
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkyl-nitrile adlayers as probes of plasmonically induced electric fields†
Daniel T.
Kwasnieski
,
Hao
Wang
and
Zachary D.
Schultz
 *
*
Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: Schultz.41@nd.edu
First published on 4th June 2015
Abstract
Vibrational Stark shifts observed from mercaptoalkyl monolayers on surface enhanced Raman (SERS) active materials are reported to provide a direct measurement of the local electric field around plasmonic nanostructures. Adlayers of CN−, p-mercaptobenzonitrile, and n-mercaptobutylnitrile were adsorbed to a heterogeneous nanostructured Ag surface. The frequency of the CN moiety was observed to shift in a correlated fashion with the SERS intensity. These shifts are attributed to a vibrational Stark shift arising from rectification of the optical field, which gives rise to a DC potential on the surface. All three molecules showed CN Stark shifts on the plasmonic surfaces. p-Mercaptobenzonitrile is observed to be a well-behaved probe of the electric field, providing a narrow spectral line, suggesting a more uniform orientation on the surface. The utility of p-mercaptobenzonitrile was demonstrated by successfully assessing the electric field between gold nanoparticles adsorbed to a monolayer of the nitrile on a flat gold surface. A model is presented where the Stark shift of the alkyl-nitrile probe can be correlated to optical field, providing an intensity independent measurement of the local electric field environment.
Introduction
The excitation of plasmon resonances in metal nanostructures results in confined electric fields that have been exploited in research areas ranging from chemical catalysis to trace detection.1–4 Plasmonic nanostructures increase the rate of reactions on their surfaces when illuminated at their plasmon resonance frequency.4–9 On nanostructure arrays, heterogeneous reactivity has been observed, implicating “hotspots”, areas of intense electric fields, as important for catalysis.10 The excitation of plasmons has long been associated with nanostructure enhanced spectroscopies,11 such as surface enhanced (SERS) and tip enhanced (TERS) Raman scattering. The increased electric field promote optical processes by increasing the magnitude of the excitation field (Eexc) and the emitted electric field (Eemm) leading to an increased response as shown in eqn (1):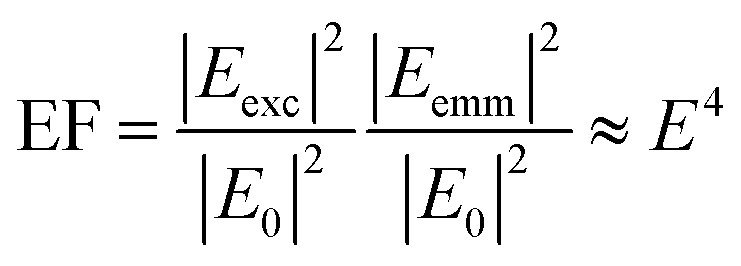 | (1) |
Experimental measurement of the electric fields that arise from plasmonic excitation is challenging and has been determined in different ways. In surface enhanced Raman scattering, the relationship between the observed Raman scattering intensity and the electromagnetic enhancements provides an indirect method to assess the electric fields using the E4 approximation.12 If one can calculate the number of molecules on the plasmonic surface, an analytical enhancement factor can be determined from the increased Raman scattering observed from the molecules relative to the normal Raman cross-section.3,15 It has been shown that hotspots dominate the SERS signal,16 and from eqn (1), the electric field can be backed out. Uncertainty with molecular orientation and surface coverage are problematic for this intensity-based determination of the electric field.17–20 An alternative approach was demonstrated by combining theory with electron energy loss spectroscopy (EELS) to indirectly probe electromagnetic hotspots.21 The use of finite element models with experiment is a common approach. These calculations seem to oversimplify the electric fields in small junctions, and to accurately model the electric fields at the nanoparticle surface requires the inclusion of quantum effects.22
A measurement directly associated with the electric field would provide increased understanding of the electric fields at the surface of nanoparticles arising from plasmon excitation. It has been shown that the intense electric fields associated with excited plasmon resonances can drive nonlinear optical phenomena.23 Specifically, optical rectification results in a DC field at interfaces under the influence of high intensity electric fields.24 This second order nonlinear process has been associated with the variation of second harmonic generation intensities with applied potential.25,26 It has also been shown that exciting a plasmonic junction at the resonant frequency induces a tunneling current,23 and that biasing these plasmonic junctions will modulate nonlinear optical processes.27
The DC field arising from plasmonic excitation has been reported by several groups to generate a Stark shift in a molecule located in the plasmonic junction.28,29 The vibrational Stark effect is a change in the vibrational frequency that arises from a perturbation to the electronic environment of a chemical bond.30 Measurement of the change in vibrational frequency of the bond can thus be used to determine the magnitude of the electric field. The vibrational Stark effect has been used to measure electric fields in a variety of chemical systems including the electrochemical double layer,31–33 catalysts,34 proteins,30,35–37 biomembranes,38 and the energy levels of molecules.39–41
Apkarian and coworkers correlated the vibrational Stark shift from a CO molecule between two spherical particles to an electric field enhancement on the order of 1012, substantiating single molecule detection.29 Results from our laboratory showed a 130 cm−1 Stark shift in the CN stretch frequency when adsorbed CN was detected in a plasmonic junction formed between a nanoparticle TERS tip and a roughened Au surface.28 Plasmonic coupling between the nanoparticle and the gold surface generates a combined plasmon that oscillates in phase preserving the break in symmetry required for the second order nonlinear process. Only the CN in the confined region is observed to shift, and the CN outside the plasmonic junction was observed to persist at the unshifted frequency.
The ability to determine electric fields on plasmonic surfaces suggests a direct method to map electric field distributions on these materials. In this manuscript we demonstrate the use of adsorbed alkyl-nitrile molecules as Stark reporters of the local electric fields. We use the observed Stark shift to determine the local electric field that gives rise to optical rectification. In particular, we note that p-mercaptobenzonitrile appears to be a well-suited probe that can be readily adsorbed to metal structures.
Experimental
Materials and reagents
The copper wire used was of 99.995% purity (Alfa Aesar). The silver electroplating solution used was 1025 RTU (Technic, Inc.). Ethanol (90%) and n-mercaptobutylnitrile (98%) were obtained from Sigma Aldrich (St. Louis, MO) and were used as received. p-Mercaptobenzonitrile was synthesized by the Notre Dame Chemical Synthesis Core and verified to be of >98% pure by NMR. Gold nanoparticle colloid (80 nm citrate NanoXact™ gold) was purchased from nanoComposix (San Diego, CA). Template stripped gold chips used as ultraflat gold surface were from Platypus (100 nm Au thickness, RMS roughness = 3.6 Å). UltraNanopure water (18.2 MΩ cm) was obtained from a Barnstead Nanopure filtration system (Thermo Scientific).Raman
Raman maps were acquired using a Renishaw inVia Raman microscope. The excitation laser was either a 632.8 nm HeNe laser (ThorLabs) for the n-mercaptobutylnitrile and p-mercaptobenzonitrile monolayer experiments or a 660 nm diode laser (Laser Quantum) for the CN− adsorbed to Ag experiments. The acquisition time per spectrum was adjusted with the incident laser power to generate spectra with sufficient signal to noise ratios for analysis. For CN− experiments, the scattering between 1940–2348 cm−1 Raman shift was collected; otherwise, the spectral range was 2016–2470 cm−1. Raman spectra for gold nanoparticles on ultraflat gold films were taken using a homebuilt Raman microscope equipped with dark field microscopy (BD objective, Olympus, LMPlanFLN, NA = 0.5). A 632.8 nm HeNe laser (Melles Griot) was used to irradiate the sample in a top illumination geometry and a Horiba Jobin Yvon iHR320 spectrometer was used to resolve the Raman scattering. The laser power measured at the sample was 0.75 mW, and the acquisition time was 1 s. The spectral data was analyzed using MATLAB and an open-source peak-fitting routine.42 Spectra were fit to a Gaussian lineshape 5 times, and the fit with the lowest % RMS error was selected.Sample preparation
The SERS substrate was fabricated using a modified procedure for embedding electrodes in polystyrene.43 A 100 cm length of approximately 25 μm diameter copper wire was held vertically so that the tip was in contact with a glass disk with copper tape edges. Polystyrene powder was added to the mold and melted at a temperature of 300 °C.43 The filled mold was allowed to cool and harden, after which the polystyrene disk with embedded wire was removed from the mold. The polystyrene was polished using successively finer grit alumina-embedded paper to expose and polish the Cu surface; the dark field scattering of the surface was observed before continuing with the sample preparation, indicating a relatively flat surface with intermittent small, rough areas.Silver was electrodeposited onto the Cu surface using a commercial plating solution containing CN−. A deposition potential of −1 V vs. Ag/AgCl was applied to the wire for 150–300 s, controlled with a CHI660D potentiostat (CH Instruments). Following electroplating, the substrates were washed sequentially with water and ethanol. The deposited surface had CN adsorbed to the surface. The Ag substrates were sized using optical microscopy in the range of 25–50 μm in diameter and were roughly circular.
Monolayers of n-mercaptobutylnitrile and p-mercaptobenzonitrile were prepared by standard monolayer deposition chemistry. First to remove the adsorbed CN−, 10 oxidation–reduction cycles (ORC) were performed in 0.1 M NaOH.44 In the ORC, the potential was swept from −0.6 V to 0.4 V at rate of 10 mV s−1. The removal of CN− was confirmed by Raman spectroscopy, after which the substrate was soaked for 24 h in a 0.01 M ethanolic solution of the appropriate thiol. Following this, the substrates were sequentially washed with water and ethanol. The substrates were stored covered under ambient conditions when not in use.
On template stripped gold films, a layer of p-mercaptobenzonitrile was self-assembled by immersing the ultraflat surface in the p-mercaptobenzonitrile ethanolic solution overnight. The film was then rinsed with ethanol and ultrapure water for 3 cycles each. The gold film with the self-assembled monolayer was subsequently immersed in 2 mL of a mono-dispersed gold nanoparticle colloid (80 nm, 0.05 mg mL−1) suspension in a Petri dish (35 × 10 mm) for 1 hour, after which the gold film was dipped into ultrapure water multiple cycles to remove weakly bound nanoparticles. The prepared film was then dried and used for dark-field microscopy and Raman spectroscopy.
Results
To assess the electric fields present on the electrodeposited silver surfaces, three different CN containing molecules were adsorbed to freshly prepared surfaces: cyanide, p-mercaptobenzonitrile, and n-mercaptobutylnitrile.Fig. 1 shows the results obtained from an electrodeposited Ag surface, with CN− detectable on it. The bright field image (Fig. 1A) indicates a heterogeneous surface topography, which correspond to regions of high and low Raman scattering from the adsorbed CN (Fig. 1B). Previously, it was shown that the frequency of the CN stretch on a Au surface correlated with the observed SERS intensity.28 By fitting the CN stretch mode to a Gaussian lineshape (Fig. 1C), we are able to extract changes in the mode across the silver surface. In Fig. 1D we see that the CN frequency varies by ∼10 to 12 cm−1 across the mapped region. The correlation between the CN frequency shift and Raman intensity is less apparent than in previous reports; however, the magnitude of the shift is consistent with low levels of enhancement. In our previous work on Au surfaces, shifts of this magnitude were also observed at low enhancement levels, thus variations in the amount of CN adsorbed may also generate significant signal changes in addition to the plasmonic enhancement. It should be pointed out that atomic level defects may impact the observed rectification associated with tunneling. It has been reported that tunneling can diminish the observed electric fields and resulting SERS response,45 that may account for some variation. It is well established that SERS enhancements are sensitive to nanoscale structure, which is quite heterogeneous in our system (Fig. S-1†).
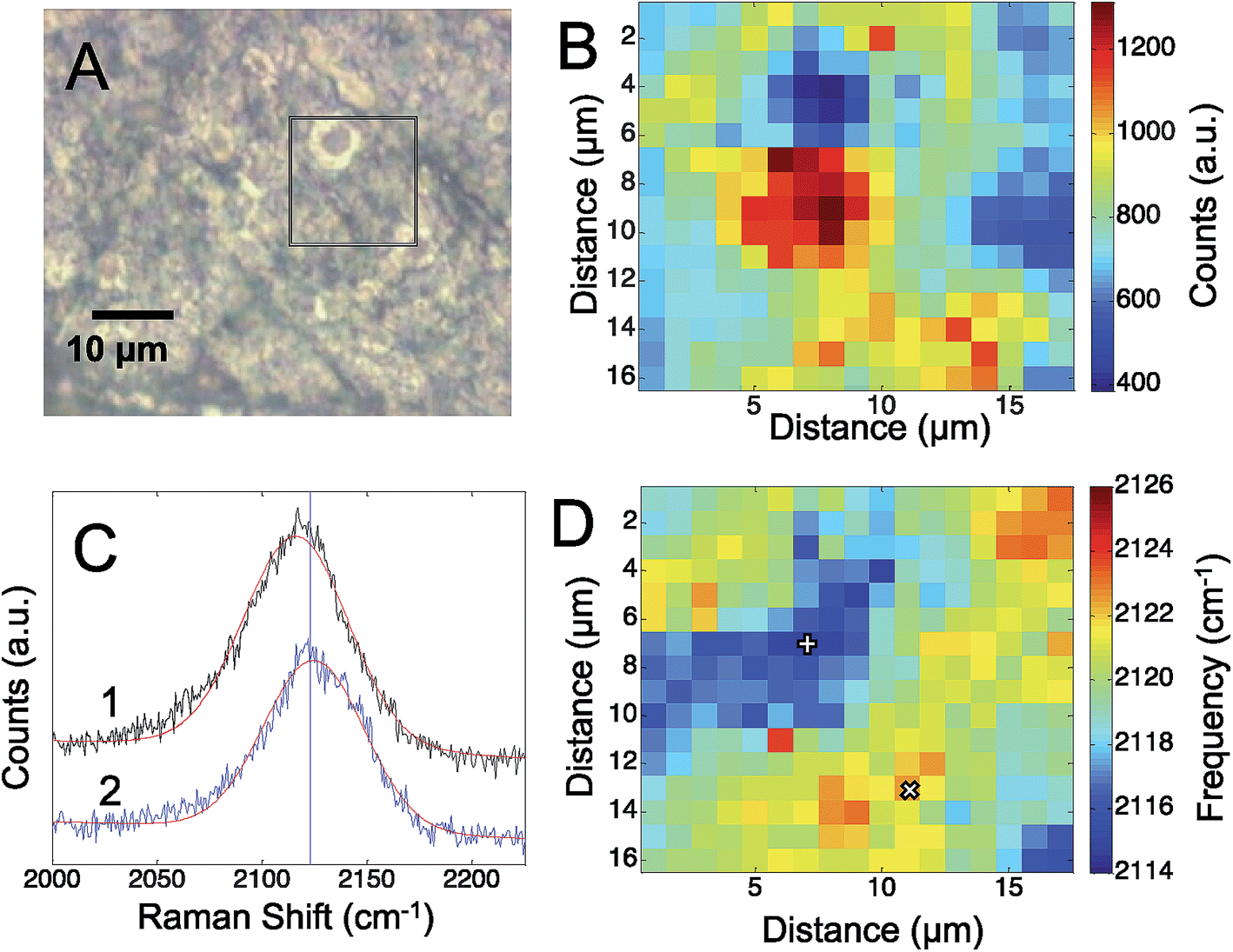 | ||
| Fig. 1 (A) The optical image of the roughened silver surface is shown. The box indicates the approximate area where the Raman map of the CN stretch intensity (B) was acquired. The CN stretch frequency was determined by fitting a Gaussian lineshape to spectrum at each pixel. Sample spectra from areas with high and low Raman intensity are shown (C). The frequency of the CN stretch is plotted (D), showing a variation of up to 12 cm−1 across the electrode. The pixels plotted in C are marked by + = 1 and × = 2. | ||
To further assess the observed Stark shift on the electrode surface, the region was mapped at increasing laser power. We mapped the Ag surface at five different excitation powers, from 0.02 mW to 2.0 mW, using 660 nm laser excitation. Fig. 2 shows three representative maps obtained at 0.1 mW (Fig. 2A), 0.2 mW (Fig. 2B), and 2.0 mW (Fig. 2C), which display a general trend of increasing CN frequency with increasing laser power.
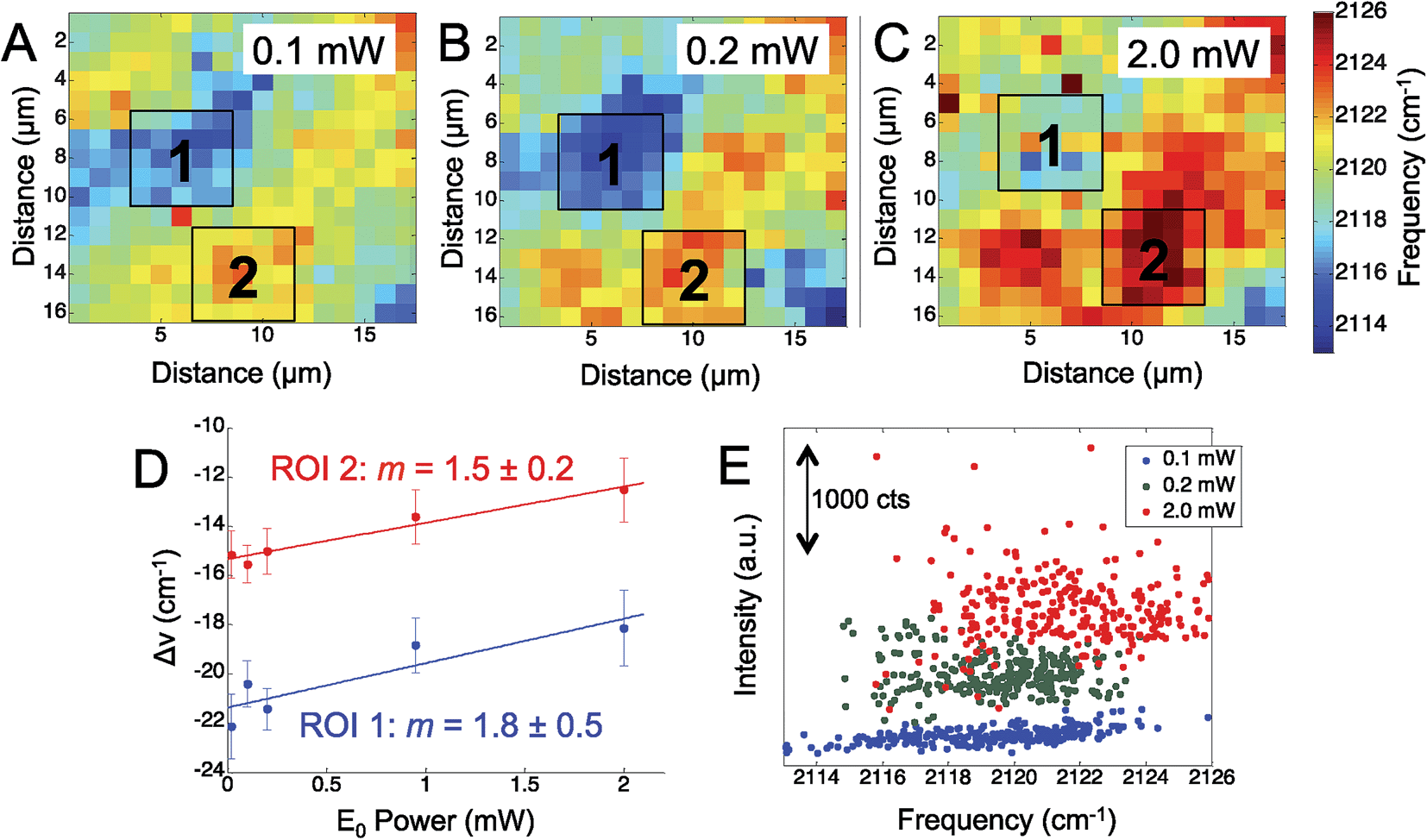 | ||
| Fig. 2 The observed CN stretch frequency observed on a roughened silver surface is plotted at increasing incident laser power: (A) 0.1 mW. (B) 0.2 mW, and (C) 2.0 mW. (D) The average frequency shift observed in each region of interest (ROI) was plotted against the incident laser power (E0). The slope (m) for each is reported. (E) The intensity of the CN mode was plotted against the observed CN frequency. | ||
While the CN stretching frequency was observed to vary across the mapped region of the Ag surface, we chose two distinct regions: one with a lower stretching frequency (blue to light blue region), denoted (1) and one region of higher stretching frequency (yellow to red region), denoted (2). The average frequency shift was calculated over these two regions of interest in the power dependent maps and plotted against the incident laser power (Fig. 2D). The shift in the CN frequency, ΔνCN, was calculated by comparing the observed frequency to a reference value of crystalline AgCN at 2139 cm−1.46 A positive linear relationship was observed between ΔνCN and the laser power in agreement with the visual inspection of the maps. The slope in each ROI was determined to be 1.8 ± 0.5 cm−1 mW−1 and 1.5 ± 0.2 cm−1 mW−1, suggesting a similar change in the CN stretch frequency is observed in both regions. The positive slope observed for the CN frequency versus incident power suggests increasing positive charge on the surface. It is worth noting that the isotropic frequency for CN is greater than the frequencies observed in Fig. 1 and 2. This suggests an induced positive DC field that shifts the CN stretch to a higher frequency. These data suggest the Ag surface potential is initially negative and may affect the Ag–CN interaction compared to CN adsorbed on Au.
Fig. 2E shows a map of the SERS intensity versus frequency of the CN band at the indicated incident field powers. The overall intensity of the CN stretch mode across the entire Raman map is observed to increase with the increased laser power (Fig. 2E). In agreement with the data in Fig. 2D, the range of high and low observed CN frequencies also trend positive with increasing incident power. The SERS intensity is expected to show a nonlinear, E4, correlation to the electric field; however, the data show increasing spread in the intensity, further suggesting the adsorbed CN is located in heterogeneous plasmonic environments.
The CN stretch frequency is highly dependent upon the bonds formed to the nitrile. Earlier work reported difficulty establishing a Stark tuning rate for CN on metals, arising from coupling of the metal–carbon and nitrile bond.47 However, alkyl nitriles have been used successfully as probes of local electric fields.30,37,38,48–50 To further investigate CN Stark shifts in plasmonic environments, we adsorbed p-mercaptobenzonitrile and n-mercaptobutylnitrile to SERS active surfaces.
Fig. 3 shows the results obtained from a self-assembled monolayer of p-mercaptobenzonitrile on the silver SERS surface. To remove any CN from the silver surface, the electrochemical potential was cycled in flowing NaOH.44 The CN band was monitored before and after to insure CN was removed. Similar to the results with adsorbed CN, a heterogeneous structured Ag surface is obtained that shows SERS activity. The Raman map shows areas that suggest hotspots based on the intensity of the adsorbed CN. The greatest SERS intensity is observed in the region between two Ag hemispherical structures. A single Gaussian lineshape was fit to the CN band (Fig. 3C) to determine variation in the CN frequency over the surface (Fig. 3D).
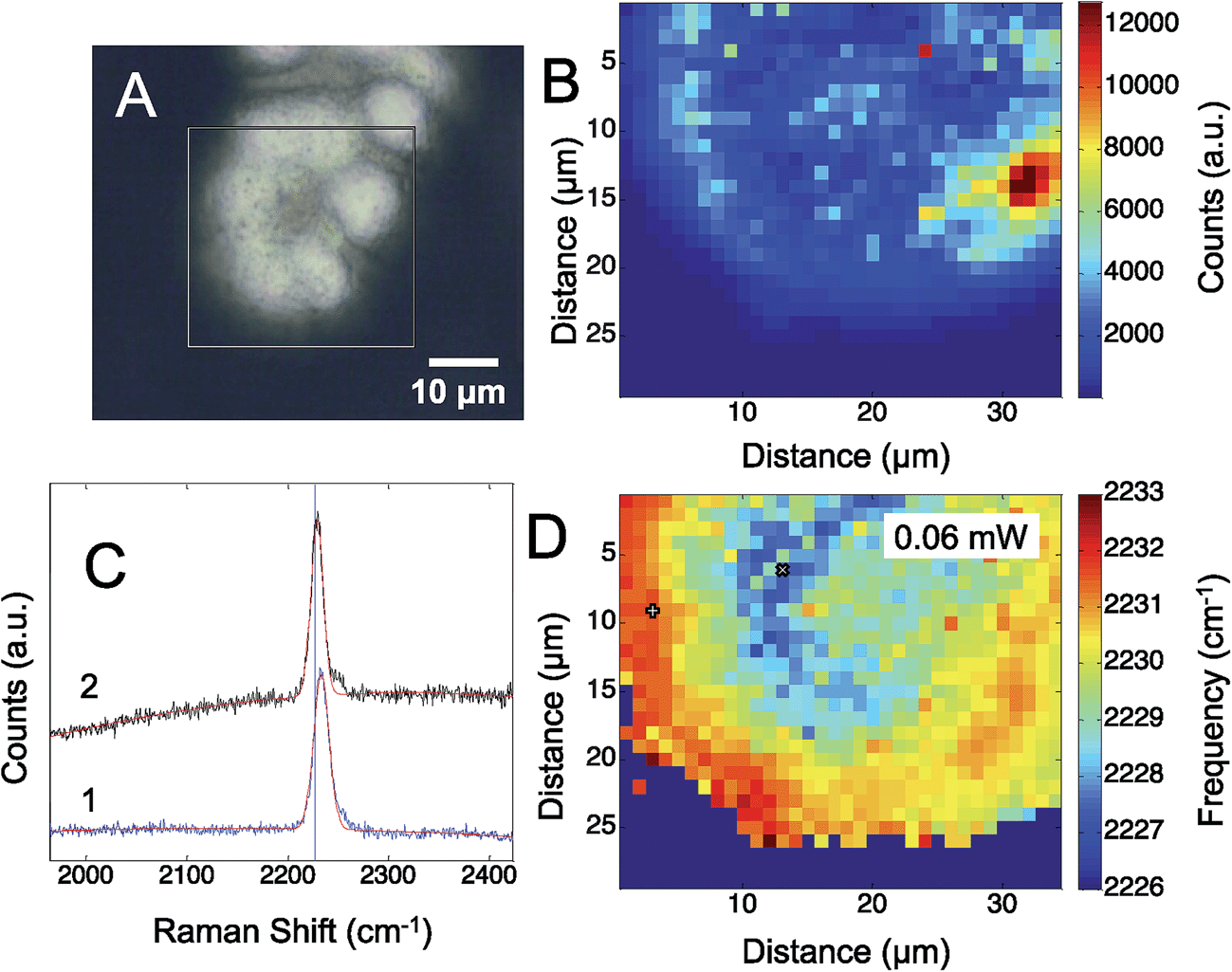 | ||
| Fig. 3 (A) The optical image of the electrodeposited silver surface with p-mercaptobenzonitrile is shown. The box indicates the area where the Raman map (B) was acquired. The CN stretch frequency was determined by fitting a Gaussian lineshape to each pixel. Sample spectra from areas with high and low Raman intensity are shown (C). The observed CN stretch frequency is plotted (D), showing a variation of up to 7 cm−1 across the electrode. The pixels plotted in C are marked by + = 1 and × = 2. | ||
The CN stretch frequency observed from p-mercaptobenzonitrile is different from adsorbed CN−. In Fig. 3C the FWHM of the CN stretch frequency is observed to be 18 and 19 cm−1, whereas adsorbed CN− exhibited a FWHM of 55–58 cm−1. Additionally, the frequency of p-mercaptobenzonitrile (Fig. 3D) is centered around 2230 cm−1, versus 2120 cm−1 in Fig. 1D. The shift in the absolute frequency is consistent with CN frequency of neat p-mercaptobenzonitrile, which we observe at 2223 cm−1 (Fig. S-2†). The change in CN frequency supports the formation of the benzonitrile adlayer on the surface. The narrower peak width observed suggests a more homogeneous environment for nitrile.
Similarly to the CN− adsorbed to Ag experiment above, the CN stretching mode of p-mercaptobenzonitrile was mapped across the Ag surface at five excitation laser powers, ranging from 0.06 mW to 6.4 mW. In these experiments a 633 nm HeNe laser was used. Fig. 4 shows representative maps of the CN frequency at laser powers of 0.06 mW (Fig. 4A), 0.65 mW (Fig. 4B), and 6.4 mW (Fig. 4C). At the lowest laser power, the CN frequency appeared to cluster into three ROIs. The shift in the CN frequency was calculated against the CN frequency of neat p-mercaptobenzonitrile (2223 cm−1). The averaged ΔνCN of three ROI were plotted versus the laser power (Fig. 4D). The trend with increased incident power is different than that observed for adsorbed CN−. Initially, a positive trend was observed for the first three maps, suggesting that the Stark shift was increasing with the incident power as before with CN− on Ag; however, at higher incident power the CN stretch frequency levels off and decreases.
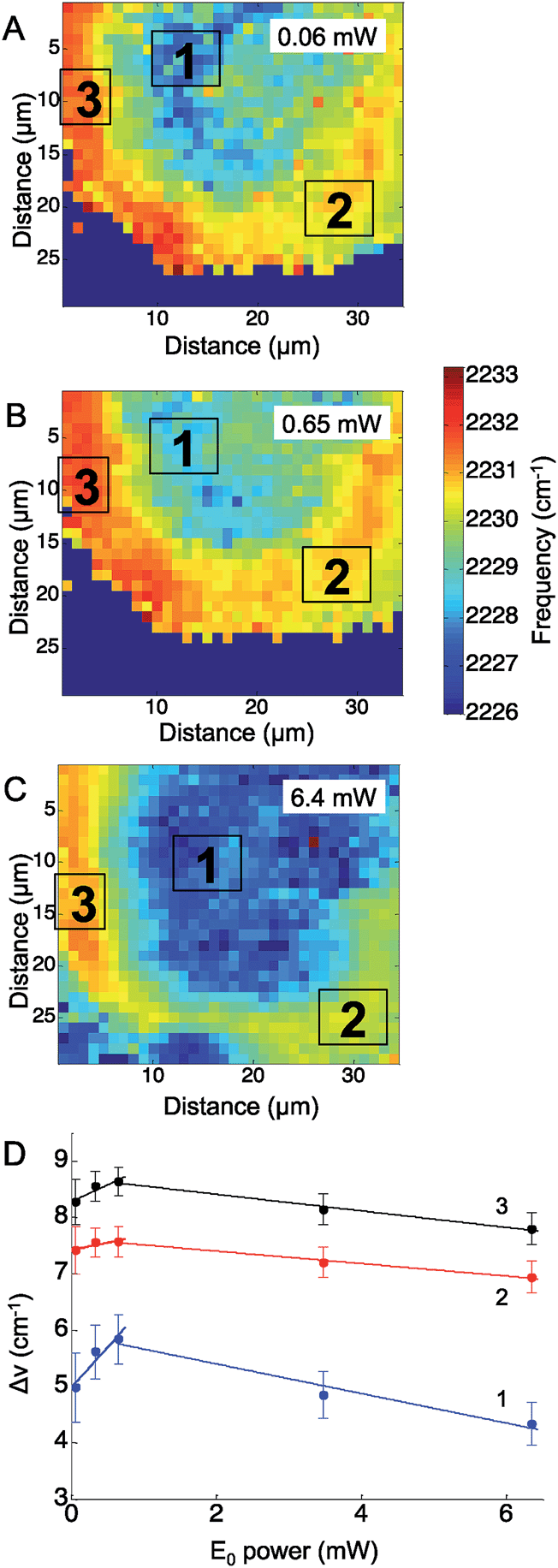 | ||
| Fig. 4 The observed CN stretch frequency of p-mercaptobenzonitrile on a roughened silver surface is plotted at increasing incident laser power: (A) 0.06 mW. (B) 0.65 mW, and (C) 6.4 mW. (D) The average frequency shift observed in each region of interest (ROI) was plotted against the incident laser power (E0). The lines in D are guides for the eye. | ||
Examining the change in the CN stretch frequency suggests at least two different trends. Fitting a line to the initial rise for each ROI, the calculated slopes were 1.4 ± 0.5 cm−1 mW−1 (ROI 1), 0.3 ± 0.1 cm−1 mW−1 (ROI 2), and 0.6 ± 0.2 cm−1 mW−1 (ROI 3). The slope observed in ROI 1 is similar to that observed with changes in incident power for adsorbed CN− (Fig. 2). The slope observed in ROI 2 and ROI 3 is significantly lower, and suggests something else is involved. It is known that organic molecules can be photodecomposed at higher laser powers.16,51 Photodamage may explain the decrease in the observed CN stretch at higher laser powers. This trend correlates with laser powers >1 mW, which are known to damage molecules. The lower initial slopes in ROIs 2 and 3 are also the areas that have the highest initial CN stretch frequencies, which would correlate with the greatest plasmonic fields and increased likelihood of damage. This agrees with work by Dlott and coworkers that suggests molecules in the highest enhancement region are most likely to be photodamaged.16 The p-mercaptobenzonitrile experiment explored higher powers than in the adsorbed CN− experiments, and at excitation powers >1 mW, all three regions show a decrease in average CN stretch frequency, which suggests only molecules adsorbed in lower enhancement regions have survived.
In Fig. 5, the CN stretch frequency shows excellent agreement with our previous results from adsorbed CN− on Au.28 There is a nonlinear trend in intensity that correlates with the observed CN stretch frequency. Interestingly, the intensity data shows a minimum in Fig. 5 near 2227 cm−1, which may indicate the isoelectric point for the CN on the Ag surface.
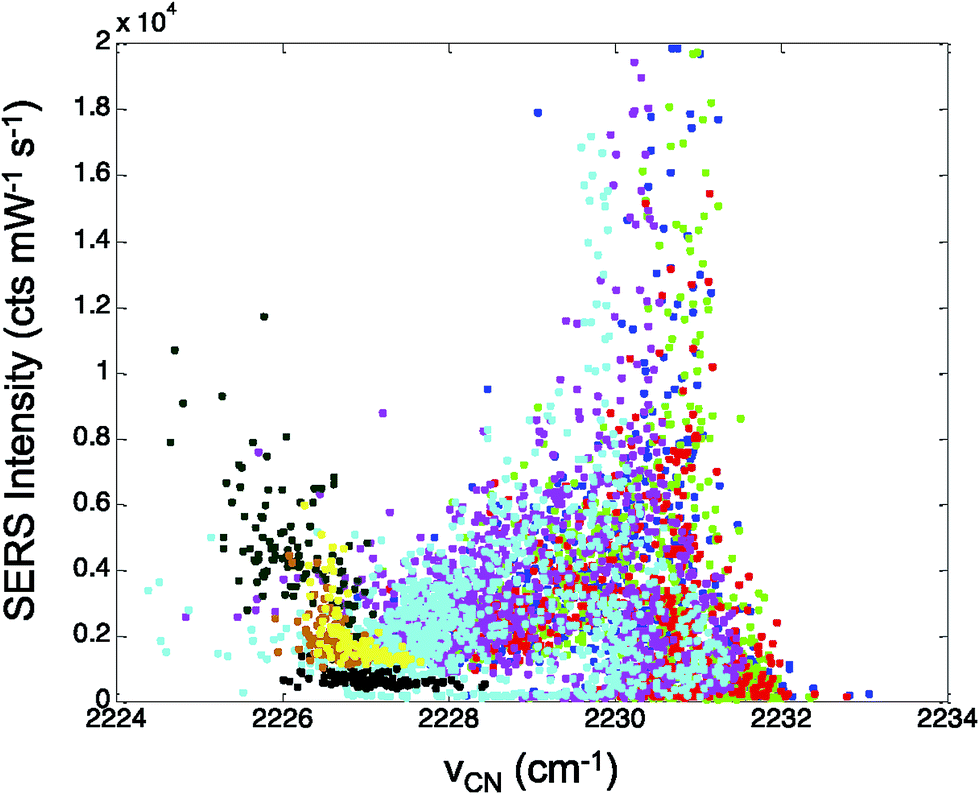 | ||
| Fig. 5 The intensity of the CN mode in mercaptobenzonitrile was plotted against the observed CN frequency across multiple data sets. The colors represent data sets at different incident powers. The reported intensity has been normalized for power and acquisition time. | ||
Fig. 6 compares the spectra obtained from p-mercaptobenzonitrile with those from n-mercaptobutylnitrile. The SERS activity of the Ag surface appears similar to that observed above; however, the observed nitrile spectrum is different than observed for either adsorbed CN− or p-mercaptobenzonitrile. The spectrum of n-mercaptobutylnitrile shows 2 peaks in the CN stretch region centered at 2245 cm−1. This is in contrast to the single peak observed in the spectrum of the crystalline compound (Fig. S-2†). A lower energy band has been previously attributed to a Fermi resonance in n-butylnitrile;52 however, the band observed here is at a considerably lower frequency. As shown in Fig. 6, we attribute the spectral variation observed in n-mercaptobutylnitrile to the flexible C–C backbone which enables rotation and multiple interactions between the CN and the surface. Such interactions are not observed with p-mercaptobenzonitrile because of the rigid phenyl ring structure.
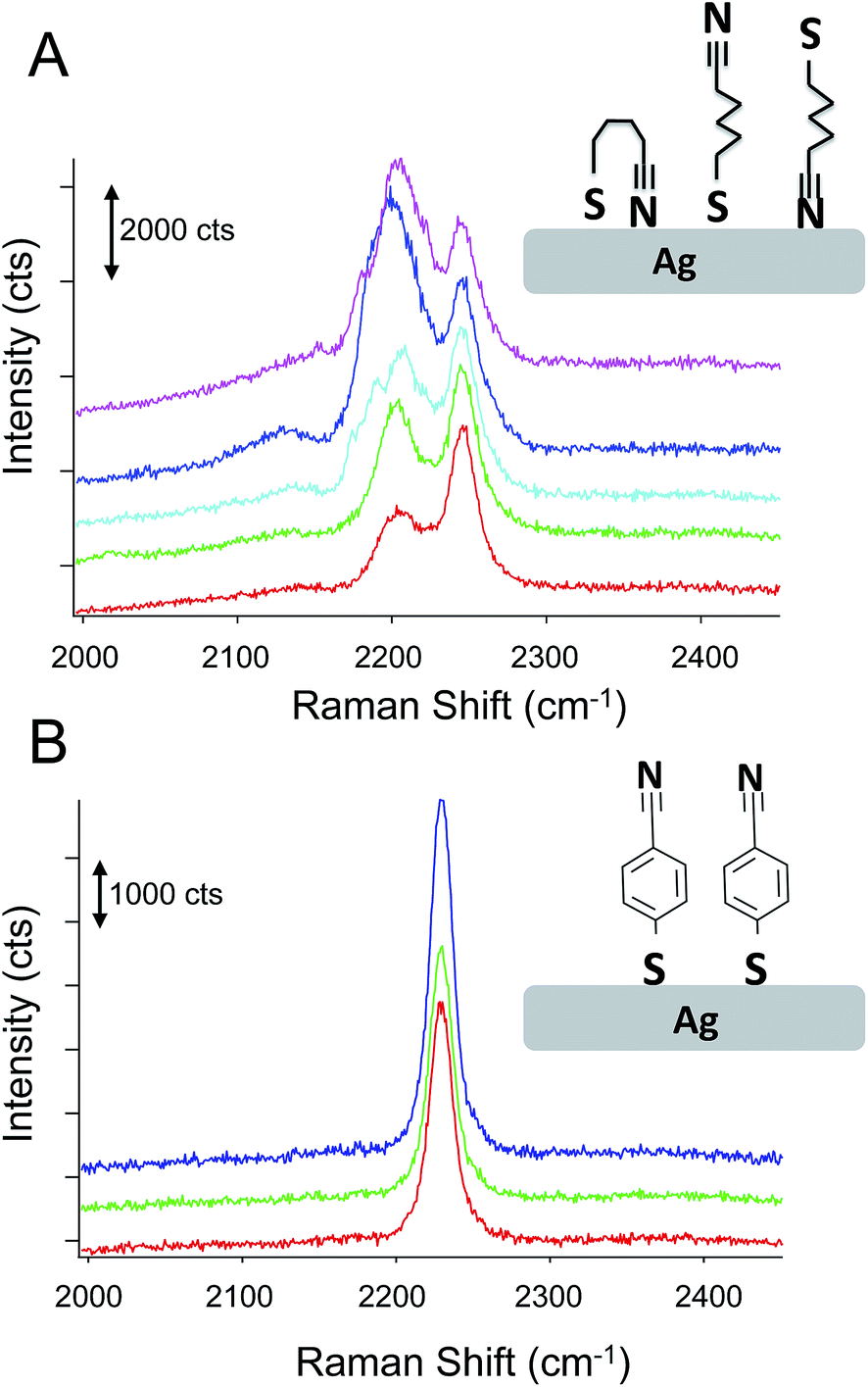 | ||
| Fig. 6 The SERS spectra observed from an adsorbed layer of n-mercaptobutylnitrile (A) show significant spectra to spectra variation compared to the spectra observed from p-mercaptobenzonitrile (B). The insets show possible binding configurations that may explain the differences in observed lineshape. | ||
Analysis of the n-mercaptobutylnitrile spectra was performed (Fig. S-3 and S-4†) and the higher intensity peak appears to shift in manner consistent with a vibrational Stark shift. Interestingly, the spectral variation observed for n-mercaptobutylnitrile appears to be time dependent. Fig. S-5† shows the time varying fluctuations of the CN signal on the SERS substrate. This was not observed in the n-mercaptobenzylnitrile, further implicating the flexible C–C backbone and multiple surface interactions as the origin of the spectral heterogeneity. This interpretation is also consistent with the broader full-width half maximum observed for adsorbed CN, which may also interact in a variety of conformations.
These results suggest that p-mercaptobenzonitrile is the optimal probes of those examined. To further evaluate this, a monolayer of p-mercaptobenzonitrile was adsorbed onto a thin Au film. Citrate capped Au nanoparticles were then deposited onto this film. Fig. 7 shows the dark field image and SERS results obtained from isolated NPs. Brighter scattering particles gave rise to more intense SERS spectra, which may indicate dimers or aggregates on the surface, which would give rise to larger field enhancements.
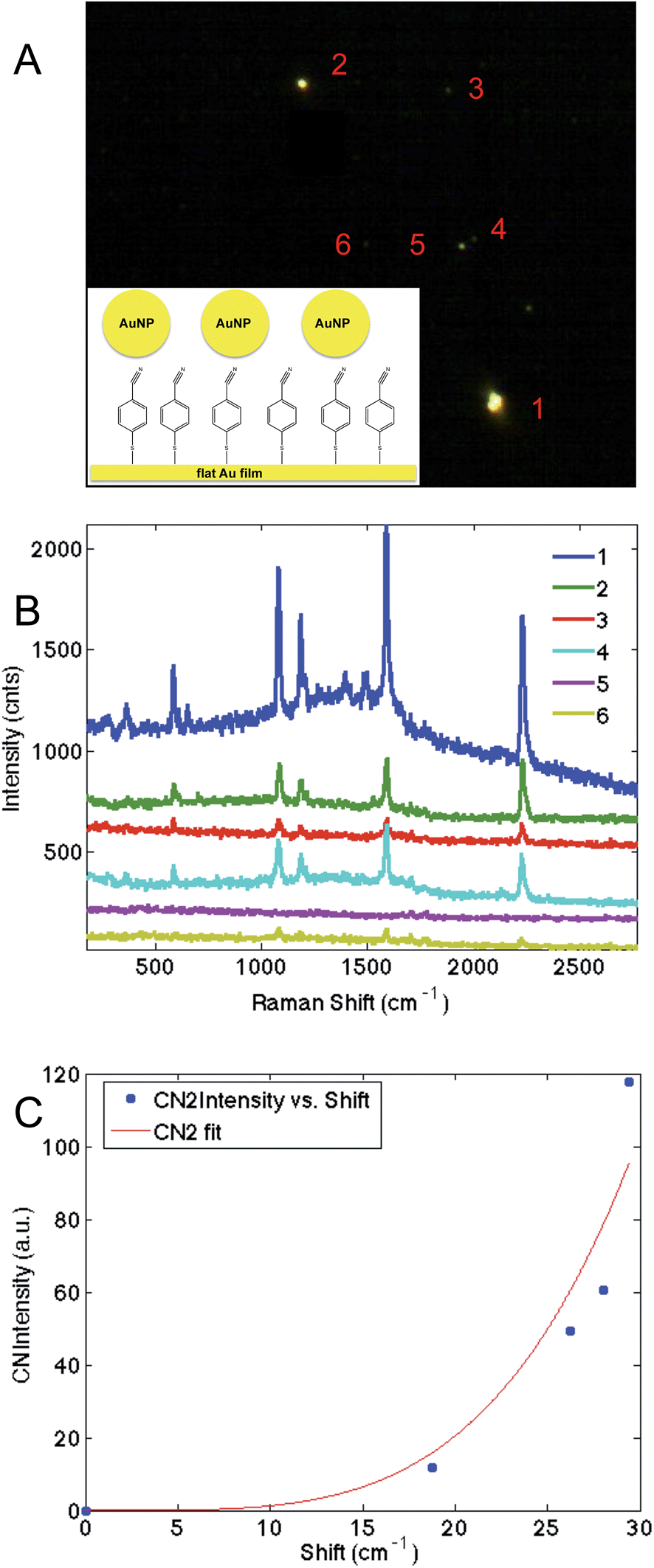 | ||
| Fig. 7 The dark-field image (A) shows the location of nanoparticles, from which SERS spectra (B) were obtained. The numbers in A correspond to the spectra in (B). The CN region was fit to two Gaussian lineshapes, and the higher energy component's frequency is plotted against its intensity, which shows a nonlinear increase (C). The line in (C) is guide for the eye. | ||
The CN region was fit (Fig. S-6†) and a higher energy feature is observed in the CN stretching regions whose frequency correlates with intensity (Fig. 7C). Similar to our previous TERS experiment,28 only a portion of the molecules are shifted, implying that the hotspot is localized. Nonetheless, the agreement between SERS intensity and CN frequency in this more controlled experiment further supports the utility of p-mercaptobenzonitrile as an electric field reporter.
Discussion
The observed shift in the CN stretch frequency is attributed to a Stark effect arising from optical rectification of the optical field to produce a DC component experienced by the adsorbed molecules. The linear dependence on the incident power observed in Fig. 2, and at low powers in Fig. 4 and S-4,† are consistent with optical rectification. This linear trend supports the hypothesis of an optically rectified DC field acting on the CN bond and giving rise to a vibrational Stark shift.A DC field arising from optical rectification suggests a means to calculate the optical field generated by the localized plasmon resonance and determine an enhancement factor independent of signal intensity. It has been shown that the static, or DC, electric field (EDC) experienced by a chemical bond can be related to the change in frequency (Δν) through the Stark tuning coefficient (μ) as follows:30
| EDC = Δνμ | (2) |
Using the vibrational frequency of the neat, or crystalline, nitrile as the reference frequency, the Δν observed on a plasmonic surface is straightforward to calculate.
In optical rectification, the magnitude of the DC field (EDC) is proportional to the time invariant component of the second order polarizability:53
| P(0)(2) ≈ 2χ(2)EE | (3) |
The exact values for the χ(2) tensor for nanostructured plasmonic surfaces are not well documented in the literature.27 However, there are approaches that let us approximate values for χ(2). It is straightforward to estimate χ(2) from literature values of χ(3).54 A first order approximation is:
| χ(2) = χ(3)EAU | (4) |
There are reports of the hyperpolarizabilities for silver and gold;60,61 however, these values are determined on per atom basis. Determining the number of metal atoms that give rise to the enhanced field is unclear. For instance, how do the electrons from adjacent nanostructures alter the hyperpolarizability? Additionally, other factors are needed to properly convert molecular hyperpolerizabilities into susceptibilities for determining the nonlinear response of the larger material.62
The calculation of χ(2) further indicates the importance of the localized plasmon resonance for generating the intense local field that drives the optical rectification. Nonlinear susceptibility measurements performed as a function of wavelength show the susceptibility increases dramatically at wavelengths associated with plasmon resonances.56,58,63–65 Interestingly previous work measuring second harmonic excitation on Ag nanoparticle arrays exhibited a maximum response near 650 nm, similar to the laser wavelengths used in this experiment.64 We have previously measured the extinction spectrum of heterogeneous Ag surface and observed a resonance at these wavelengths.66 These observations are consistent with our hypothesis that the enhanced local fields drive the optical rectification that gives rise to the observed Stark shift.
Using an approximate χ(2) = 104 cm MV−1, the enhanced electromagnetic fields that generate the SERS response, and thus the electromagnetic enhancement factor can be calculated for this system. Literature values for the Stark tuning coefficient for butylnitrile [0.5 cm−1 (MV cm−1)−1] and benzonitrile [0.6 cm−1 (MV cm−1)−1] can convert the frequency shift into a field intensity.67,68 The exact Stark tuning coefficient for CN− is not known, but the value 2.9 cm−1 (MV cm−1)−1 is consistent with our previous work.28 Recognizing the EDC (eqn (2)) is equivalent to the P(0)(2) (eqn (3)), we can establish:
| EDC = χ(2)EE | (5) |
From the measured power and laser spot size, the electric field incident on the surface can be calculated. For low laser powers (0.06 mW, 0.06 mW, and 0.02 mW for p-mercaptobenzonitrile, n-mercaptobutylnitriles, and CN−, respectively) we observe electric field enhancements of 4-6× (p-mercaptobenzonitrile), 2-3× (n-mercaptobutylnitriles), and 6-9× (CN−). Using the standard E4 approximation, these local electric fields correspond to quite modest SERS enhancement factors of ∼102 to 103, which seem low given the observed SERS intensity. Interestingly, the shift observed from gap modes formed between Au nanoparticles and a Au surface are twice the magnitude observed on the Ag substrates, which is consistent with the large electric fields found in this geometry.
There are sources of uncertainty that may explain the low fields calculated. For example, one possible source of difference is the distance of CN bond from the surface. The plasmonic field is reported to decay quickly from the surface, on the length scale of a few nanometers.69,70 Another source of uncertainty is the orientation of the CN bond with respect to the electric field. However, it is worth noting that the calculated electric fields agree fairly well for all three probes given the heterogeneous surface examined in these experiments. As mentioned above, the heterogeneous nanostructure may result in differences between the SERS enhancement, related to the AC optical field, and the observed Stark Shift, which we attribute to the rectified field.
A more fundamental source of uncertainty involves how χ(2) is determined. In the methods above, χ(2) is determined from an experimental measurement of χ(3). With planar Ag substrates, the enhancement of second order processes involves both an increase in the susceptibility associated with the plasmon resonance of the nanostructure but also enhanced local fields arising from exciting this LSPR. Similarly, the χ(3) values measured from Ag nanoparticles determine χ(3) from the response and the incident electric fields. Again, local field enhancements associated with excited LSPRs are convoluted into the response. This suggests the methods we have used to determine χ(2) underestimate the fields giving rise to the optically rectified field. A more accurate value for χ(2) would clearly clarify possible discrepancies.
Conclusions
Vibrational Stark shifts in nitriles are shown to correlate with the electric field present on plasmonically excited structures, thus providing a direct indicator of the enhanced electric field's magnitude. Optical rectification gives rise to a DC field that perturbs the CN stretch frequency. This optical rectification also arises from the optically enhanced field and thus can be used to determine electric field enhancements associated with plasmons. Mercaptobenzonitrile was shown to be an advantageous probe of electric field strength for several reasons. First, sulfur-metal bonding makes it straightforward to assemble the probe on many plasmonic surfaces in contrast to CN−, which requires electrochemical deposition. The p-mercaptobenzonitrile appears to adsorb in a more uniform fashion, which is evidenced by a narrower FWHM for the CN stretch. As shown above, the CN stretch mode for adsorbed CN− exhibited a FWHM of ∼55 cm−1, while FWHM associated with p-mercaptobenzonitrile probe was approximately 20 cm−1. By physically separating the nitrile bond from the metal surface, a more reliable Stark tuning coefficients can be determined. The rigid linker provided by the phenyl ring maintains the CN group in a consistent orientation with respect to the surface, which results in a spectrum with a clear nitrile stretch, unlike n-mercaptobutylnitrile's spectrum that is obscured by multiple spectroscopic features. Additionally, the higher Stark tuning coefficient of p-mercaptobenzonitrile relative to n-mercaptobutylnitrile means larger frequency shifts are observed from small changes in the electric field. The results with Au nanoparticles further demonstrate that this probe can be used on different metals and provides a measurement of the electric field strength associated with plasmonic excitation.Acknowledgements
The award R01 GM109988GM109988 from the National Institutes of Health, a Cottrell Scholar Award from Research Corporation for Science Advancement, and the Advanced Diagnostics and Therapeutics Initiative at the University of Notre Dame supported this work.References
- Z. D. Schultz, J. M. Marr and H. Wang, Tip Enhanced Raman Scattering: Plasmonic Enhancements for Nanoscale Chemical Analysis, Nanophotonics, 2014, 3, 91–104 CrossRef CAS PubMed.
- N. J. Halas, S. Lal, W. S. Chang, S. Link and P. Nordlander, Plasmons in Strongly Coupled Metallic Nanostructures, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. R. Van Duyne, Surface-Enhanced Raman Spectroscopy, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Plasmonic-Metal Nanostructures for Efficient Conversion of Solar to Chemical Energy, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- S. Navalon, M. de Miguel, R. Martin, M. Alvaro and H. Garcia, Enhancement of the Catalytic Activity of Supported Gold Nanoparticles for the Fenton Reaction by Light, J. Am. Chem. Soc., 2011, 133, 2218–2226 CrossRef CAS PubMed.
- P. Christopher, H. L. Xin and S. Linic, Visible-Light-Enhanced Catalytic Oxidation Reactions on Plasmonic Silver Nanostructures, Nat. Chem., 2011, 3, 467–472 CAS.
- H. Y. Zhu, X. B. Ke, X. Z. Yang, S. Sarina and H. W. Liu, Reduction of Nitroaromatic Compounds on Supported Gold Nanoparticles by Visible and Ultraviolet Light, Angew. Chem., Int. Ed., 2010, 49, 9657–9661 CrossRef CAS PubMed.
- X. Chen, Z. F. Zheng, X. B. Ke, E. Jaatinen, T. F. Xie, D. J. Wang, C. Guo, J. C. Zhao and H. Y. Zhu, Supported Silver Nanoparticles as Photocatalysts under Ultraviolet and Visible Light Irradiation, Green Chem., 2010, 12, 414–419 RSC.
- X. Chen, H. Y. Zhu, J. C. Zhao, Z. T. Zheng and X. P. Gao, Visible-Light-Driven Oxidation of Organic Contaminants in Air with Gold Nanoparticle Catalysts on Oxide Supports, Angew. Chem., Int. Ed., 2008, 47, 5353–5356 CrossRef CAS PubMed.
- L. Kubus, H. Erdogan, S. S. Cetin, E. Biskin and G. Demirel, Plasmon-Enhanced Photocatalysis on Anisotropic Gold Nanorod Arrays, ChemCatChem, 2013, 5, 2973–2977 CrossRef CAS PubMed.
- M. Moskovits, Surface Roughness and the Enhanced Intensity of Raman Scattering by Molecules Adsorbed on Metals, J. Chem. Phys., 1978, 69, 4159–4161 CrossRef CAS PubMed.
- D. S. Wang and M. Kerker, Enhanced Raman-Scattering by Molecules Adsorbed at the Surface of Colloidal Spheroids, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 1777–1790 CrossRef CAS.
- S. Nie and S. R. Emory, Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. Dasari and M. S. Feld, Single Molecule Detection Using Surface-Enhanced Raman Scattering (SERS), Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- S. L. Kleinman, R. R. Frontiera, A. I. Henry, J. A. Dieringer and R. P. Van Duyne, Creating, Characterizing, and Controlling Chemistry with SERS Hot Spots, Phys. Chem. Chem. Phys., 2013, 15, 21–36 RSC.
- Y. Fang, N.-H. Seong and D. D. Dlott, Measurement of the Distribution of Site Enhancements in Surface-Enhanced Raman Scattering, Science, 2008, 321, 388–392 CrossRef CAS PubMed.
- E. C. Le Ru, J. Grand, N. Felidj, J. Aubard, G. Levi, A. Hohenau, J. R. Krenn, E. Blackie and P. G. Etchegoin, Experimental Verification of the SERS Electromagnetic Model Beyond the |E|4 Approximation: Polarization Effects, J. Phys. Chem. C, 2008, 112, 8117–8121 CAS.
- E. C. Le Ru and P. G. Etchegoin, Rigorous Justification of the |E|4 Enhancement Factor in Surface Enhanced Raman Spectroscopy, Chem. Phys. Lett., 2006, 423, 63–66 CrossRef CAS PubMed.
- P. G. Etchegoin, C. Galloway and E. C. Le Ru, Polarization-Dependent Effects in Surface-Enhanced Raman Scattering (SERS), Phys. Chem. Chem. Phys., 2006, 8, 2624–2628 RSC.
- S. Franzen, Intrinsic Limitations on the |E|4 Dependence of the Enhancement Factor for Surface-Enhanced Raman Scattering, J. Phys. Chem. C, 2009, 113, 5912–5919 CAS.
- N. W. Bigelow, A. Vaschillo, V. Iberi, J. P. Camden and D. J. Masiello, Characterization of the Electron- and Photon-Driven Plasmonic Excitations of Metal Nanorods, ACS Nano, 2012, 6, 7497–7504 CrossRef CAS PubMed.
- R. Esteban, A. G. Borisov, P. Nordlander and J. Aizpurua, Bridging Quantum and Classical Plasmonics with a Quantum-Corrected Model, Nat. Commun., 2012, 3, 825 CrossRef PubMed.
- D. R. Ward, F. Huser, F. Pauly, J. C. Cuevas and D. Natelson, Optical Rectification and Field Enhancement in a Plasmonic Nanogap, Nat. Nanotechnol., 2010, 5, 732–736 CrossRef CAS PubMed.
- Y. R. Shen, The Principles of Nonlinear Optics, J. Wiley, New York, 1984, p xii, p. 563 Search PubMed.
- R. M. Corn, M. Romagnoli, M. D. Levenson and M. R. Philpott, The Potential Dependence of Surface Plasmon-Enhanced 2nd-Harmonic Generation at Thin-Film Silver Electrodes, Chem. Phys. Lett., 1984, 106, 30–35 CrossRef CAS.
- G. L. Richmond, Adsorption of Ions on Smooth and Roughened Silver Surfaces - a Comparative-Study by Optical 2nd Harmonic-Generation, Chem. Phys. Lett., 1985, 113, 359–363 CrossRef CAS.
- W. Cai, A. P. Vasudev and M. L. Brongersma, Electrically Controlled Nonlinear Generation of Light with Plasmonics, Science, 2011, 333, 1720–1723 CrossRef CAS PubMed.
- J. M. Marr and Z. D. Schultz, Imaging Electric Fields in SERS and TERS Using the Vibrational Stark Effect, J. Phys. Chem. Lett., 2013, 4, 3268–3272 CrossRef CAS PubMed.
- M. Banik, P. Z. El-Khoury, A. Nag, A. Rodriguez-Perez, N. Guarrottxena, G. C. Bazan and V. A. Apkarian, Surface-Enhanced Raman Trajectories on a Nano-Dumbbell: Transition from Field to Charge Transfer Plasmons as the Spheres Fuse, ACS Nano, 2012, 6, 10343–10354 CrossRef CAS PubMed.
- S. G. Boxer, Stark Realities, J. Phys. Chem. B, 2009, 113, 2972–2983 CrossRef CAS.
- V. Oklejas and J. M. Harris, Potential-Dependent Surface-Enhanced Raman Scattering from Adsorbed Thiocyanate for Characterizing Silver Surfaces with Improved Reproducibility, Appl. Spectrosc., 2004, 58, 945–951 CrossRef CAS PubMed.
- V. Oklejas, C. Sjostrom and J. M. Harris, SERS Detection of the Vibrational Stark Effect from Nitrile-Terminated SAMs to Probe Electric Fields in the Diffuse Double-Layer, J. Am. Chem. Soc., 2002, 124, 2408–2409 CrossRef CAS PubMed.
- D. K. Lambert, Vibrational Stark Effect of Adsorbates at Electrochemical Interfaces, Electrochim. Acta, 1996, 41, 623–630 CrossRef CAS.
- M. McEntee, A. Stevanovic, W. Tang, M. Neurock and J. T. Yates, Electric Field Changes on Au Nanoparticles on Semiconductor Supports – the Molecular Voltmeter and Other Methods to Observe Adsorbate-Induced Charge-Transfer Effects in Au/Tio2 Nanocatalysts, J. Am. Chem. Soc., 2015, 137, 1972–1982 CrossRef CAS PubMed.
- A. J. Stafford, D. L. Ensign and L. J. Webb, Vibrational Stark Effect Spectroscopy at the Interface of Ras and Rap1a Bound to the Ras Binding Domain of Ralgds Reveals an Electrostatic Mechanism for Protein-Protein Interaction, J. Phys. Chem. B, 2010, 114, 15331–15344 CrossRef CAS PubMed.
- L. J. Webb and S. G. Boxer, Electrostatic Fields near the Active Site of Human Aldose Reductase: 1. New Inhibitors and Vibrational Stark Effect Measurements, Biochemistry, 2008, 47, 1588–1598 CrossRef CAS PubMed.
- I. T. Suydam and S. G. Boxer, Vibrational Stark Effects Calibrate the Sensitivity of Vibrational Probes for Electric Fields in Proteins, Biochemistry, 2003, 42, 12050–12055 CrossRef CAS PubMed.
- W. H. Hu and L. J. Webb, Direct Measurement of the Membrane Dipole Field in Bicelles Using Vibrational Stark Effect Spectroscopy, J. Phys. Chem. Lett., 2011, 2, 1925–1930 CrossRef CAS.
- W. A. Bryan, C. R. Calvert, R. B. King, J. B. Greenwood, W. R. Newell and I. D. Williams, Controlled Redistribution of Vibrational Population by Few-Cycle Strong-Field Laser Pulses, Faraday Discuss., 2011, 153, 343–360 RSC.
- S. Y. Zou, Q. H. Ren, G. G. Balint-Kurti and F. R. Manby, Analytical Control of Molecular Excitations Including Strong Field Polarization Effects, Phys. Rev. Lett., 2006, 96, 243003 CrossRef.
- K. Hermansson and H. Tepper, Electric-Field Effects on Vibrating Polar Molecules: From Weak to Strong Fields, Mol. Phys., 1996, 89, 1291–1299 CAS.
- T. O'Haver, Interactive Peak Fitter 9.2, Mathworks, Natick, MA, 2013 Search PubMed.
- A. Selimovic and R. S. Martin, Encapsulated Electrodes for Microchip Devices: Microarrays and Platinized Electrodes for Signal Enhancement, Electrophoresis, 2013, 34, 2092–2100 CrossRef CAS PubMed.
- M. R. Bailey, A. M. Pentecost, A. Selimovic, R. S. Martin and Z. D. Schultz, Sheath-Flow Microfluidic Approach for Combined Surface Enhanced Raman Scattering and Electrochemical Detection, Anal. Chem., 2015, 87, 4347–4355 CrossRef CAS PubMed.
- K. J. Savage, M. M. Hawkeye, R. Esteban, A. G. Borisov, J. Aizpurua and J. J. Baumberg, Revealing the Quantum Regime in Tunnelling Plasmonics, Nature, 2012, 491, 574–577 CrossRef CAS PubMed.
- A. A. Isab, M. I. M. Wazeer, M. Fettouhi, B. A. Al-Maythalony, A. R. Al-Arfaj and N. O. Al-Zamil, Solid State and Solution Nmr, X-Ray and Antimicrobial Studies of 1 : 1 and 2 : 1 Complexes of Silver(I) Cyanide with Alkanediamine Ligands, Inorg. Chim. Acta, 2007, 360, 3719–3726 CrossRef CAS PubMed.
- P. S. Bagus, C. J. Nelin, W. Müller, M. R. Philpott and H. Seki, Field-Induced Vibrational Frequency Shifts of Co and Cn Chemisorbed on Cu(100), Phys. Rev. Lett., 1987, 58, 559–562 CrossRef CAS.
- N. M. Levinson, S. D. Fried and S. G. Boxer, Solvent-Induced Infrared Frequency Shifts in Aromatic Nitriles Are Quantitatively Described by the Vibrational Stark Effect, J. Phys. Chem. B, 2012, 116, 10470–10476 CrossRef CAS PubMed.
- E. S. Park, S. S. Andrews, R. B. Hu and S. G. Boxer, Vibrational Stark Spectroscopy in Proteins: A Probe and Calibration for Electrostatic Fields, J. Phys. Chem. B, 1999, 103, 9813–9817 CrossRef CAS.
- V. Oklejas, C. Sjostrom and J. M. Harris, Surface-Enhanced Raman Scattering Based Vibrational Stark Effect as a Spatial Probe of Interfacial Electric Fields in the Diffuse Double Layer, J. Phys. Chem. B, 2003, 107, 7788–7794 CrossRef CAS.
- T. Schmid, A. Messmer, B. S. Yeo, W. H. Zhang and R. Zenobi, Towards Chemical Analysis of Nanostructures in Biofilms Ii: Tip-Enhanced Raman Spectroscopy of Alginates, Anal. Bioanal. Chem., 2008, 391, 1907–1916 CrossRef CAS PubMed.
- S. S. Andrews and S. G. Boxer, Vibrational Stark Effects of Nitriles Ii. Physical Origins of Stark Effects from Experiment and Perturbation Models, J. Phys. Chem. A, 2002, 106, 469–477 CrossRef CAS.
- R. W. Boyd, Nonlinear Optics, Academic Press, San Diego, CA, 2nd edn, 2003, p xvii, p. 578 Search PubMed.
- F. Brown and M. Matsuoka, Effect of Adsorbed Surface Layers on Second-Harmonic Light from Silver, Phys. Rev., 1969, 185, 985–987 CrossRef CAS.
- Y. B. Band, Light and Matter : Electromagnetism, Optics, Spectroscopy and Lasers, John Wiley, Chichester, Hoboken, NJ, 2006, p xv, p. 640 Search PubMed.
- N. Okada, Y. Hamanaka, A. Nakamura, I. Pastoriza-Santos and L. M. Liz-Marzan, Linear and Nonlinear Optical Response of Silver Nanoprisms: Local Electric Fields of Dipole and Quadrupole Plasmon Resonances, J. Phys. Chem. B, 2004, 108, 8751–8755 CrossRef CAS.
- N. Faraji, W. M. M. Yunus, A. Kharazmi, E. Saion, M. Shahmiri and N. Tamchek, Synthesis, Characterization and Nonlinear Optical Properties of Silver/Pva Nanocomposites, J. Eur. Opt. Soc., 2012, 7, 12040 CrossRef.
- L. A. Gomez, C. B. de Araujo, A. M. B. Silva and A. Galembeck, Influence of Stabilizing Agents on the Nonlinear Susceptibility of Silver Nanoparticles, J. Opt. Soc. Am. B, 2007, 24, 2136–2140 CrossRef CAS.
- J. Renger, R. Quidant and L. Novotny, Enhanced Nonlinear Response from Metal Surfaces, Opt. Express, 2011, 19, 1777–1785 CrossRef CAS PubMed.
- Z. Hu, J. Autschbach and L. Jensen, Simulation of Resonance Hyper-Rayleigh Scattering of Molecules and Metal Clusters Using a Time-Dependent Density Functional Theory Approach, J. Chem. Phys., 2014, 141, 124305 CrossRef PubMed.
- E. C. Hao, G. C. Schatz, R. C. Johnson and J. T. Hupp, Hyper-Rayleigh Scattering from Silver Nanoparticles, J. Chem. Phys., 2002, 117, 5963–5966 CrossRef CAS PubMed.
- H. Reis, Problems in the Comparison of Theoretical and Experimental Hyperpolarizabilities Revisited, J. Chem. Phys., 2006, 125, 014506 CrossRef CAS PubMed.
- Y. Hamanaka, N. Hayashi, A. Nakamura and S. Omi, Dispersion of Third-Order Nonlinear Optical Susceptibility of Silver Nanocrystal-Glass Composites, J. Lumin., 2000, 87–89, 859–861 CrossRef CAS.
- A. M. Moran, J. H. Sung, E. M. Hicks, R. P. Van Duyne and K. G. Spears, Second Harmonic Excitation Spectroscopy of Silver Nanoparticle Arrays, J. Phys. Chem. B, 2005, 109, 4501–4506 CrossRef CAS PubMed.
- D. Faccio, P. Di Trapani, E. Borsella, F. Gonella, P. Mazzoldi and A. M. Malvezzi, Measurement of the Third-Order Nonlinear Susceptibility of Ag Nanoparticles in Glass in a Wide Spectral Range, Europhys. Lett., 1998, 43, 213–218 CrossRef CAS.
- S. M. Asiala and Z. D. Schultz, Characterization of Hotspots in a Highly Enhancing SERS Substrate, Analyst, 2011, 136, 4472–4479 RSC.
- S. H. Brewer and S. Franzen, A Quantitative Theory and Computational Approach for the Vibrational Stark Effect, J. Chem. Phys., 2003, 119, 851–858 CrossRef CAS PubMed.
- S. S. Andrews and S. G. Boxer, Vibrational Stark Effects of Nitriles I. Methods and Experimental Results, J. Phys. Chem. A, 2000, 104, 11853–11863 CrossRef CAS.
- S. Lal, N. K. Grady, G. P. Goodrich and N. J. Halas, Profiling the near Field of a Plasmonic Nanoparticle with Raman-Based Molecular Rulers, Nano Lett., 2006, 6, 2338–2343 CrossRef CAS PubMed.
- J. A. Dieringer, A. D. McFarland, N. C. Shah, D. A. Stuart, A. V. Whitney, C. R. Yonzon, M. A. Young, X. Zhang and R. P. Van Duyne, Surface Enhanced Raman Spectroscopy: New Materials, Concepts, Characterization Tools, and Applications, Faraday Discuss., 2006, 132, 9–26 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S-1–S-5, are freely available. See DOI: 10.1039/c5sc01265a |
| This journal is © The Royal Society of Chemistry 2015 |