the change in SPR response is almost instantaneous with the change in bulk refractive index. In the current experiments, with the microporous membrane, diffusion must take place through the pores and then from the pores to the sensor surface (1 mm distance). Thus, the initial change in the SPR response was monitored for nearly 2 minutes following the injection of the sucrose solution (Fig. SI1†), before the SPR response was then a function of the flux of sucrose molecules arriving at the surface until equilibrium was reached.
The SPR response can thus be decomposed into two independent factors: the diffusion time of molecules through the pores and the passive mixing through diffusion in the sensing chamber. Diffusion of molecules through microporous membranes has been extensively studied and reported in the literature.27,28 The Renkin equation predicts the effective diffusion coefficient through a membrane and can be express as:29
| Deff/D0 = (1 − RH/RP)2(1 − 2.1RH/RP + 2.1RH/RP3 − 0.95RH/RP5) | (1) |
Sucrose has a significantly smaller hydrodynamic radius (approximately 0.47 nm) than the 200 nm membrane pore radius (RH/RP smaller than 0.025). The Renkin equation predicts that Deff for sucrose will correspond to approximately 99% of the diffusion coefficient in bulk solution.28 While the diffusion coefficient remains essentially constant, the presence of the microporous membrane in the fluidic cell will restrict the equilibration of the concentration on both sides of the chamber. The time required for solutions between the reservoir and the sensing chamber to reach equilibrium can be characterized with the effective time of diffusion though a membrane (teff) in a system under continuous stirring:28
| c = ceq(1 − e−t/teff) | (2) |
| Deff = A/teff | (3) |
The experimental SPR response was correlated to the concentration of sucrose in the sensing chamber, and so by tracking the change in SPR wavelength, the diffusion process in the dialysis chamber was monitored in real-time. Fitting the experimental data with eqn (2) led to the estimation of the experimental effective diffusion times. While the theoretical diffusion time was 96 s for chambers under stirring (using a spacer of 1 mm), the experimental effective time was calculated at 15.5 hours. This large difference was a consequence of the passive mixing in the dialysis chamber.
Diffusion times are generally proportional to the square of the distance, thus the spacer thickness was incrementally reduced to 150 μm in order to obtain shorter diffusion times. Reducing the thickness of the spacer also had the advantage of reducing the volume of the sensing chamber, decreasing the dilution factor of the sample. The volume of the reservoir chamber was set at 135 μL, while the total volumes for the sensing chambers were 6.9, 14, 28 and 46 μL for the spacers of 150, 300, 600 and 1000 μm, respectively. As expected, the effective diffusion times decreased with the spacer thickness (Table 1). The theoretical effective times for sucrose ranged from 18 to 96 s (directly proportional to the spacer thicknesses of 150 to 1000 μm), while the experimental effective times ranged from 1.5 to 15.5 h. Thus, the absence of mixing resulted in a 300-fold increase in equilibration time of the sensing chamber for the 150 μm spacer, a direct consequence of the diffusion time sucrose required to pass through the membrane and reach the sensing surface. While the influence of the spacer thickness was linear for the theoretical teff, the experimental values followed a second power exponential due to the influence of the passive mixing with diffusion following the Stokes–Einstein equation ([x]2 = 4Dt/π where x = distance, D = diffusion coefficient, t = time).
The sensing performance of SPR was established with sucrose solutions of different concentrations and the dialysis chamber composed of the microporous membrane and the 150 μm spacer (Fig. SI2†). It is important to note that a concentration gradient existed in the sensing chamber due to the absence of mixing. The concentration of sucrose was higher near the porous membrane and lower at the SPR sensor. The sensitivity decreased when using the fully functional dialysis chamber (1764 nm/RIU) in comparison to analysis in the absence of the microporous membrane (2221 nm/RIU). The equilibrium SPR signal obtained for a sucrose solution of 1.34569 RIU with each spacer thickness was calculated and normalized with the SPR signal obtained without the microporous membrane (Table 1). The normalized SPR signal decreased from 74% to 39% as the spacer thickness increased from 0.15 to 1 mm. A smaller volume of the sensing chamber decreased the dilution factor of the solution explaining the larger relative signal obtained with thinner spacers. In addition, a smaller volume of the sensing chamber (V2) led to a smaller value of the A term and teff decreased proportionally to the spacer thickness (eqn (3)). The shorter teff were advantageous due to lesser dilution and a greater SPR response with the 150 μm thick spacer, therefore this spacer was used for further analysis.
Diffusion of larger biomolecules
The equilibration time of the sensing chamber depended on the diffusion properties and the size of the molecules (Table 2). Thus, the impact of diffusion coefficient and molecular size on the effective diffusion time in the dialysis chamber was verified with different biomolecules, including sucrose (342 Da; Rh = 0.47 nm), poly(acrylic acid) (2000 Da; Rh = 0.96 nm), hemoglobin (64.0 kDa; Rh = 3.1 nm), BSA (66.4 kDa; Rh = 3.5 nm) and IgG (∼150 kDa; Rh = 5.4 nm). Larger, high molecular weight biomolecules such as proteins have a slower diffusion coefficient than small molecules such as sucrose. Larger biomolecules increased the RH/RP ratio leading to greater steric hindrance in the pores and slower effective diffusion coefficients through the pores (eqn (1)). The passive mixing through diffusion in the sensing chamber was also slower for larger biomolecules. In consequence, the equilibration time increased for large biomolecules in comparison to small molecules (Fig. 2).| Spacer (mm) | t eff (s) | ||||
|---|---|---|---|---|---|
| Sucrose | PAA | Hemoglobin | BSA | IgG | |
| 0.15 | 18 | 48 | 130 | 167 | 272 |
| 0.30 | 35 | 92 | 248 | 319 | 519 |
| 0.60 | 64 | 169 | 453 | 584 | 949 |
| 1.00 | 96 | 253 | 678 | 874 | 1420 |
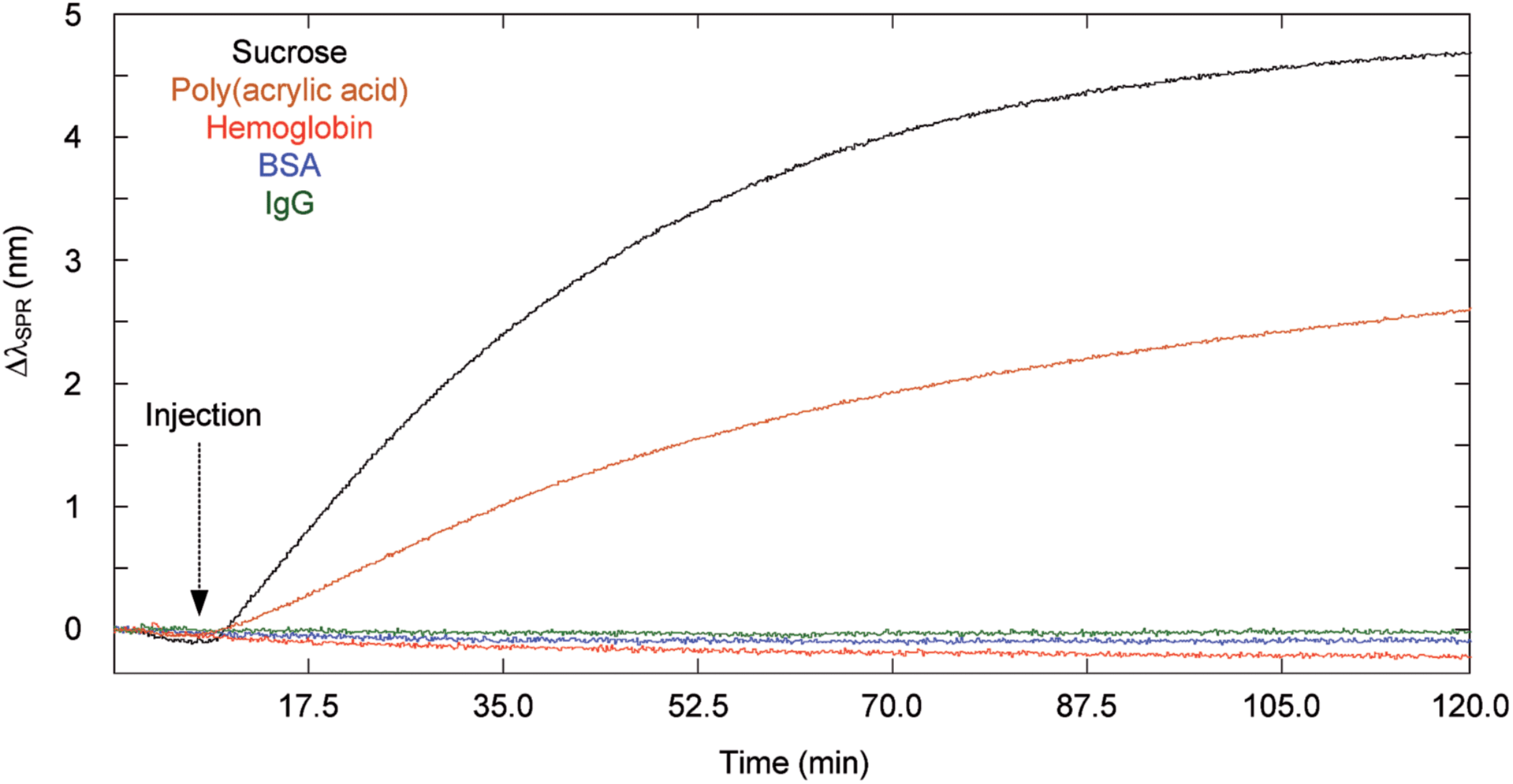 | ||
| Fig. 2 Diffusion of sucrose (MW = 342 Da), poly(acrylic acid) (MW = 2 kDa), hemoglobin (MW = 64.0 kDa), BSA (MW = 66.4 kDa) and IgG (MW = 150 kDa) through the microporous membrane of 0.4 μm pore size (concentration of 10 mg mL−1 for sucrose and poly(acrylic acid) solution and 1 mg mL−1 for each protein solution). | ||
The diffusion experiments with large biomolecules clearly demonstrated that the analysis of proteins did not result in an SPR response when using the microporous membrane over the course of the experiment (Fig. 2). Sucrose reached equilibrium, while the poly(acrylic acid) solution resulted in an SPR response that was still increasing after 2 h. The magnitude of the SPR response thus followed the rate of diffusion of molecules (Dproteins < Dshort chain polymer < Dsmall molecules). Importantly, the initial rise in the SPR response after injection was delayed by a few minutes due to the diffusion time required by the molecules to travel through the membrane and towards the SPR sensor. In absence of the microporous membrane, every solution led to a significant SPR response at the moment of injection (Fig. SI3† for IgG). A suppression factor was thus calculated to show the decrease in background protein concentration at the sensor's surface. The suppression percentage for BSA and IgG was greater than 99%, such that proteins would not interfere throughout the duration of a binding experiment for small molecules within the dialysis chamber. In these experiments, the signal was not corrected with the reference channel, thus the suppression of the response for BSA and IgG was absolute. Nonspecific interaction was thus minimal.
Although the analysis time is longer than a typical SPR experiment, the dialysis chamber provided a better discrimination between molecules with different size. The effective diffusion times for the molecules investigated were calculated for a theoretical system under stirring and with different spacers thicknesses (Table 2). Again, teff was shown to increase linearly with increasing spacer thicknesses and ranged from 18 s for sucrose to 272 s for IgG with a 150 μm thick spacer. Considering that SPR experiments typically run for 10 to 20 minutes (600 to 1200 seconds), every molecule, including proteins, would reach equilibrium in that period of time if mixing was performed. The influence of the passive diffusion was clear from these calculations and demonstrated that this method of diffusion is required for the dialysis chamber to work effectively as a diffusion gate. The thickness of the spacer controlled the time and thus, the molecular weight or diffusion constant range of molecules reaching the SPR sensor over the course of an experiment. The 150 μm spacer thus facilitated the design of a biosensing assay for small analytes (∼1 kDa) contained in a highly concentrated protein solution such as blood-based fluid.30,31
Biosensing with a dialysis chamber SPR instrument
Biosensing with the dialysis chamber was demonstrated with a model system of hexapeptide ligands binding to the cluster of differentiation 36 (CD36). The construction of the biosensor involved the immobilization of a His-tagged type B scavenger receptor CD36, as previously reported in the literature.20 The receptor CD36 is an 88 kDa integral membrane protein that is highly glycosylated and found in platelets, macrophages and microvascular endothelium.32 It has been shown that this protein interacts with a variety of different biomolecules, for example collagen and thrombospondin,32 and is involved in the modulation of angiogenesis and in the scavenging of oxidized low-density lipoproteins.33 The receptor CD36 is also a receptor involved in atherosclerosis, which can be inhibited by peptides with anti-atherosclerotic activity.33 There is a significant interest in the development of therapeutic ligands with anti-atherosclerotic activities since atherosclerosis plays a role in heart diseases, common in industrial countries.34 In addition, this model system was perfectly suited for the demonstration of the performance of the dialysis chamber SPR instrument.This CD36 based biosensor was competent for the detection of therapeutic hexapeptides such as His-D-Trp-Ala-AzaTyr-D-Phe-Lys-NH2 (named DBG178 with a KD = 5 μM in saline solution). This peptide belongs to the growth hormone-releasing peptide (GHRP) family,33,35 which interacts with CD36. DBG178 possess a molecular weight (MW 850.97 g mol−1) intermediate to sucrose and PAA, and this peptide should therefore have a similar mechanism of diffusion, reaching the SPR sensor within a comparable time. The CD36 based biosensor was validated within the dialysis chamber by injecting either 10 μM of DBG178 or CP-2B(i) (His-D-Trp-AzaLeu-Trp-D-Phe-Ala-NH2; MW = 858.98 Da), where DBG178 served as the positive control (KD = 5 μM) and CP-2B(i) (KD = 31 μM) served as a negative control. A signal change of 0.28 ± 0.11 nm and −0.073 ± 0.030 nm was obtained for DBG178 and CP-2B(i), respectively. A chip-to-chip variation of 9% was calculated with this system by comparing the SPR signal for the immobilization of CD36 on the surface prior to the addition of the porous membrane. It is also important to note that the calibration curves and data reported here were constructed from data collected with several SPR chips. The successful detection of DBG178 in buffer through the porous membrane thus confirmed the suitability of the dialysis chamber for monitoring biomolecular interactions.
The CD36 biosensor within the dialysis chamber was calibrated with injection of varying concentrations of DBG178 between 5 and 30 μM (Fig. 3) using analysis period of 90 minutes per concentration. The dilution of the sample in the process of dialysis leads to slightly smaller responses for DBG178 than could be obtained using classical SPR and as a result concentrations below 5 μM could not be detected, and a KD in saline solution could not be estimated with the dialysis chamber. Improvement in the fluidic design by using fabrication method leading to thinner spacer could also reduce the dilution factor observed between the reservoir and the sensing chamber, providing increased signal for biosensing. The concentrations reported in Fig. 3 do not account for the dilution factor reported above. Nonetheless, the detection of several concentrations of DBG178 was achieved within 90 minutes for each concentration with the dialysis chamber and SPR analysis.
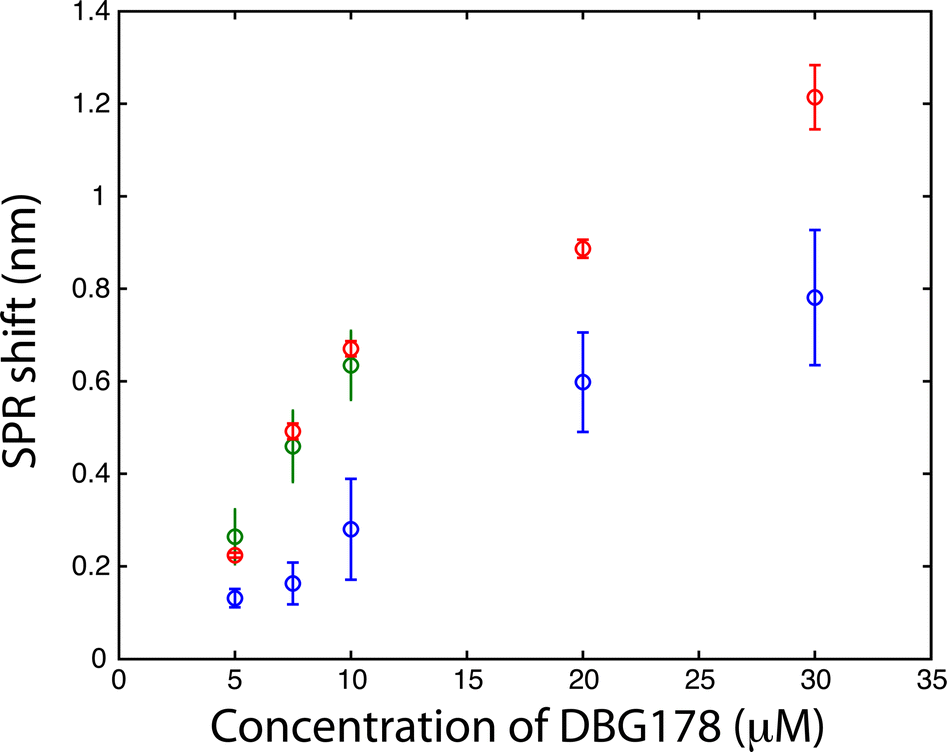 | ||
| Fig. 3 Detection of a small peptide (DBG178) in PBS (blue), in human serum (red) and in human whole blood (green) by using a biosensor based on CD36 (each data point reported above are triplicate measurements; n = 3). | ||
Protein sensing is of high importance for the diagnosis of several diseases. As a proof-of-concept experiment, IgG detection at nanomolar concentrations was performed with the dialysis chamber. It should be stated that the analysis was carried out over approx. 8 hours since the IgG diffused slowly as a result of its molecular weight (150 kDa), an advantage when detecting small molecules via the dialysis chamber. Protein detection was successfully achieved with the 150 μm spacer, however, the detection time is currently prohibitive for a useful assay. Analysis time could be improved by reducing the thickness of the spacer and therefore one can envision the adaptation of this dialysis chamber for a variety of surface-based sensors to combat the challenges associated with sensing in biofluids.
Diffusion gated detection of small peptide ligands in crude biofluids with a CD36 biosensor
Similarly to BSA and IgG, the suppression of the background signal from blood and serum was approximately 99% with the microporous membrane (Table 3) for an analysis time of 2 hours. Again, the suppression of the background signal was absolute, as the data was not compensated with the reference channel. If longer analysis times are used, the background signal will indeed rise due to the diffusion of proteins from serum and blood diffusing to the SPR sensor. For BSA and IgG, a diffusion time of nearly 8 hours was required to observe a significant SPR response. Thus, the protein concentrations (a major contributor to the background SPR signal from serum and blood) was about 1% of the reservoir concentration at the SPR sensor surface after more than 6 hours of dialysis. The dialysis chamber provided an extended period of time during which biodetection of small molecules can be performed in a biofluid with limited interference from the matrix.| Biomolecule/fluid | ΔλSPR with dialysis chamber (nm) | ΔλSPR without dialysis chamber (nm) | SPR response reduction (%) |
|---|---|---|---|
| a Solution concentration: 1 mg mL−1 for BSA and IgG; 10 mg mL−1 for sucrose and poly(acrylic acid). | |||
| Sucrose | 18.3 ± 1.6 | 24.7 ± 0.6 | 26% |
| PAA | 1.8 ± 0.9 | 3.9 ± 0.1 | 54% |
| BSA | <LOD | 3.7 ± 0.4 | >99% |
| IgG | <LOD | 10.7 ± 0.4 | >99% |
| Serum | 0.26 ± 0.05 | 23.3 ± 1.9 | 99% |
| Blood | 0.44 ± 0.06 | 38.5 ± 0.2 | 99% |
To confirm the competence of the dialysis chamber for biosensing in a complex biological fluid, human serum was spiked with DBG178 and analyzed with the SPR instrument. No treatment nor dilution of the serum was done before its injection. Despite higher viscosity of blood compared to buffer, the analysis time of 90 minutes was still sufficient to reach equilibrium of the SPR sensor in whole blood (see Fig. SI4†). The analysis of DBG178 in human serum was successfully performed in the same concentration range as reported for PBS (Fig. 3), and the calibration curve obtained showed an increased in signal compared to PBS. Albeit at a 1% of its original concentration in blood, the presence of serum proteins in the sensing chamber might explain the slight increase in sensitivity compared to PBS measurements. The presence of 1% of protein concentration is insufficient to induce significant nonspecific adsorption on the SPR sensor (Table 3). Data were background subtracted to remove the small contribution of nonspecific adsorption. It is thus suspected that the increase of the response in biofluids is due to the bound fraction of DBG178 in serum and blood. The KD was measured at 17 μM in human serum, lower than the value reported in PBS. However, this could be expected as DBG178 is diluted slightly in the sensing chamber from the original concentration in serum and that the free fraction of DBG178 may not be 100%. The total refractive index or mass change induced by DBG178 bound to serum proteins would increase the SPR response at identical DBG178 concentrations in comparison to PBS, and thus could explain the larger sensitivity in serum and whole blood. DBG178 was also injected at concentrations in the nanomolar range. However, the SPR response for these concentrations was below the detection limit of the SPR sensor and thus, was omitted in Fig. 3 for clarity.
Finally, whole human blood was spiked with DBG178 and injected into the SPR instrument with the dialysis chamber at concentrations of 5, 7.5 and 10 μM DBG178. The SPR system configuration allowed a triplicate measurement in separate sensing chambers and a fourth sensing chamber was dedicated to a reference measurement. In the reference channel, a blank blood or serum (unspiked with DBG178) was injected to correct for bulk refractive index and remaining nonspecific adsorption of blood component over the sensor. CD36 was also immobilized on the reference channel, and thus the reference channel was identical to the sensing channel to ensure close correlation between the background response of the sensing and the reference channels. The SPR responses in serum or blood were subtracted with the reference channel to compensate for these fluctuations. The SPR response obtained directly in whole blood was in great agreement with human serum (Fig. 3). The SPR responses for 7.5 μM DBG178 in serum and blood were statistically identical at 0.49 ± 0.02 nm and 0.46 ± 0.08 nm respectively. These results supported the hypothesis that the porous membrane was efficient in blocking cells and platelets from entering the sensing chambers. No clogging of the porous membrane was observed following analysis of DBG178 in whole blood. Anticoagulant was added to blood to prevent clogging (see ESI† for the source of blood and details about the anticoagulant). In comparison, the SPR response without the dialysis chamber was measured at 0.71 ± 0.10 nm for 8.75 μM DBG178, indicating that the magnitude of the SPR response is reduced with the dialysis chamber for lower concentrations. For the highest concentration, the difference was reduced to 12%, as the response for DBG178 without the microporous membrane was 0.89 nm in comparison to 0.78 nm with the microdialysis chamber. To the best of our knowledge, this is the first report of a plasmonic biosensor working in whole human blood.
Conclusions
The microporous membrane of the dialysis chamber created a size exclusion filter and a diffusion gate for blood. The membrane had pore size of 0.4 μm diameter, smaller than the average diameter of a red blood cell (∼8 μm),36 white cell (∼6 to 10 μm)37 and platelets (∼3.9 μm).38 These three major components of blood were not transferred from the reservoir chamber to the sensing chamber and will not interfere with analysis, as would be the case when using a conventional SPR fluidic chamber. The molecules in blood including the proteins, metabolites and small molecules were able to cross the membrane since they all have a smaller hydrodynamic radius than the pores, which at 400 nm diameter were perfectly suited to filter cells and platelets, without impeding the diffusion of small molecules and biomolecules. The microporous membrane also served as a diffusion gate since molecules crossing the membrane will reach the sensor according to their size and diffusion coefficient. In the experiments reported, molecules or small biomolecule such as sucrose, DBG178 and PAA diffused at a faster rate in comparison to large biomolecules like albumin or IgG. Due to high concentration of protein in serum or blood,30 a large shift could still be observed if enough diffusion time is allowed (∼7–8 hours after serum or blood injection). The biosensing of smaller molecules can therefore be performed before larger biomolecules that interfere with the SPR response can reach the sensor, and over the course of the reported experiments, the dialysis chamber was able to suppress the bulk refractive index change from blood-based components. Improvement in the fluidic design by using fabrication method leading to thinner spacer could also reduce the detection times and the dilution factor observed between the reservoir and the sensing chamber, providing increased SPR signal and faster response for biosensing in crude biofluids. The potential sensing capabilities of this SPR diffusion gated biosensor could provide a rapid, label-free platform for direct ligand screening in untreated blood samples from patients.Acknowledgements
The authors thank William D. Lubell and Huy Ong of the Université de Montréal for providing the peptides and CD36, and Joachim Wegener of the University of Regensburg for fruitful discussions. The author also acknowledge financial support of the National Science and Engineering Research Council (NSERC) of Canada, the Canadian foundation for innovation (CFI), the Fonds québécois de recherche-Nature et technologies (FQR-NT) and the Centre for self-assembled chemical structures (CSACS).Notes and references
- J. R. Pappenheimer, Physiol. Rev., 1953, 33, 387–423 CAS.
- M. Polanski and N. L. Anderson, Biomarker Insights, 2007, 1, 1–48 Search PubMed.
- E. C. Kohn, N. Azad, C. Annunziata, A. S. Dhamoon and G. Whiteley, Dis. Markers, 2007, 23, 411–417 CrossRef CAS PubMed.
- U. Kraghhansen, Pharmacol. Rev., 1981, 33, 17–53 CAS.
- M. Fasano, S. Curry, E. Terreno, M. Galliano, G. Fanali, P. Narciso, S. Notari and P. Ascenzi, IUBMB Life, 2005, 57, 787–796 CrossRef CAS PubMed.
- J. J. Vallner, J. Pharm. Sci., 1977, 66, 447–465 CrossRef CAS PubMed.
- V. T. G. Chuang, U. Kragh-Hansen and M. Otagiri, Pharm. Res., 2002, 19, 569–577 CrossRef.
- C. F. Hogman, Vox Sang., 1998, 74, 177–187 CrossRef PubMed.
- M. Toner and D. Irimia, in Annual Review of Biomedical Engineering, Annual Reviews, Palo Alto, 2005, vol. 7, pp. 77–103 Search PubMed.
- X. D. Fan and I. M. White, Nat. Photonics, 2011, 5, 591–597 CrossRef CAS PubMed.
- J. G. E. Gardeniers and A. van den Berg, Anal. Bioanal. Chem., 2004, 378, 1700–1703 CrossRef CAS PubMed.
- J. D. Chen, D. Chen, T. Yuan, X. Chen, Y. Xie, H. L. Fu, D. X. Cui, X. D. Fan and M. K. K. Oo, Microelectron. Eng., 2014, 128, 36–41 CrossRef CAS PubMed.
- K. Aran, A. Fok, L. A. Sasso, N. Kamdar, Y. L. Guan, Q. Sun, A. Undar and J. D. Zahn, Lab Chip, 2011, 11, 2858–2868 RSC.
- C. Blaszykowski, S. Sheikh and M. Thompson, Chem. Soc. Rev., 2012, 41, 5599–5612 RSC.
- K. Bjorhall, T. Miliotis and P. Davidsson, Proteomics, 2005, 5, 307–317 CrossRef PubMed.
- M. Zellner, W. Winkler, H. Hayden, M. Diestinger, M. Eliasen, B. Gesslbauer, I. Miller, M. Chang, A. Kungl, E. Roth and R. Oehler, Electrophoresis, 2005, 26, 2481–2489 CrossRef CAS PubMed.
- M. Couture, S. S. Zhao and J.-F. Masson, Phys. Chem. Chem. Phys., 2013, 15, 11190–11216 RSC.
- N. P. Huang, J. Voros, S. M. De Paul, M. Textor and N. D. Spencer, Langmuir, 2002, 18, 220–230 CrossRef CAS.
- H. Vaisocherova, W. Yang, Z. Zhang, Z. Q. Cao, G. Cheng, M. Piliarik, J. Homola and S. Y. Jiang, Anal. Chem., 2008, 80, 7894–7901 CrossRef CAS PubMed.
- O. R. Bolduc, P. Lambert-Lanteigne, D. Y. Colin, S. S. Zhao, C. Proulx, D. Boeglin, W. D. Lubell, J. N. Pelletier, J. Fethiere, H. Ong and J. F. Masson, Analyst, 2011, 136, 3142–3148 RSC.
- P. U. Abel and T. von Woedtke, Biosens. Bioelectron., 2002, 17, 1059–1070 CrossRef CAS.
- R. Knake, P. Jacquinot, A. W. E. Hodgson and P. C. Hauser, Anal. Chim. Acta, 2005, 549, 1–9 CrossRef CAS PubMed.
- M. E. Montecillo, T. Yoshidome, T. Yamagata, T. Yamasaki, M. Mitsushio, B. J. Sarno and M. Higo, Bull. Chem. Soc. Jpn., 2010, 83, 1531–1533 CrossRef CAS.
- R. C. Stevens, S. D. Soelberg, S. Near and C. E. Furlong, Anal. Chem., 2008, 80, 6747–6751 CrossRef CAS PubMed.
- S. S. Zhao, N. Bukar, J. L. Toulouse, D. Pelechacz, R. Robitaille, J. N. Pelletier and J. F. Masson, Biosens. Bioelectron., 2015, 64, 664–670 CrossRef CAS PubMed.
- M. Ratel, A. Provencher-Girard, S. S. Zhao, J. Breault-Turcot, J. Labrecque-Carbonneau, M. Branca, J. N. Pelletier, A. R. Schmitzer and J. F. Masson, Anal. Chem., 2013, 85, 5770–5777 CrossRef CAS PubMed.
- I. A. Kathawalla and J. L. Anderson, Ind. Eng. Chem. Res., 1988, 27, 866–871 CrossRef CAS.
- G. Guillot, L. Leger and F. Rondelez, Macromolecules, 1985, 18, 2531–2537 CrossRef CAS.
- E. M. Renkin, J. Gen. Physiol., 1954, 38, 225–243 CAS.
- N. L. Anderson and N. G. Anderson, Mol. Cell. Proteomics, 2002, 1, 845–867 CAS.
- J. M. Jacobs, J. N. Adkins, W. J. Qian, T. Liu, Y. F. Shen, D. G. Camp and R. D. Smith, J. Proteome Res., 2005, 4, 1073–1085 CrossRef CAS PubMed.
- D. E. Greenwalt, R. H. Lipsky, C. F. Ockenhouse, H. Ikeda, N. N. Tandon and G. A. Jamieson, Blood, 1992, 80, 1105–1115 CAS.
- A. Demers, N. McNicoll, M. Febbraio, M. Servant, S. Marleau, R. Silverstein and H. Ong, Biochem. J., 2004, 382, 417–424 CrossRef CAS PubMed.
- S. N. Bhupathiraju and K. L. Tucker, Clin. Chim. Acta, 2011, 412, 1493–1514 CrossRef CAS PubMed.
- V. Bodart, M. Febbraio, A. Demers, N. McNicoll, P. Pohankova, A. Perreault, T. Sejlitz, E. Escher, R. L. Silverstein, D. Lamontagne and H. Ong, Circ. Res., 2002, 90, 844–849 CrossRef CAS.
- P. Aarts, P. A. Bolhuis, K. S. Sakariassen, R. M. Heethaar and J. J. Sixma, Blood, 1983, 62, 214–217 CAS.
- G. W. Schmidschonbein, Y. Y. Shih and S. Chien, Blood, 1980, 56, 866–875 CAS.
- P. Noris, G. Biino, A. Pecci, E. Civaschi, A. Savoia, M. Seri, F. Melazzini, G. Loffredo, G. Russo, V. Bozzi, L. D. Notarangelo, P. Gresele, P. G. Heller, N. Pujol-Moix, S. Kunishima, M. Cattaneo, J. Bussel, E. De Candia, C. Cagioni, U. Ramenghi, S. Barozzi, F. Fabris and C. L. Balduini, Blood, 2014, 124, E4–E10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and supplementary figures. See DOI: 10.1039/c5sc00716j |
| This journal is © The Royal Society of Chemistry 2015 |