
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Significant improvement of oxidase activity through the genetic incorporation of a redox-active unnatural amino acid†
Yang
Yu‡
c,
Qing
Zhou‡
b,
Li
Wang‡
ab,
Xiaohong
Liu
b,
Wei
Zhang
b,
Meirong
Hu
b,
Jianshu
Dong
b,
Jiasong
Li
ab,
Xiaoxuan
Lv
b,
Hanlin
Ouyang
c,
Han
Li
b,
Feng
Gao
b,
Weimin
Gong
b,
Yi
Lu
*c and
Jiangyun
Wang
*b
aSchool of Life Sciences, University of Science and Technology of China, Hefei, Anhui 230026, China
bLaboratory of RNA Biology, Institute of Biophysics, Chinese Academy of Sciences, 15 Datun Road, Chaoyang District, Beijing, 100101, China. E-mail: jwang@ibp.ac.cn
cCenter of Biophysics and Computational Biology and Department of Chemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: yi-lu@illinois.edu
First published on 13th April 2015
Abstract
While nature employs various covalent and non-covalent strategies to modulate tyrosine (Y) redox potential and pKa in order to optimize enzyme activities, such approaches have not been systematically applied for the design of functional metalloproteins. Through the genetic incorporation of 3-methoxytyrosine (OMeY) into myoglobin, we replicated important features of cytochrome c oxidase (CcO) in this small soluble protein, which exhibits selective O2 reduction activity while generating a small amount of reactive oxygen species (ROS). These results demonstrate that the electron donating ability of a tyrosine residue in the active site is important for CcO function. Moreover, we elucidated the structural basis for the genetic incorporation of OMeY into proteins by solving the X-ray structure of OMeY specific aminoacyl-tRNA synthetase complexed with OMeY.
Introduction
Designing artificial enzymes with higher activity and selectivity can reveal important features responsible for tuning enzymatic activities, and result in efficient catalysts for practical applications.1–9 One key mechanism which accounts for the high activity of many natural enzymes is the fine-tuning of the redox potential of tyrosine residues. In order to optimize the electron transfer rate to enable enzymatic turnover with high efficiency and selectivity, nature has exploited various strategies, such as post-translational modifications including topa quinone in copper amine oxidases, the Tyr-His crosslink in CcO, the Tyr-Cys crosslink in galactose oxidases (GO), and histidine base association in photosystem II (PSII).10–16 Such strategies are highly effective in modulating the redox potential (from 0.1 V to 1.1 V vs. NHE) and pKa of specific tyrosine residues to suit the specific needs of various reactions, thereby greatly enhancing enzyme activity.10–16 However, it remains difficult to perform rational tuning of the redox potential and pKa of specific tyrosine residues in designed metalloproteins. Here we show that, through the genetic incorporation of redox-active unnatural amino acids with desirable redox potential, a significant improvement in oxidase activity can be achieved.During the final stage of aerobic respiration, CcO catalyzes the efficient reduction of O2 to H2O, which requires rapid transfer of four electrons and four protons to the oxygen substrate, preventing the release of toxic reactive oxygen species (ROS).15 The key step in oxygen reduction is the scission of the O–O bond in the ferric-superoxo intermediate, leading to the formation of an intermediate P in the heme a3/CuB binuclear active site.17 The donor of a proton and electron for this reaction has been suggested to be a unique tyrosine residue covalently cross-linked to one of the histidine ligands of CuB. This Tyr-His crosslink is thought to lower the pKa and redox potential of the tyrosine residue, thus facilitating proton and electron donation to the oxygen substrate.15,18
In previous studies, we have reported the introduction of various tyrosine analogs into a myoglobin-based functional oxidase,19,20 including imiTyr,19 which mimics the Tyr-His cross-link in CcO, and a series of halogenated Tyr analogs with decreasing pKa.21 By replacing Tyr33 with imiTyr and halogenated tyrosine analogs, the activity and selectivity of the functional oxidase increases. Moreover, the oxidase activity is correlated with the pKa of the phenol ring of Tyr or its halogenated analogs, indicating the active role of Tyr in the oxidase reaction. However, since the reduction potentials of halogenated Tyr analogs are closely related to their pKa, we have not yet addressed whether fine-tuning the redox potential of the tyrosine residue influences oxidase activity. Herein we report genetic incorporation of a tyrosine analog, 3-methoxy tyrosine (OMeY), that has a lower reduction potential but similar pKa compared to Tyr, to provide evidence that tuning the reduction potential of Tyr is also important for oxidase activity.
Results and discussion
We first attempted to genetically incorporate two previously reported Tyr analogs, 3,4-dihydroxy-L-phenylalanine (Dopa)22 and 3-amino-tyrosine (NH2Y),23 which have lower redox potentials than tyrosine, into the 33rd position of CuBMb. However, yields of these mutant proteins were quite low, preventing further characterization. One explanation for these results is that Dopa and NH2Y can both undergo an irreversible two-electron oxidation reaction to afford dopaquinone, which is well-known to be highly reactive and to play pivotal roles in melanogenesis (Fig. S1†).24 This problem may be exacerbated when an oxidase or oxidase-mimicking enzyme is over-expressed in E. coli. In order to circumvent this problem, we decided to genetically incorporate OMeY, because its redox potential at pH 7 is 179 mV lower than that of Tyr (Fig. S2†), but it cannot easily undergo the two-electron oxidation reaction.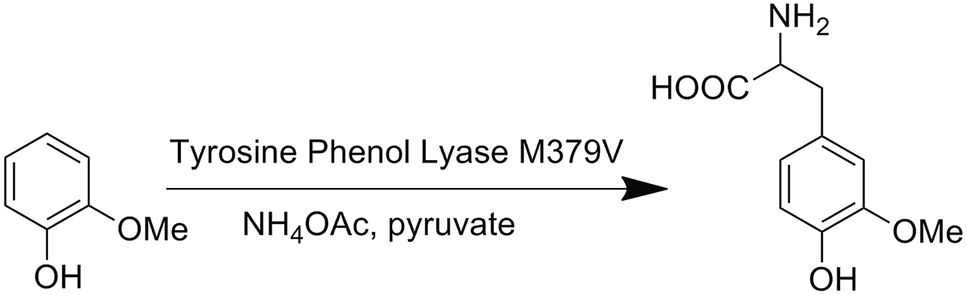 | ||
| Scheme 1 A biosynthetic route to OMeY, catalyzed by the TPL mutant M379V. | ||
The difficulty of synthesizing unnatural amino acid analogs of Tyr, such as OMeY, is often a limiting factor for the systematic investigation of the role of Tyr in different enzymes. Enzymatic transformation by native or engineered tyrosine phenol lyase (TPL), as demonstrated herein, has been proven to be a powerful tool for synthesizing tyrosine analogs (Scheme 1).25,26 To synthesize OMeY, we first tried to transform 2-methoxyphenol to OMeY by using the wild type Symbiobacterium sp. SC-1 TPL, which is more thermostable than Citrobacter freundii (ATCC8090).27 However, we could not detect any OMeY by ninhydrin thin-layer chromatography (TLC) assay. To evolve a TPL mutant that could efficiently catalyze this transformation, we screened a TPL library pEt-SymbTPL, which harbors random mutations at sites Phe36, Met288, Met379, and Phe448 as previously reported,26,28 and found that one clone efficiently catalyzed the synthesis of OMeY, as confirmed by mass spectrometry after purification of the product by HPLC (Fig. S3–S4†). DNA sequencing revealed that this clone contains the Met379Val mutation. Molecular modeling indicated that the Met379Val mutation results in a significant enlargement of the enzyme pocket to allow for optimal interaction between the enzyme and the OMeY substrate (Fig. S5†). The reduction potential of OMeY is 179 mV lower than that of Tyr at pH 7, whereas the pKa values of OMeY and tyrosine are similar (Fig. S2 and S6†).
To selectively incorporate OMeY at defined sites in proteins, a mutant Methanocaldococcus jannaschii tyrosyl amber suppressor tRNA (MjtRNACUATyr)/tyrosyl-tRNA synthetase (MjTyrRS) pair was evolved that uniquely specifies OMeY in response to the TAG codon, as previously reported.19 The evolved TyrRS (Fig. S7–S8†), named OMeYRS, has six mutations: Tyr32Glu, Leu65Ser, His70Gly, Tyr109Gly, Asp158Asn, and Leu162Val.
To determine if OMeY could be incorporated into proteins with high efficiency and fidelity, an amber stop codon was substituted for Ser4 in sperm whale myoglobin (Mb). Protein production was carried out in E. coli in the presence of the selected synthetase (OMeYRS), MjtRNACUATyr and 1 mM OMeY, or in the absence of OMeY as a negative control. Analysis of the purified protein by SDS-PAGE showed that full-length myoglobin was expressed only in the presence of OMeY (Fig. 1A), indicating that OMeYRS was specifically active with OMeY but inactive with natural amino acids. The yield for this mutant myoglobin was 10 mg L−1. By comparison, the yield of wild-type sperm whale myoglobin (WTMb) was 50 mg L−1. ESI-MS analysis of the Ser4 OMeY mutant myoglobin gave an observed average mass of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 461.6 Da, in agreement with the calculated mass of 18461.1 Da (Fig. 1B).
461.6 Da, in agreement with the calculated mass of 18461.1 Da (Fig. 1B).
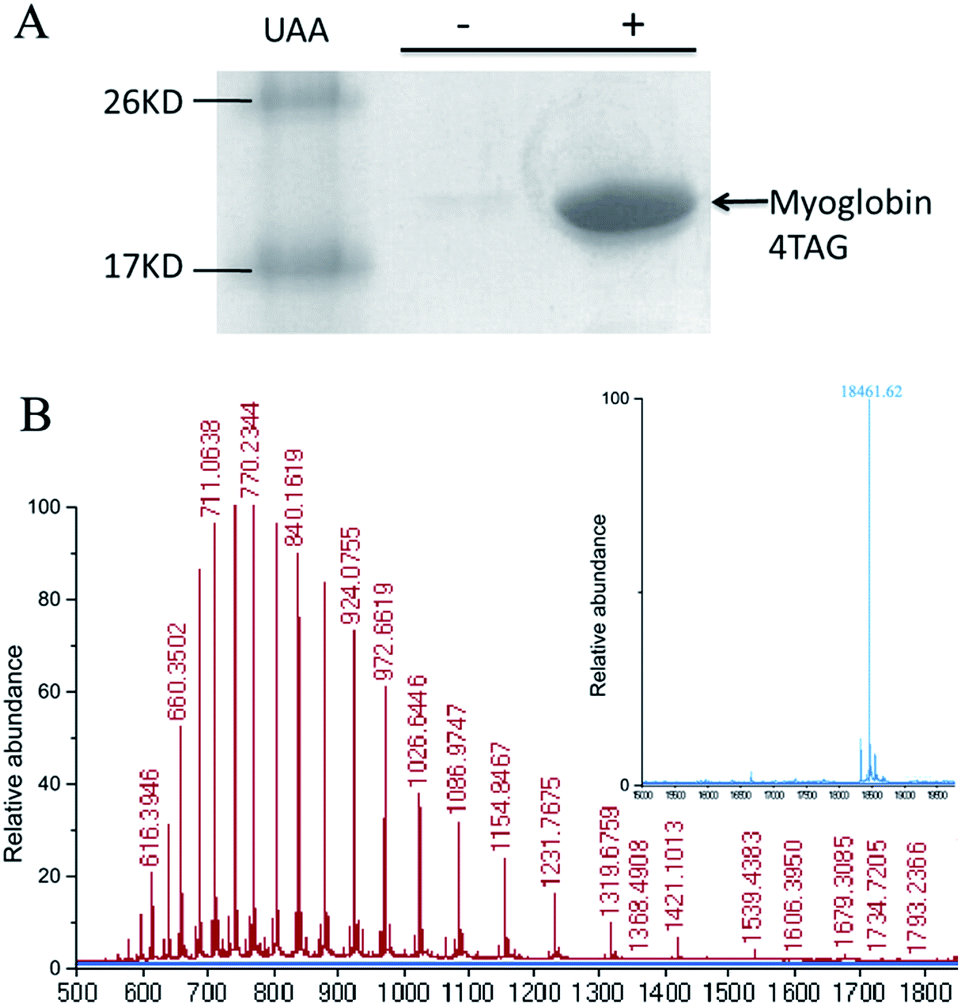 | ||
| Fig. 1 (A) SDS-PAGE of expression of TAG4 myoglobin mutant in the presence (right lane) and absence (middle lane) of 1 mM unnatural amino acid OMeY (UAA). (B) ESI-MS of the TAG4 mutant. The inset shows the deconvoluted spectrum; expected mass: 18461 Da, found: 18461.62 Da. | ||
To test whether the catalytic activity could be improved through the genetic incorporation of unnatural amino acids, we replaced Phe33 in CuBMb with OMeY, generating Phe33OMeY-CuBMb (Fig. 2). This mutant showed a similar UV-vis spectrum to that of Phe33Tyr-CuBMb (Fig. S9†), indicating that the overall environment around the heme center should also be similar.20
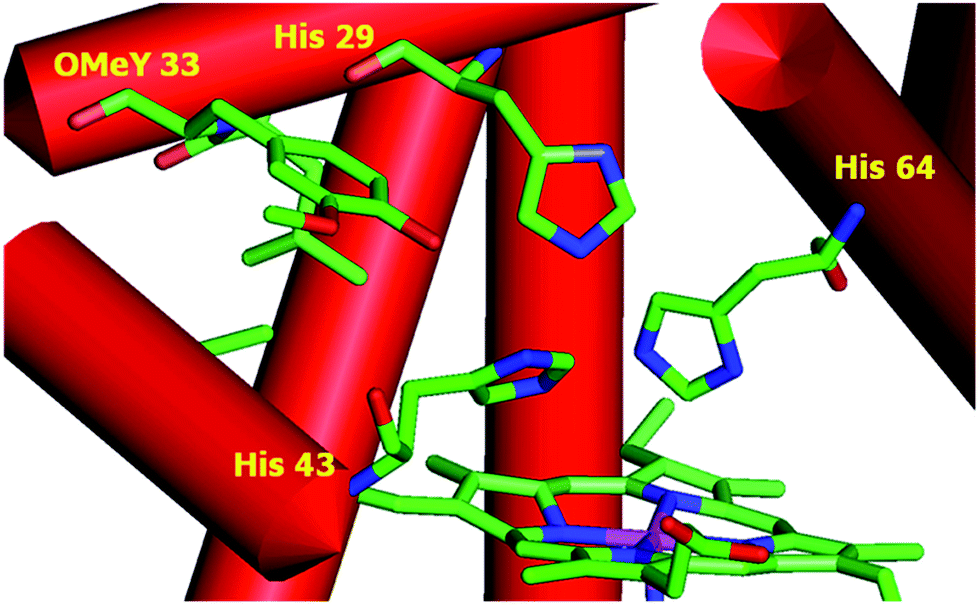 | ||
| Fig. 2 Structural model of OMeY myoglobin mutant, constructed based on the crystal structure of Phe33Tyr-CuBMb (pdb code 4FWX). | ||
We then measured the rates of oxygen reduction catalyzed by 6 μM myoglobin mutants with an O2 electrode in 20 mM tris(hydroxymethyl)aminomethane (Tris) buffer at pH 7.4. Ascorbate (1000 equivalents) and tetramethyl-p-phenylenediamine dihydrochloride (TMPD, 100 equivalents) were used as reductant and redox mediator, respectively.29 To differentiate between reactive oxygen species (ROS) and water products, we used catalase and superoxide dismutase (SOD), which catalyze the disproportionation of hydrogen peroxide or superoxide into oxygen and water. If O2 consumption results in the formation of ROS but not water, the O2 reduction rate should decrease in the presence of catalase and SOD, because they will convert ROS to O2. By comparing the rates of reduction in the presence and absence of ROS scavenger, the portions of O2 reduction due to water formation (in blue) and due to ROS formation (in red) can be calculated (Fig. 3A and Table S2†). Our results show that Phe33Tyr-CuBMb was able to reduce O2 at a rate of 6.5 μM min−1, with 51% of O2 being converted into water. In contrast, Phe33OMeY-CuBMb exhibited significantly higher oxidase activity at 15.0 μM min−1 for O2 reduction, with 82% conversion of O2 into water. Similarly to the case of Phe33Tyr-CuBMb, addition of copper to Phe33OMeY-CuBMb did not increase oxidase activity.20 Since the pKa of OMeY (Fig. S6†) is similar to that of Tyr, the lower redox potential of OMeY is likely responsible for the increased oxidase activity and O2 reduction selectivity of Phe33OMeY-CuBMb.
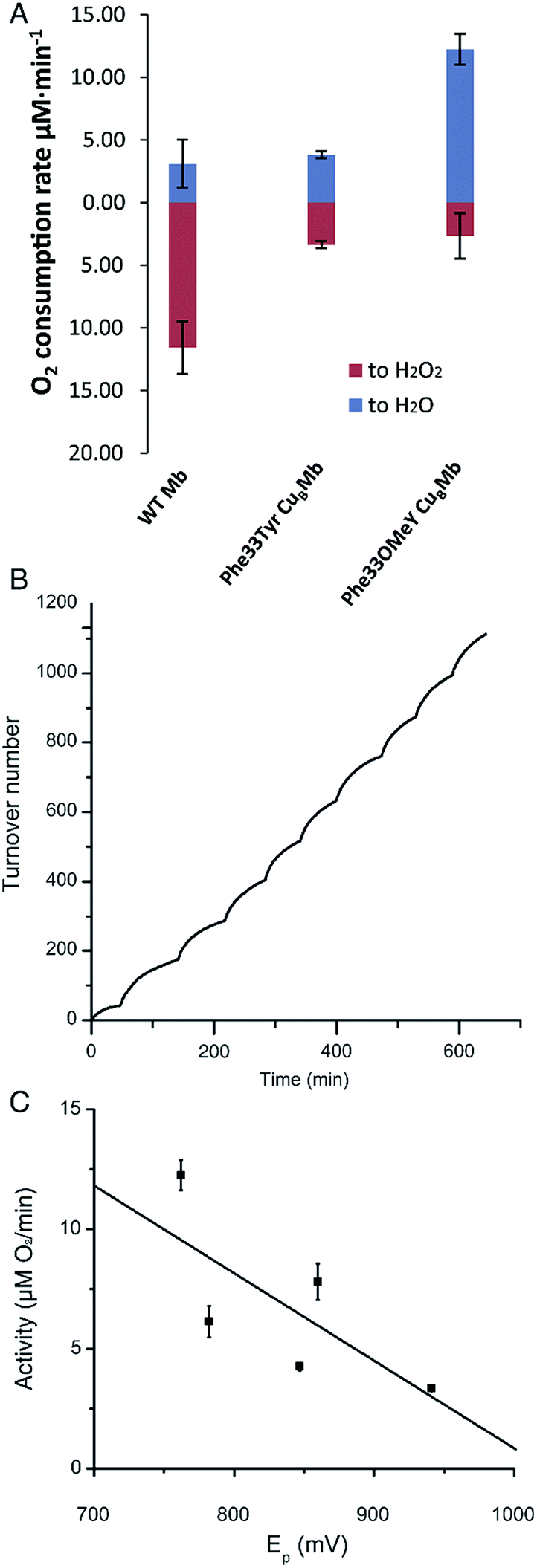 | ||
| Fig. 3 (A) Rates of oxygen reduction to form either water (blue) or ROS (red) catalyzed by 6 μM WTMb, Phe33Tyr-CuBMb or Phe33OMeY-CuBMb. (B) O2 reduction turnover number for Phe33OMeYCuBMb, measured during the stepwise addition of O2. (C) Plot of oxidase activity of Phe33Tyr-CuBMb, Phe33ClY-CuBMb, Phe33F2Y-CuBMb, Phe33F3Y-CuBMb, and Phe33OMeY-CuBMb vs. the peak potential at pH 7 (Ep) of the corresponding Tyr and Tyr analogs. Ep values measured by cyclic voltammetry were 941 mV for Tyr, 847 mV for ClY, 782 mV for F2Tyr, 860 mV for F3Y, and 762 mV for OMeY. Abbreviations: ClY, 3-chlorotyrosine; F2Y, 3,5-difluorotyrosine; F3Y, 2,3,5-trifluorotyrosine. | ||
To further demonstrate the robustness of the best oxidase-mimicking enzyme, Phe33OMeY-CuBMb, we carried out multiple turnover experiments (Fig. 3B). Phe33OMeY-CuBMb was able to catalyze O2 reduction for more than 1100 turnovers without significant reduction of catalytic rate. Under similar conditions, Phe33Tyr-CuBMb could only catalyze the reaction for fewer than 500 turnovers.20
Previous studies with various halogenated Tyr analogs in an oxidase model have shown that oxidase activity is correlated with the pKa of Tyr or its analogs, however, correlation between oxidase activity and reduction potential at pH 7 is weak. Since Tyr oxidation at neutral pH, when Tyr is protonated, is a process coupled with the loss of a proton, the reduction potential of Tyr is influenced by pKa. It is hard to separate the effect of pKa from the reduction potential of Tyr. As OMeY has a much lower reduction potential but similar pKa to Tyr, it is clear that decreasing the reduction potential, similar to decreasing the pKa, also enhances oxidase activity (Fig. 3C). The correlation of reduction potential/pKa with oxidase activity is consistent with the active role of Tyr in the oxidase reaction, as previous studies have shown that a tyrosyl radical is formed during the oxygen reduction reaction of the Mb-based functional oxidase.30
Unnatural Tyr analogs as spectroscopic or functional probes have been developed to study the function of Tyr in different enzymes. Halogenated Tyr analogs have different pKa values, as well as distinct EPR signals,21,25 making them useful for pin-pointing the location of the tyrosyl radical intermediate and the proton donating ability of Tyr. Dopa and NH2Y have decreased reduction potential.22,23 They are used in reductive enzymes as they are susceptible to oxidative damage. OMeY has a lower reduction potential than Tyr, yet is relatively stable under oxidative conditions, making it suitable for studying oxidative enzymes, such as cytochrome c oxidase, galactose oxidase, and lytic polysaccharide monooxygenase.
Conclusions
In summary, by incorporation of OMeY, an analog with a 179 mV lower reduction potential and similar pKa to Tyr, into a Mb-based functional oxidase, we found that the oxidase activity of the protein is correlated with the reduction potential of active site Tyr or its analogs. This further reveals the active role of Tyr in the oxidase reaction.Tyr is an important residue for electron transfer31 as well as catalysis,14 due to its redox activity and proton-coupled electron transfer ability. Nature has evolved different ways of conducting post-translational modifications,32–34 along with manipulating hydrogen bonding and π–π stacking to fine-tune the properties of Tyr. OMeY, with its low reduction potential while being relatively stable to O2, has been added as a unique member to the toolbox of Tyr analogs35–37 for studying and engineering Tyr-containing enzymes.
Acknowledgements
We thank the Shanghai Synchrotron Radiation Facility beamline scientists for their technical support during crystal diffraction data collection. We gratefully acknowledge the Major State Basic Research Program of China (2015CB856203, 2011CBA00800), National Science Foundation of China (21325211, 90913022, 31270859 21473237) and MOST innovation funds (14C26211100178) to J.W., and National Institutes of Health (GM06221) to Y.L.Notes and references
- R. Das and D. Baker, Annu. Rev. Biochem., 2008, 77, 363–382 CrossRef CAS PubMed.
- J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309–313 CrossRef CAS PubMed.
- W. F. DeGrado, C. M. Summa, V. Pavone, F. Nastri and A. Lombardi, Annu. Rev. Biochem., 1999, 68, 779–819 CrossRef CAS PubMed.
- G. Grigoryan, Y. H. Kim, R. Acharya, K. Axelrod, R. M. Jain, L. Willis, M. Drndic, J. M. Kikkawa and W. F. DeGrado, Science, 2011, 332, 1071–1076 CrossRef CAS PubMed.
- R. L. Koder, J. L. Anderson, L. A. Solomon, K. S. Reddy, C. C. Moser and P. L. Dutton, Nature, 2009, 458, 305–309 CrossRef CAS PubMed.
- T. K. Hyster, L. Knorr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- Y. Li, B. Wang, Y. Luo, D. Yang, P. Tong, J. Zhao, L. Luo, Y. Zhou, S. Chen, F. Cheng and J. Qu, Nat. Chem., 2013, 5, 320–326 CrossRef CAS PubMed.
- A. J. Reig, M. M. Pires, R. A. Snyder, Y. Wu, H. Jo, D. W. Kulp, S. E. Butch, J. R. Calhoun, T. Szyperski, E. I. Solomon and W. F. DeGrado, Nat. Chem., 2012, 4, 900–906 CrossRef CAS PubMed.
- M. S. Rogers, E. M. Tyler, N. Akyumani, C. R. Kurtis, R. K. Spooner, S. E. Deacon, S. Tamber, S. J. Firbank, K. Mahmoud, P. F. Knowles, S. E. Phillips, M. J. McPherson and D. M. Dooley, Biochemistry, 2007, 46, 4606–4618 CrossRef CAS PubMed.
- K. Kano, T. Mori, B. Uno, M. Goto and T. Ikeda, Biochim. Biophys. Acta, Gen. Subj., 1993, 1157, 324–331 CrossRef CAS.
- J. W. Whittaker, Chem. Rev., 2003, 103, 2347–2363 CrossRef CAS PubMed.
- P. A. Frey, A. D. Hegeman and G. H. Reed, Chem. Rev., 2006, 106, 3302–3316 CrossRef CAS PubMed.
- J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98, 705–762 CrossRef CAS PubMed.
- V. R. Kaila, M. I. Verkhovsky and M. Wikstrom, Chem. Rev., 2010, 110, 7062–7081 CrossRef CAS PubMed.
- B. Diner and R. D. Britt, in Photosystem II, ed. T. Wydrzynski, K. Satoh and J. Freeman, Springer, Netherlands, 2005, vol. 22, ch. 10, pp. 207–233 Search PubMed.
- M. Wikstrom, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 468–475 CrossRef CAS PubMed.
- D. A. Pratt, R. P. Pesavento and W. A. van der Donk, Org. Lett., 2005, 7, 2735–2738 CrossRef CAS PubMed.
- X. Liu, Y. Yu, C. Hu, W. Zhang, Y. Lu and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 4312–4316 CrossRef CAS PubMed.
- K. D. Miner, A. Mukherjee, Y.-G. Gao, E. L. Null, I. D. Petrik, X. Zhao, N. Yeung, H. Robinson and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 5589–5592 CrossRef CAS PubMed.
- Y. Yu, X. Lv, J. Li, Q. Zhou, C. Cui, P. Hosseinzadeh, A. Mukherjee, M. J. Nilges, J. Wang and Y. Lu, J. Am. Chem. Soc., 2015, 137, 4594–4597 CrossRef CAS PubMed.
- L. Alfonta, Z. Zhang, S. Uryu, J. A. Loo and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 14662–14663 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, J. Xie, C. T. Chan, P. G. Schultz and J. Stubbe, J. Am. Chem. Soc., 2007, 129, 15060–15071 CrossRef CAS PubMed.
- S. Ito and K. Wakamatsu, Photochem. Photobiol., 2008, 84, 582–592 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, S. Y. Reece, D. G. Nocera and J. Stubbe, J. Am. Chem. Soc., 2006, 128, 1569–1579 CrossRef CAS PubMed.
- X. Liu, J. Li, C. Hu, Q. Zhou, W. Zhang, M. Hu, J. Zhou and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 4805–4809 CrossRef CAS PubMed.
- S. G. Lee, S. P. Hong, Y. H. Choi, Y. J. Chung and M. H. Sung, Protein Expression Purif., 1997, 11, 263–270 CrossRef CAS PubMed.
- Q. Zhou, M. Hu, W. Zhang, L. Jiang, S. Perrett, J. Zhou and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 1203–1207 CrossRef CAS PubMed.
- A. S. Pawate, J. Morgan, A. Namslauer, D. Mills, P. Brzezinski, S. Ferguson-Miller and R. B. Gennis, Biochemistry, 2002, 41, 13417–13423 CrossRef CAS PubMed.
- Y. Yu, A. Mukherjee, M. J. Nilges, P. Hosseinzadeh, K. D. Miner and Y. Lu, J. Am. Chem. Soc., 2014, 136, 1174–1177 CrossRef CAS PubMed.
- J. J. Warren, J. R. Winkler and H. B. Gray, FEBS Lett., 2012, 586, 596–602 CrossRef CAS PubMed.
- Y. K. Lee, M. M. Whittaker and J. W. Whittaker, Biochemistry, 2008, 47, 6637–6649 CrossRef CAS PubMed.
- T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono and S. Yoshikawa, Science, 1996, 272, 1136–1144 CAS.
- S. Yoshikawa, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, E. Yamashita, N. Inoue, M. Yao, M. J. Fei, C. P. Libeu, T. Mizushima, H. Yamaguchi, T. Tomizaki and T. Tsukihara, Science, 1998, 280, 1723–1729 CrossRef CAS.
- J. A. Cotruvo and J. Stubbe, Annu. Rev. Biochem., 2011, 80, 733–767 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413 CrossRef CAS PubMed.
- C. Hu, S. I. Chan, E. B. Sawyer, Y. Yu and J. Wang, Chem. Soc. Rev., 2014, 43, 6498–6510 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures. See DOI: 10.1039/c5sc01126d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |