
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification, classification, and signal amplification capabilities of high-turnover gas binding hosts in ultra-sensitive NMR†
Martin
Kunth
 a,
Christopher
Witte
a,
Andreas
Hennig
b and
Leif
Schröder
*a
a,
Christopher
Witte
a,
Andreas
Hennig
b and
Leif
Schröder
*a
aERC Project BiosensorImaging, Leibniz-Institut für Molekulare Pharmakologie (FMP), 13125 Berlin, Germany. E-mail: lschroeder@fmp-berlin.de; Tel: +49 30 947 93 121
bJacobs University Bremen, Department of Life Sciences and Chemistry, Campus Ring 1, 28759 Bremen, Germany
First published on 6th July 2015
Abstract
Nuclear Magnetic Resonance (NMR) can be a powerful tool for investigating exchange kinetics of host–guest interactions in solution. Beyond conventional direct NMR detection, radiofrequency (RF) saturation transfer can be used to enhance the study of such chemical exchange or to enable signal amplification from a dilute host. However, systems that are both dilute and labile (fast dissociation/re-association) impose specific challenges to direct as well as saturation transfer detection. Here we investigate host–guest systems under previously inaccessible conditions using saturation transfer techniques in combination with hyperpolarized nuclei and quantitative evaluation under different RF exposure. We further use that information to illustrate the consequences for signal amplification capabilities and correct interpretation of observed signal contrast from comparative exchange data of different types of hosts. In particular, we compare binding of xenon (Xe) to cucurbit[6]uril (CB6) with binding to cryptophane-A monoacid (CrA) in water as two different model systems. The Xe complexation with CB6 is extremely difficult to access by conventional NMR due to its low water solubility. We successfully quantified the exchange kinetics of this system and found that the absence of Xe signals related to encapsulated Xe in conventional hyperpolarized 129Xe NMR is due to line broadening and not due to low binding. By introducing a measure for the gas turnover during constant association–dissociation, we demonstrate that the signal amplification from a dilute pool of CB6 can turn this host into a very powerful contrast agent for Xe MRI applications (100-fold more efficient than cryptophane). However, labile systems only provide improved signal amplification for suitable saturation conditions and otherwise become disadvantageous. The method is applicable to many hosts where Xe is a suitable spy nucleus to probe for non-covalent interactions and should foster reinvestigation of several systems to delineate true absence of interaction from labile complex formation.
Introduction
The noble gas xenon (Xe) undergoes non-covalent interactions with hydrophobic cavities of natural biomacromolecular and artificial supramolecular structures.1–4 It has consequently been used in a large variety of contexts, for example, to explore protein surfaces5–9 and the structure of bacterial spores,10 to study gas diffusion through nanotubes11 and gas adsorption by metal–organic frameworks,12–14 as well as a probe for understanding the driving forces of supramolecular host–guest systems.15,16 In addition, biosensing using 129Xe nuclear magnetic resonance (NMR) is an emerging field with great potential for medically important applications17–19 and has led to the development of Xe contrast agents for magnetic resonance imaging (MRI). Hence, the characterization of such Xe–host interactions is extremely important to supramolecular chemists, materials scientists and biochemists, as it allows access to the driving forces of non-covalent interactions.Popular methods to characterize Xe binding include X-ray crystallography and isothermal titration calorimetry (ITC). They provide structural information in the solid state and thermodynamic parameters in solutions, respectively, and thus complement each other. An alternative method to study both dynamic and structural aspects of Xe host–guest systems in solution is 129Xe NMR spectroscopy. The high polarizability of the large Xe electron cloud renders the chemical shift of the NMR-active 129Xe isotope extremely sensitive to its molecular environment,20 and 129Xe NMR can thus report on small structural changes. Moreover, 129Xe NMR spin labeling allows the study of exchange kinetics and thereby provides dynamic parameters of Xe binding such as binding constants, association and dissociation rate constants. In addition, Xe can be hyperpolarized, which leads to a 105-fold NMR signal enhancement and enables the direct detection of micromolar quantities of Xe-binding hosts.21,22
However, the study of dynamic Xe binding and exchange in aqueous solution by conventional NMR detection often remains problematic. Known binding sites were typically determined from NMR observations under conditions where either (a) complex formation and dissociation is slow enough to yield a sharp NMR resonance of the (at least temporarily) bound Xe or (b) in the case that there is no unique observable peak from the complex, the existence of a complex can be inferred from (small) shifts in the signal of free Xe when high concentrations of the host are present. Utilizing these methods is particularly challenging for hosts with poor solubility. In addition, new labile systems or known hosts under conditions with accelerated Xe exchange might easily be overlooked, even though a high exchange rate might be of considerable interest. For instance, Xe can be used as a probe for oxygen binding pockets related to reactive centers in biological systems but exchange in such systems is expected to be rapid. To overcome these limitations, the Hyper-CEST technique was developed19 by combining hyperpolarized Xe with chemical exchange saturation transfer (CEST).23 The Hyper-CEST method also utilizes the Xe–host as an MRI contrast agent and enhances its detection sensitivity down to the picomolar range using the dynamic nature of the Xe–host interaction;9,24–26 however, it does not per se provide quantitative information about Xe complexation. For example, the recent study of bacterial spores reported in this journal10 used Hyper-CEST to confirm the existence of a Xe complex but detailed quantification of the Xe exchange remained elusive. To address this shortcoming, we recently introduced a concept to comprehensively analyze the Hyper-CEST signal of a host where a clear NMR signal from bound Xe is easy to identify for a system well within the slow exchange regime. Data acquired at varying saturation pulse strengths and durations provide a quantitative method for Hyper-CEST (qHyper-CEST).27
Herein, we quantify the Xe exchange kinetics for two different Xe–host systems, cryptophane-A monoacid (CrA) and cucurbit[6]uril (CB6), with unprecedented sensitivity in water. Thus, we demonstrate that qHyper-CEST is not only applicable in the slow regime (Xe in CrA), but becomes even more valuable in the intermediate regime (Xe in CB6). In particular, the interaction of Xe with CB6 is otherwise impractical to access owing to its inherently low water solubility in combination with its labile complex formation, which is a typical property of many gas binding substances. By characterizing the previously unquantified, labile and barely soluble version of the Xe–CB6 system we demonstrate that such systems are indeed detectable by NMR and, as a consequence, we believe that other Xe–host systems may be worth reinvestigating. We then compare these two systems with each other. To enhance the discussion, we additionally include a third Xe–host system that has been previously quantified (CrA in dimethyl sulfoxide, DMSO, which is in the slow exchange regime).27 To facilitate an objective comparison between these different systems we propose to rank them according to their gas turnover rate. We find that in water, CB6 exhibits a ca. 100-fold increased gas turnover rate in comparison to CrA and is therefore, under appropriate conditions, a significantly more sensitive Hyper-CEST biosensor and superior Xe-MRI contrast agent. Finally, we discuss which saturation conditions are needed to fully realize the potential of the superior gas turnover rate of CB6.
Results and discussion
Quantifying exchange kinetics with qHyper-CEST
In Hyper-CEST, an abundant pool (free Xe in solution, pool A, see Fig. 1) is used to detect a dilute pool (bound Xe, pool B) by application of a (selective) radiofrequency (RF) saturation pulse with a specific strength, B1, and for a certain duration, tsat, on-resonant with pool B. In this study, we used continuous-wave (cw) saturation. The induced spin depolarization is transferred to pool A via chemical exchange, which results in a cumulative decrease in the large detection pool A. A reference measurement with off-resonant RF saturation reveals the intensity of the saturation transfer. The qHyper-CEST technique takes advantage of the spectral dimension by iterative variation of the saturation frequency to cover a whole range of chemical shifts. The normalized Hyper-CEST signal with respect to the saturation pulse frequency is referred to as a z-spectrum. In qHyper-CEST, multiple z-spectra acquired with different saturation pulse conditions are fit with the full Hyper-CEST (FHC) solution28 which is an analytical solution of the Bloch–McConnell equations.29,30 This enables the sensitive and simultaneous quantification of Xe exchange kinetic and binding parameters such as: the ratio of bound to free Xe, fB, the Xe exchange rate, kBA, the chemical shifts of free and bound Xe, δA,B, the Xe association (binding) constant, KA, and the Xe host occupancy, β (see Table 1).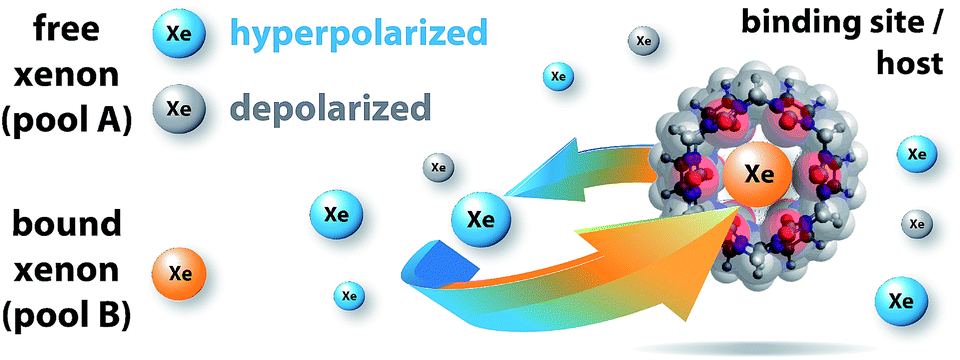 | ||
| Fig. 1 Probing reversible, labile binding in a molecular cavity with xenon: free xenon atoms (pool A, where blue indicates hyperpolarized Xe and gray indicates depolarized Xe) undergo constant exchange with the binding site/host (i.e., cucurbit[6]uril, CB6; transparent overlay of the ball-stick-model of the molecule with its van der Waals radius representation; molecular modeling in Fig. S1 in the ESI†). The NMR signal of bound Xe shows a remarkably large chemical shift (indicated by orange xenon atoms; pool B). The host geometry with the two opposing portals facilitates fast dissociation of the complex. This causes detrimental line broadening, precluding conventional NMR detection. | ||
| Solvent | [Xe]a (μM) | host | [hosttot] (μM) | Δδ (ppm) | f B (10−4) | k BA (s−1) | β (%) | K A (M−1) | [hostocc] (μM) | β k BA (% s−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Calculation given in the Experimental Section. b As given by eqn (3) in ref. 27. c As given by eqn (4) in ref. 27. | ||||||||||
| Water | 390 | CB6 | 3.4 | −96.1 ± 0.1 | 43 ± 1 | 2100 ± 300 | 49 | 2500 ± 400 | 1.7 | 1029 |
| Water | 390 | CrA | 11 | −132.06 ± 0.02 | 70 ± 11 | 38 ± 6 | 29 | 850 ± 250 | 3.2 | 11 |
| DMSO | 2340 | CrA | 50 | −166.37 ± 0.04 | 18 ± 1 | 250 ± 130 | 9 | 38 ± 4 | 4.5 | 23 |
Xenon exchange kinetics for cryptophane-A in water
One prominent synthetic host often used in 129Xe NMR studies is CrA. Its Xe binding constant has been measured using ITC31 and the kinetics has been studied by direct detection of the caged Xe in organic solvent32,33 and in water.18,34 We have recently used CrA in an organic solvent to validate qHyper-CEST,27 and now apply quantitative saturation transfer to this system in water to have a reference for the subsequently studied more labile system and to illustrate the consequences in saturation transfer performance under different RF exposures. A direct 129Xe NMR spectrum (see Fig. S2a in the ESI†) and qHyper-CEST analysis for [CrA] = 11 μM can be found in the S2 section in ESI†. The determined exchange rate kBA of (38 ± 6) s−1 is in excellent agreement with previous studies.18,34 As such, the Xe–CrA complexation in water is well within the slow exchange regime (kBA/Δω = 0.0024 ≪ 1). The results for the other parameters are listed in Table 1.Xenon exchange kinetics for cucurbit[6]uril in water
Another class of macrocycles where 129Xe NMR spectroscopy can be used to probe Xe–host binding are cucurbit[n]urils.35–38 Cucurbiturils are highly important in the fields of molecular self-assembly and nano-technology.39 Due to their nontoxicity40 new biological applications are currently emerging for cucurbiturils, such as drug carriers, as molecular recognition units for insulin or β-amyloid fibers,41 as well as pH-responsive supramolecular nanovalves42 or in fluorescence assays.43,44 A cucurbit[n]uril homologue with promising Xe binding capabilities is CB6, which, however, suffers from a low water solubility (<30 μM (ref. 45)). Without qHyper-CEST, high contents of inorganic salt are compulsory to solubilize sufficient amounts for CB6 detection. This complicates the quantification of actual binding constants45 because of competitive binding of cations to the carbonyl portals of the macrocycle. However, we now demonstrate that reversible occupation of the cavity happens already at fairly low concentrations.To exemplify, we recorded a direct 129Xe NMR spectrum of CB6 (3.4 μM) with 64 signal averages in pure water, which, contrary to CrA at comparable concentration, shows no signal from the Xe–CB6 complex (Fig. 2a). In contrast, Hyper-CEST z-spectra at varying saturation pulse conditions obtained with CB6 at the same concentration clearly revealed a distinct signature of the Xe–CB6 complex at −95.6 ppm upfield from free Xe (Fig. 2b), which facilitated the corresponding qHyper-CEST analysis (Table 1). This clearly demonstrates that studying hosts at the low concentrations used here may be misleading with direct NMR and easily lead to the wrong assumption of no complexation.
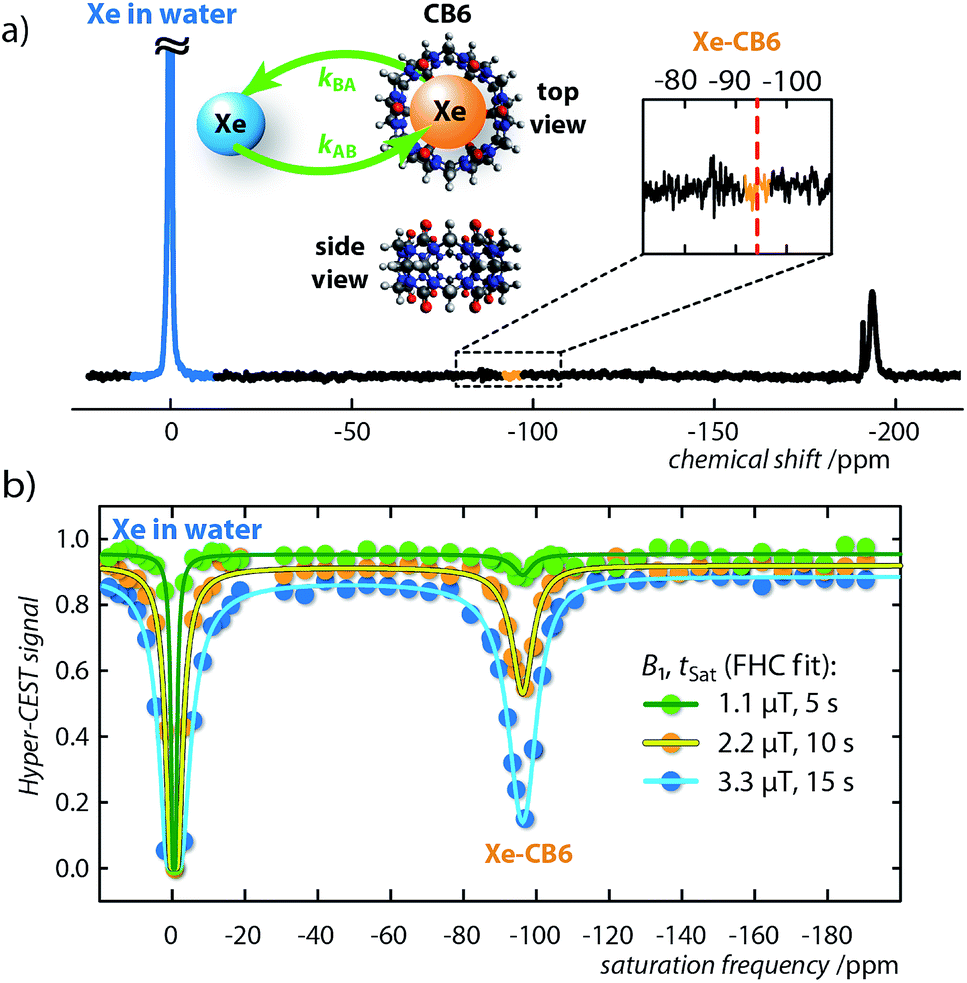 | ||
| Fig. 2 Direct and indirect (Hyper-CEST) 129Xe NMR measurements for cucurbit[6]uril (CB6) at a concentration of 3.4 μM dissolved in pure water. (a) 129Xe NMR spectrum with 64 averages at T = 295 K. Retrospectively, the Xe–CB6 resonance is expected to appear at ca. δB = −95 ppm (red dashed line). The insert shows the CB6 structure as top and side view including the Xe exchange, kAB,BA. (b) Hyper-CEST z-spectra (dots) for continuous-wave (cw) saturation of B1/tsat = {1.1/5 (green), 2.2/10 (orange), 3.3/15 (blue)} μT/s including fitting curves of the full Hyper-CEST (FHC) solution (solid lines); results are listed in Table 1. | ||
Both the exchange rate, kBA, and association constant, KA, obtained by the qHyper-CEST analysis are in good agreement with expectations based on literature results with a water-soluble CB6 derivative, which has been measured by conventional hyperpolarized 129Xe NMR spectroscopy at 103-fold higher concentration.37,46–48 Most strikingly, only the qHyper-CEST analysis was able to reveal that CB6 has a higher occupancy (β = 49%) than CrA (β = 29%) under the given experimental conditions suggesting that Xe binding to CB6 is more efficient than to CrA in pure water where the portals are freely accessible. Moreover, the Xe–CB6 system enters the intermediate exchange regime on the NMR time scale (kBA/Δω = 0.2 < 1; the corresponding value for CrA in water is 100-fold lower and in the slow regime), which should additionally contribute to a higher Hyper-CEST signal compared to CrA. As a consequence, the signal of bound Xe in the direct 129Xe NMR spectrum is most likely only below the noise level because of extreme line broadening and not because of insufficient binding (for more details see section S3 in the ESI†).
Overall, we believe that it might be worthwhile to revisit some previously studied host systems with qHyper-CEST, since their performance may have been similarly overlooked as is the case for CB6. This extends also to systems where Xe is used as a probe for other guests by competitive binding studies. Our findings apply in particular to numerous low binding, but rapidly exchanging host–guest systems, in which Xe NMR was so far restricted to organic solvents (or at least admixtures thereof) to achieve sufficient signal. Since results from organic solvents have limited transferability for conditions in aqueous solution,27,49 this corroborates the strong motivation for the use of carefully designed Hyper-CEST, which now provides the sensitivity for the re-investigation of labile/dilute Xe–host systems under more realistic conditions.
Comparison of supramolecular Xe-binding hosts
In addition to the optimization of the saturation pulse conditions50 the choice of the Xe–host can also greatly influence the sensitivity of Hyper-CEST detection. Intuitively, the saturation transfer increases by both a large number of Xe atoms continuously moving through the host during the saturation period, i.e., a high Xe exchange rate, kBA, and moreover, a large amount of Xe atoms that bind in chemical equilibrium to the hosts, i.e., a high Xe association constant, KA. Unfortunately, an increase in one parameter is often associated with a decrease in the other and, as a consequence, make the sensitivity improvement insignificant. Consider for example the Xe exchange kinetics for CrA in the two solvents water and previously quantified dimethyl sulfoxide (DMSO; Fig. S4 in the ESI†)27 in Table 1. While the Xe exchange rate can be improved by more than 6-fold when switching from water to DMSO, the binding constant, in contrast, decreases by a factor of 22. Thus, the Xe exchange rate of CrA improves at the cost of the binding constant. The question of which system provides superior 129Xe depolarization becomes even more complex considering that in DMSO, 6 times more free Xe atoms are in solution (Table 1) and the manipulation of this large signal via saturation transfer becomes less sensitive. Moreover, as a surprising result, we found that CB6 in pure water has both a superior Xe exchange rate of, kBA = 2100 s−1, and a superior Xe binding constant, KA = 2500 M−1.To allow an objective comparison of these systems in terms of their Hyper-CEST performance and to specifically classify and engineer Xe–host systems with highly optimized properties for Hyper-CEST detection, we propose to use the maximum depolarization rate per host molecule for a given Xe concentration, i.e., the gas turnover rate, β kBA, as an appropriate measure for the performance of a given Xe–host system (see Table 1; S5 in the ESI†). Here, the host occupancy, β, can be related to the binding constant, KA = [Xe]−1{β/(1 − β)}.27 The gas turnover rate gives the maximum possible Hyper-CEST effect for an infinitely strong saturation pulse. In terms of this measure, CB6 is 100 times more efficient than CrA. Hence, it has the potential to be a much better Hyper-CEST agent. Yet the question remains, if CB6 has superior Hyper-CEST performance under all saturation conditions?
Signal amplification strategies
Care has to be taken to consider what saturation parameters and Xe host were used when interpreting Hyper-CEST contrast. Only saturation pulses that are strong enough to depolarize the Xe magnetization during the rather short residence time within CB6 will fully take advantage of its signal amplification potential (see Fig. S5a in the ESI†). Having said that, this aspect at the same time has consequences for the image contrast similar to the film speed in optical detection but with a somewhat inverse behavior: efficient amplification build-up at high RF “exposure” comes along with poor sensitivity at low exposure (for 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Xe–host complexation and occupancy of ≤ 100 %). To illustrate this, we compare the gas turnover of two dilute host structures side by side in an inhomogeneous setup. Two solutions were prepared and studied under different exposure conditions: one of CB6 and the other CrA (both at [CB6] = [CrA] = 12.9 μM). These solutions were placed into separate, concentric compartments, CrA in the inner compartment and CB6 in the outer compartment (Fig. 3a) and studied by MRI. As can be seen (Fig. 3b), when increasing the saturation pulse strength from 5.5 μT to 33.3 μT (both with 2 s saturation duration) there is almost no change in the CrA Hyper-CEST effect. Due to its slower exchange rate (Table 1) CrA has reached its intrinsic depolarization maximum (i.e., 60 % Hyper-CEST effect). In contrast, with its faster exchange, there is a significant amplification in the CB6 Hyper-CEST effect as the saturation strength is increased, namely from < 30 % to ∼ 100 % (Fig. 3c). Best comparison of the gas turnover and the related signal amplification is possible for high RF exposure. It should, however, be noted that CrA can still remain the host of choice, if saturation pulse strength is limited, e.g., due to specific absorption rate concerns, (Fig. S5a in the ESI†). Weak pulses are only capable of saturating Xe with longer residence times, as is the case for the slower exchange of the Xe–CrA system. In such cases, the Hyper-CEST performance as a function of the saturation pulse strength, B1, should also be considered (Fig. S5a in the ESI†).
:1 Xe–host complexation and occupancy of ≤ 100 %). To illustrate this, we compare the gas turnover of two dilute host structures side by side in an inhomogeneous setup. Two solutions were prepared and studied under different exposure conditions: one of CB6 and the other CrA (both at [CB6] = [CrA] = 12.9 μM). These solutions were placed into separate, concentric compartments, CrA in the inner compartment and CB6 in the outer compartment (Fig. 3a) and studied by MRI. As can be seen (Fig. 3b), when increasing the saturation pulse strength from 5.5 μT to 33.3 μT (both with 2 s saturation duration) there is almost no change in the CrA Hyper-CEST effect. Due to its slower exchange rate (Table 1) CrA has reached its intrinsic depolarization maximum (i.e., 60 % Hyper-CEST effect). In contrast, with its faster exchange, there is a significant amplification in the CB6 Hyper-CEST effect as the saturation strength is increased, namely from < 30 % to ∼ 100 % (Fig. 3c). Best comparison of the gas turnover and the related signal amplification is possible for high RF exposure. It should, however, be noted that CrA can still remain the host of choice, if saturation pulse strength is limited, e.g., due to specific absorption rate concerns, (Fig. S5a in the ESI†). Weak pulses are only capable of saturating Xe with longer residence times, as is the case for the slower exchange of the Xe–CrA system. In such cases, the Hyper-CEST performance as a function of the saturation pulse strength, B1, should also be considered (Fig. S5a in the ESI†).
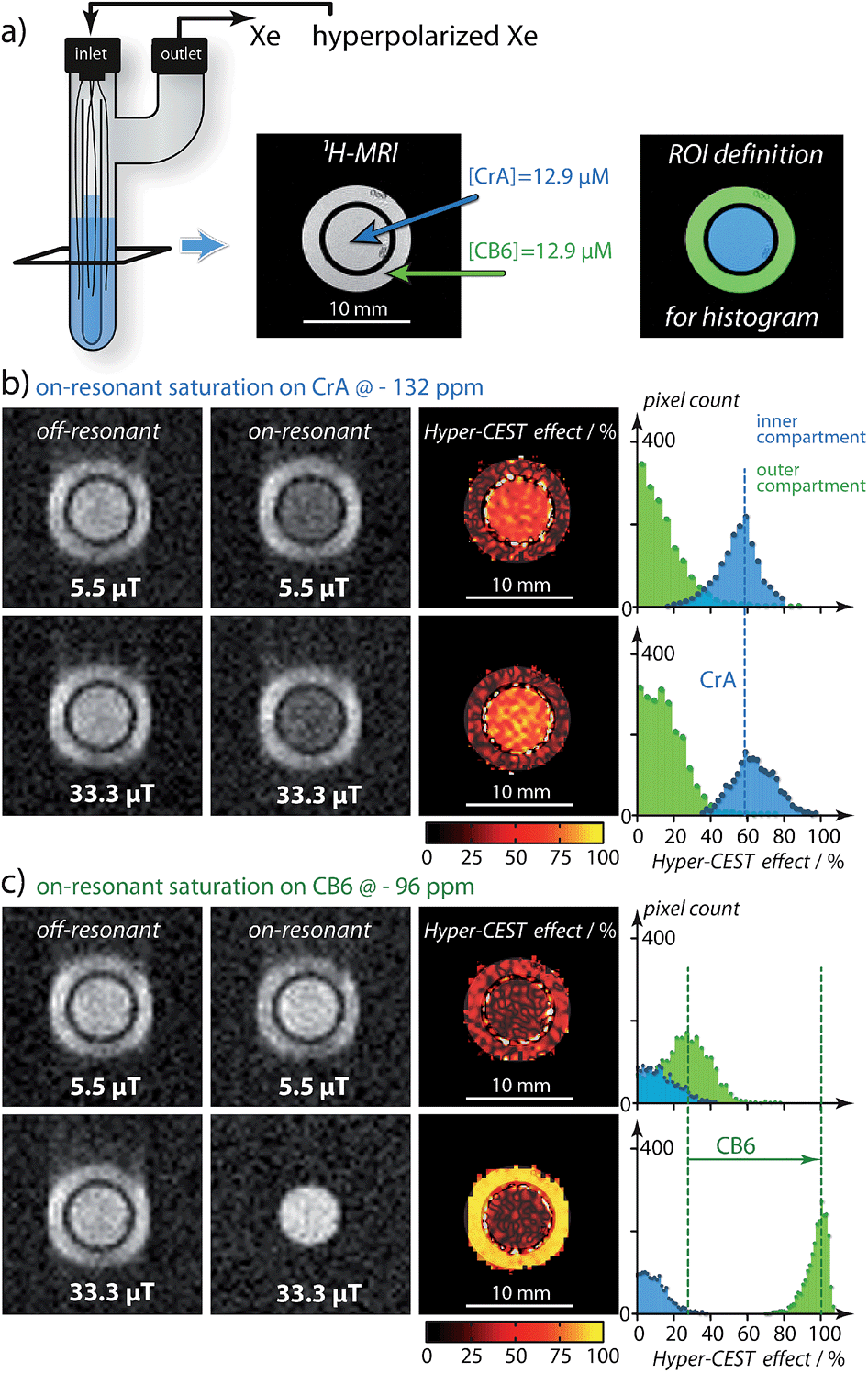 | ||
| Fig. 3 Hyper-CEST effect mapping for equal concentrations of [CB6] = [CrA] = 12.9 μT in water at T = 295 K. (a) shows the proton (1H)-MRI as a cross-section (tilted black square) of the double bubbling phantom and the region-of-interest (ROI) definition for histogram analysis. (b) Hyper-CEST effect maps of CrA for 5.5 μT (“low RF exposure”) and 33.3 μT (“high RF exposure”) both for 2 s of cw saturation calculated as the difference of the on-resonant image (saturation at − 132 ppm) and the off-resonant image (saturation at + 132 ppm) with respect to the free Xe in solution resonance. The Hyper-CEST effect of 60 % for both RF exposures is significant but unchanged. (c) CB6 Hyper-CEST effect maps for identical RF exposure but the on-resonant image (saturation at − 96 ppm) and the off-resonant image (saturation at + 96 ppm) with respect to the free Xe in solution resonance. Whereas the Hyper-CEST effect for 5.5 μT saturation strength was below 30 %, the stronger saturation resulted in ∼ 100 % Hyper-CEST effect, thus revealing significantly higher gas turnover for the Xe–CB6 complex. The Xe-MR images were acquired with 642 resolution and cubic spline interpolated to 2562. The slight blurring in phase encoding direction originated from faster T2 relaxation with CB6 in the outer compartment. | ||
Conclusion
We demonstrated that a dilute host providing a binding site for labile complex formation with Xe (in the intermediate exchange regime) can be identified and classified by saturation transfer NMR where conventional detection fails. The gas turnover rate is accessible through qHyper-CEST as a highly sensitive method for the comprehensive quantification of Xe exchange kinetics and binding affinities. In particular, we successfully quantified the Xe interaction with CB6 in pure water, a Xe–host system which is extremely difficult to access by conventional NMR owing to the exceedingly low solubility of CB6. This indicated that CB6 can be a much more efficient Hyper-CEST agent than previously presumed, because the absence of a detectable signal from the Xe–CB6 complex in direct NMR precluded its comprehensive characterization. Given proper saturation conditions, CB6 is 100 times more efficient than CrA, which is the most prominent contrast agent used so far in Xe biosensing. We could attribute the superior performance of CB6 to its fast exchange rate, kBA, and high occupancy, β. We additionally suggest that other Xe–host systems are valuable to be reinvestigated, since their performance may have similarly been overlooked, if the combination of a high exchange rate and low solubility led to the absence of a signal in direct Xe NMR. In addition, we provide the gas turnover rate, β kBA, as a simple parameter to classify the constant complex association/dissociation and to assess the prospective Hyper-CEST performance of particular Xe–host systems. This is not restricted to macrocyclic host systems, but will be similarly applicable to other emerging potential contrast agents such as bacterial spores,10 nanodroplets26 or genetically encoded gas vesicles (gas-binding protein nanostructures).9 It is also noteworthy that the different exchange parameters (Table 1) in combination with their determination at low concentrations by Hyper-CEST, paves the way for analytical multiplexing applications by exploiting these parameters as unique fingerprints assignable to different Xe–host systems.Experimental section
Data processing and fitting
All simulations, calculations and fitting routines were implemented and performed in Matlab 7 (The Mathworks, Natick, MA, USA) on a standard desktop PC (64 bit, 8 cores each at 2.80 GHz, 4 GB RAM) as described in ref. 27.Sample preparation
Cucurbit[6]uril (CB6) was synthesized as described.51 The synthesized product was characterized by 1H-NMR and ESI mass spectrometry and agreed with the literature.52 Noteworthy, qHyper-CEST analysis of a commercial CB6 sample gave a binding constant and host occupancy, which was inconsistent with results from literature and those reported herein (S6 in ESI†), which we ascribe to an unknown impurity in the commercial sample such as cations. Cryptophane-A monoacid (CrA, provided by Kang Zhao, Tianjin University, China) samples in water were prepared at room temperature followed by 25 minutes of sonication. Due to the higher solubility of CrA in dimethyl sulfoxide (DMSO), this sample could be readily prepared at room temperature.Hyperpolarization and Xe delivery
Using a continuous-flow (0.35 standard liters per minute (SLM)) custom-designed polarizer53via spin exchange optical pumping with rubidium atoms, circa 25 % Xe spin-hyperpolarization of a Xe gas mix (2/10/88 vol % Xe/N2/He, 129Xe natural abundance: 26.4 %) were obtained. At a total pressure of p = 4.5 atm, the electrons of Rb were excited using a 150 W continuous wave-laser (795 nm, 0.5 nm bandwidth, QPC Lasers, Sylmar, CA, USA). Before signal acquisition, the samples were bubbled with the hyperpolarized Xe gas mixture with the following conditions: DMSO sample: bubbling time 10 s, bubble collapse time 2 s, at a flow rate of 0.1 SLM; CrA in water sample: bubbling time 12 s, bubble collapse time 2 s, at a flow rate of 0.1 SLM; For Fig. 2: bubbling time 7 s, bubble collapse time 3 s, at a flow rate of 0.18 SLM (S7 in ESI†). For Fig. 3: bubbling time 11 s, bubble collapse time 4 s, at a flow rate of 0.07 SLM. We triggered these bubbling delays from the NMR spectrometer. The Xe concentration, assuming Xe saturation, was in DMSO [Xe] = 2340 μM, and in water [Xe] = 390 μM ([Xe] = Γ p Xepc/(0.0254 L mM−1), with the Xe Ostwald solubility coefficient in DMSO, Γ = 0.66 L atm−1, and in water, Γ = 0.11 L atm−1, and Xepc = 0.02). The shot-to-shot noise, corresponding to the reproducibility of the Xe concentration in solution, is < 1 % for our system.54NMR experiments
NMR experiments were performed on a B0 = 9.4 T NMR wide bore spectrometer (Bruker Biospin, Ettlingen, Germany) equipped with gradient coils for imaging and a variable temperature unit which was adjusted to room temperature (T = 295 K ∼ 22 °C) for all samples. A 10 mm inner-diameter double-resonant probe (129Xe and 1H) was used for excitation and detection and a flip angle calibration was performed for all samples. Since the B1 field inhomogeneities can significantly affect the CEST quantification, these must be known. As shown in ref. 27, B1 field inhomogeneities were not significant for our micro imaging system. For the DMSO sample (S4 in ESI†), 129Xe Hyper-CEST data was obtained in form of images using a Hyper-CEST echo-planar imaging24 pulse sequence with the following parameters: Fourier acceleration: 1.68, EPI echo time: 5.7 ms, acquisition time: 19.8 ms, field of view: 20 × 20 mm2, matrix size: 32 × 32; in plane resolution: 625 μm and slice thickness: 20 mm. No smoothing filter was applied to the images. The 129Xe Hyper-CEST images for water were obtained from a single-shot Cartesian rapid acquisition with relaxation enhancement pulse sequence modified for Hyper-CEST, with the following parameters: centric encoding, effective echo time: 12.17 ms, no Fourier acceleration, 90° hermite excitation pulse (length = 3.375 ms, bandwidth = 1600 Hz) and 180° mao refocusing pulse (length = 3.105 ms, bandwidth = 2000 Hz), field of view: 20 × 20 mm2, matrix size: 64 × 64; in plane resolution: 321 μm, slice thickness: 20 mm. The saturation strength and saturation length used varies and are mentioned in the figures. The exchange regime on the NMR time scale, kBA/Δω, was calculated with Δω = Δδ × 10−6 × γ/(2π) B0 = Δδ × 10−6 × 11.77 MHz T−1 × 9.4 T.Acknowledgements
This work has been supported by the European Research Council under the European Community's Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement no. 242710 and the Human Frontier Science Program no. LT000858/2010 C. The authors thank Dr. Jabadurai Jayapaul and Matthias Schnurr for the helpful discussions regarding sample preparation. We gratefully thank the World Molecular Imaging Congress (WMIC) committee for generous support enabling Martin Kunth the presentation of parts of this work at the WMIC meeting 2014 in Seoul, Korea.References
- L. Dubois, P. Da Silva, C. Landon, J. G. Huber, M. Ponchet, F. Vovelle, P. Berthault and H. Desvaux, J. Am. Chem. Soc., 2004, 126, 15738–15746 CrossRef CAS PubMed.
- H. Desvaux, L. Dubois, J. G. Huber, M. L. Quillin, P. Berthault and B. W. Matthews, J. Am. Chem. Soc., 2005, 127, 11676–11683 CrossRef CAS PubMed.
- C. Gröger, A. Möglich, M. Pons, B. Koch, W. Hengstenberg, H. R. Kalbitzer and E. Brunner, J. Am. Chem. Soc., 2003, 125, 8726–8727 CrossRef PubMed.
- J. M. Chambers, P. A. Hill, J. A. Aaron, Z. Han, D. W. Christianson, N. N. Kuzma and I. J. Dmochowski, J. Am. Chem. Soc., 2009, 131, 563–569 CrossRef CAS PubMed.
- C. R. Bowers, V. Storhaug, C. E. Webster, J. Bharatam, A. Cottone, R. Gianna, K. Betsey and B. J. Gaffney, J. Am. Chem. Soc., 1999, 121, 9370–9377 CrossRef CAS PubMed.
- S. M. Rubin, M. M. Spence, B. M. Goodson, D. E. Wemmer and A. Pines, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9472–9475 CrossRef CAS PubMed.
- S. M. Rubin, M. M. Spence, I. E. Dimitrov, E. J. Ruiz, A. Pines and D. E. Wemmer, J. Am. Chem. Soc., 2001, 123, 8616–8617 CrossRef CAS.
- S. M. Rubin, S.-Y. Lee, E. Ruiz, A. Pines and D. E. Wemmer, J. Mol. Biol., 2002, 322, 425–440 CrossRef CAS.
- M. G. Shapiro, R. M. Ramirez, L. J. Sperling, G. Sun, J. Sun, A. Pines, D. V. Schaffer and V. S. Bajaj, Nat. Chem., 2014, 6, 629–634 CrossRef CAS PubMed.
- Y. Bai, Y. Wang, M. Goulian, A. Driks and I. J. Dmochowski, Chem. Sci., 2014, 5, 3197–3203 RSC.
- C.-Y. Cheng and C. R. Bowers, J. Am. Chem. Soc., 2007, 129, 13997–14002 CrossRef CAS PubMed.
- S. Pawsey, I. Moudrakovski, J. Ripmeester, L.-Q. Wang, G. J. Exarhos, J. L. C. Rowsell and O. M. Yaghi, J. Phys. Chem. C, 2007, 111, 6060–6067 CAS.
- J. J. Perry, S. L. Teich-McGoldrick, S. T. Meek, J. A. Greathouse, M. Haranczyk and M. D. Allendorf, J. Phys. Chem. C, 2014, 118, 11685–11698 CAS.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- T. Adiri, D. Marciano and Y. Cohen, Chem. Commun., 2013, 49, 7082–7084 RSC.
- J. Roukala, J. Zhu, C. Giri, K. Rissanen, P. Lantto and V.-V. A. Telkki, J. Am. Chem. Soc., 2015, 137, 2464–2467 CrossRef CAS PubMed.
- M. M. Spence, S. M. Rubin, I. E. Dimitrov, E. J. Ruiz, D. E. Wemmer, A. Pines, S. Q. Yao, F. Tian and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10654–10657 CrossRef CAS PubMed.
- M. M. Spence, E. J. Ruiz, S. M. Rubin, T. J. Lowery, N. Winssinger, P. G. Schultz, D. E. Wemmer and A. Pines, J. Am. Chem. Soc., 2004, 126, 15287–15294 CrossRef CAS PubMed.
- L. Schröder, T. J. Lowery, C. Hilty, D. E. Wemmer and A. Pines, Science, 2006, 314, 446–449 CrossRef PubMed.
- B. M. Goodson, J. Magn. Reson., 2002, 155, 157–216 CrossRef CAS PubMed.
- L. Schröder, Phys. Med., 2013, 29, 3–16 CrossRef PubMed.
- P. Nikolaou, A. M. Coffey, L. L. Walkup, B. M. Gust, N. Whiting, H. Newton, S. Barcus, I. Muradyan, M. Dabaghyan, G. D. Moroz, M. S. Rosen, S. Patz, M. J. Barlow, E. Y. Chekmenev and B. M. Goodson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14150–14155 CrossRef CAS PubMed.
- K. Ward, A. Aletras and R. Balaban, J. Magn. Reson., 2000, 143, 79–87 CrossRef CAS PubMed.
- M. Kunth, J. Döpfert, C. Witte, F. Rossella and L. Schröder, Angew. Chem., Int. Ed., 2012, 51, 8217–8220 CrossRef CAS PubMed.
- Y. Bai, P. A. Hill and I. J. Dmochowski, Anal. Chem., 2012, 84, 9935–9941 CrossRef CAS PubMed.
- T. K. Stevens, R. M. Ramirez and A. Pines, J. Am. Chem. Soc., 2013, 135, 9576–9579 CrossRef CAS PubMed.
- M. Kunth, C. Witte and L. Schröder, J. Chem. Phys., 2014, 141, 194202 CrossRef CAS PubMed.
- M. Zaiss, M. Schnurr and P. Bachert, J. Chem. Phys., 2012, 136, 144106 CrossRef PubMed.
- H. M. McConnell, J. Chem. Phys., 1958, 28, 430–431 CrossRef CAS PubMed.
- D. E. Woessner, J. Chem. Phys., 1961, 35, 41–48 CrossRef CAS PubMed.
- P. A. Hill, Q. Wei, R. G. Eckenhoff and I. J. Dmochowski, J. Am. Chem. Soc., 2007, 129, 9262–9263 CrossRef CAS PubMed.
- P. Berthault, J. G. Huber and H. Desvaux, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 55, 35–60 CrossRef CAS PubMed.
- K. Bartik, M. Luhmer, J.-P. Dutasta, A. Collet and J. Reisse, J. Am. Chem. Soc., 1998, 120, 784–791 CrossRef CAS.
- S. Korchak, W. Kilian and L. Mitschang, Chem. Commun., 2015, 51, 1721–1724 RSC.
- Y. Miyahara, K. Abe and T. Inazu, Angew. Chem., Int. Ed., 2002, 41, 3020–3023 CrossRef CAS.
- S. Y. Jon, N. Selvapalam, D. H. Oh, J.-K. Kang, S.-Y. Kim, Y. J. Jeon, J. W. Lee and K. Kim, J. Am. Chem. Soc., 2003, 125, 10186–10187 CrossRef CAS PubMed.
- B. S. Kim, Y. H. Ko, Y. Kim, H. J. Lee, N. Selvapalam, H. C. Lee and K. Kim, Chem. Commun., 2008, 2756–2758 RSC.
- G. Huber, F.-X. Legrand, V. Lewin, D. Baumann, M.-P. Heck and P. Berthault, ChemPhysChem, 2011, 12, 1053–1055 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037–2042 CAS.
- J. M. Chinai, A. B. Taylor, L. M. Ryno, N. D. Hargreaves, C. A. Morris, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2011, 133, 8810–8813 CrossRef CAS PubMed.
- S. Angelos, Y.-W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222–2226 CrossRef CAS PubMed.
- A. Hennig, H. Bakirci and W. M. Nau, Nat. Methods, 2007, 4, 629–632 CrossRef CAS PubMed.
- M. Florea and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 9338–9342 CrossRef CAS PubMed.
- C. Marquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806–5816 CrossRef CAS PubMed.
- M. E. Haouaj, M. Luhmer, Y. H. Ko, K. Kim and K. Bartik, J. Chem. Soc., Perkin Trans. 2, 2001, 804–807 RSC.
- J. Zhao, H.-J. Kim, J. Oh, S.-Y. Kim, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 4233–4235 CrossRef CAS.
- M. I. El-Barghouthi, H. M. Abdel-Halim, F. J. Haj-Ibrahim and K. I. Assaf, Supramol. Chem., 2014, 27, 80–89 CrossRef PubMed.
- K. Bartik, M. Luhmer, S. J. Heyes, R. Ottinger and J. Reisse, J. Magn. Reson., Ser. B, 1995, 109, 164–168 CrossRef CAS.
- M. Kunth, C. Witte and L. Schröder, NMR Biomed., 2015, 28, 601–606 CrossRef CAS PubMed.
- C. Marquez, F. Huang and W. M. Nau, IEEE Trans. NanoBiosci., 2004, 3, 39–45 CrossRef.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- C. Witte, M. Kunth, J. Döpfert, F. Rossella and L. Schröder, J. Visualized Exp., 2012, 67, e4268 Search PubMed.
- C. Witte, M. Kunth, F. Rossella and L. Schröder, J. Chem. Phys., 2014, 140, 084203 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CB6 modeling, Xe–CrA exchange kinetics in water, CB6 line broadening, Xe–CrA exchange kinetics in DMSO, Xe–host properties for efficient Hyper-CEST detection, commercially CB6 sample and the Xe delivery into water and DMSO. See DOI: 10.1039/c5sc01400j |
| This journal is © The Royal Society of Chemistry 2015 |